Week 2 HW: DNA Read, Write, and Edit
Part 1: Benchling & In-silico Gel Art
I created a Benchling account, imported the Lambda phage genome (GenBank J02459, ~48.5 kb), and ran restriction simulations for all seven enzymes. Benchling calculates cut sites and spits out the expected fragment sizes for each digest, which is really all you need to start planning a gel layout.
The design side is where it gets interesting. Paul Vanouse’s Latent Figure Protocol uses the natural variation in fragment sizes across different enzyme digests to compose images — the biology does the art. I used Ronan’s gel simulation tool to quickly preview different lane arrangements without re-running everything in Benchling each time, which saved a lot of back-and-forth. The goal was to pick an enzyme combination and lane order where the resulting band pattern, viewed across lanes, suggests a recognizable shape.
Expected fragment sizes: • EcoRI: 21,226 / 7,421 / 5,804 / 5,643 / 4,878 / 3,530 bp (6 fragments) • HindIII: 23,130 / 9,416 / 6,557 / 4,361 / 2,322 / 2,027 / 564 / 125 bp (8 fragments) • BamHI: 16,841 / 7,233 / 6,770 / 6,527 / 5,626 / 5,505 bp (6 fragments) • KpnI: 6 fragments ranging from ~1,400 to ~14,000 bp • EcoRV: Multiple fragments across several cut sites • SacI: ~4 fragments • SalI: 3 large fragments (only 2 cut sites in Lambda)
Here are some screenshots of the process:
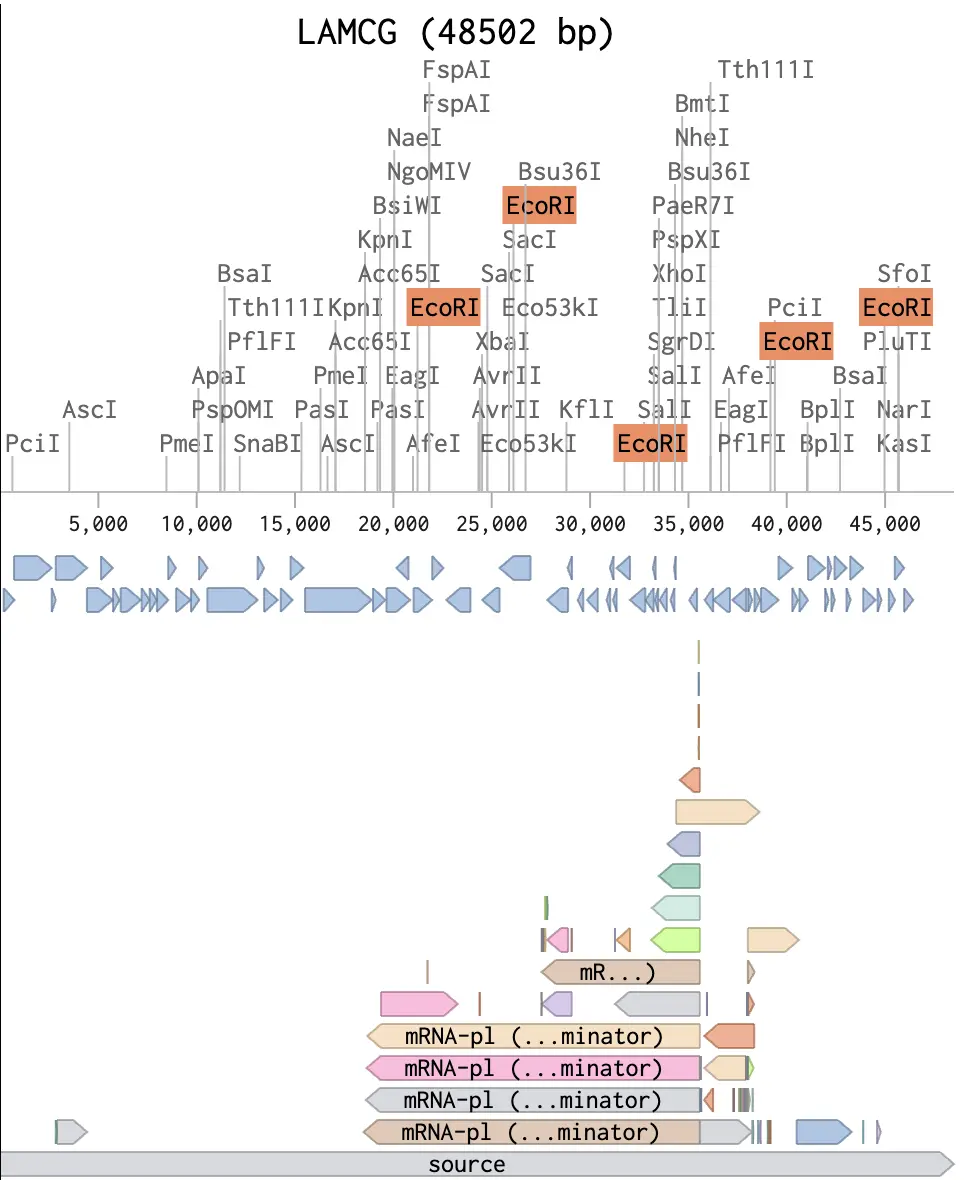
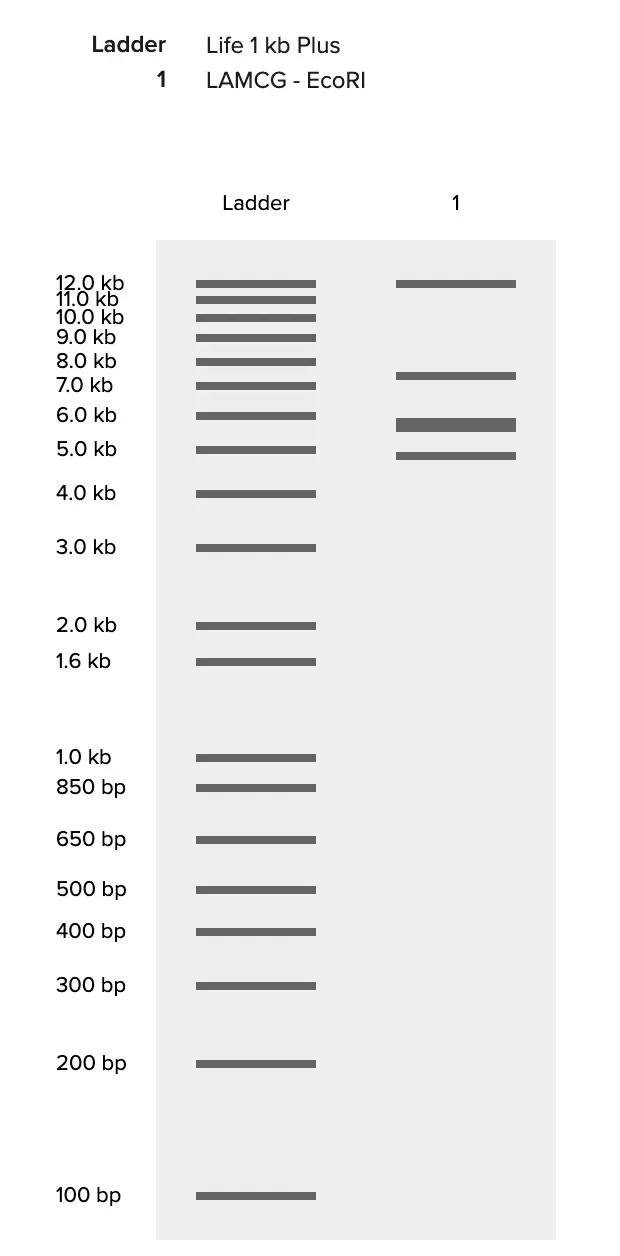
Part 3: DNA Design Challenge
3.1. Choose your protein: Allophycocyanin (APC)
I picked Allophycocyanin (APC) — a deep blue-green light-harvesting protein found in cyanobacteria and red algae. It absorbs at ~652 nm and emits at ~660 nm, it looks striking, and it has an actually interesting backstory: cyanobacteria expressing APC are responsible for a huge chunk of oceanic nitrogen fixation, making them quietly foundational to the global food supply. Spirulina, the supplement, is loaded with phycobiliproteins including APC. It also sees real use as a fluorescent label in flow cytometry. Visually interesting, agriculturally relevant, well-documented — easy choice. Protein sequence (UniProt P00299 | APC_ANASP, alpha chain, Anabaena sp. PCC 7120):
sp|P00299|APC_ANASP Allophycocyanin alpha chain OS=Anabaena sp. (strain PCC 7120) OX=103690 PE=1 SV=2 MSVTKSIVNAADKRSYVLTYVAGGTQMPYEQISQLISESDGLQGAVIEAAISQLDTFNSSRLAAAINGKLNRRNAAAQKIRDTESVYNQLGEPQNKKIASALSLFNSGEPAQLLAEHLLPNAQTSVTESLAAALRQYILAHQAAYVQKME
3.2. Reverse Translate
The central dogma runs DNA → RNA → protein. Reverse translation works backwards from the amino acid sequence to infer a plausible DNA sequence. The wrinkle is that the genetic code is degenerate — most amino acids are encoded by multiple codons — so there’s no single “correct” reverse translation, just many valid options. The native apcA gene sequence from Anabaena sp. (NCBI Gene ID: 1108771) is one of them: ATGAGTGTTACTAAATCTATTGTTAATGCAGCAGATAAGCGTTCGTATGTTCTTACGTACGTTGCAGGTGGCACGCAGATGCCTTATGAACAAATTTCTCAGCTGATTTCGGAGAGTGATGGCTTGCAAGGCGCTGTCATAGAAGCTGCCATCAGCCAGCTAGATACGTTCAACTCGAGCCGCTTGGCCGCCGCAATCAACGGCAAACTTAACCGTCGCAATGCAGCAGCTCAGAAGATCCGTGATACTGAAAGTGTATACAATCAACTCGGCGAACCTCAAAACAAAAAGATTGCTTCGGCTTTGAGCCTGTTCAACTCTGGCGAACCTGCTCAGCTCCTTGCCGAACATCTTCTCCCGAATGCTCAGACGTCTGTTACCGAAAGTCTTGCAGCTGCTTTACGTCAATATATCCTTGCACATCAAGCAGCATATGTTCAGAAGATGGAA
3.3. Codon optimization
Different organisms have strong preferences for which synonymous codons they actually use — codon usage bias. A codon that’s common in cyanobacteria might be rare in human cells, meaning the ribosome stalls, makes errors, or just produces less protein. Swapping in codons the host organism prefers dramatically improves yield and accuracy. It’s one of those steps that’s easy to skip and consistently costs you later. I optimized for human cells because the interesting downstream applications for APC — fluorescent reporter in mammalian cell biology, diagnostic tools, potential therapeutic use — all live in that space. I used the Twist Bioscience Codon Optimization Tool, with BsaI, BsmBI, and BbsI recognition sites removed as required. Human-optimized APC alpha sequence: ATGAGTGTGACCAAGTCCATCGTGAATGCAGCTGACAAGCGCTCCTACGTGCTGACCTACGTGGCTGGCGGCACCCAGATGCCCTACGAGCAGATCTCCCAGCTGATCTCCGAGAGCGACGGCCTGCAGGGCGCTGTGATCGAGGCAGCCATCTCCCAGCTGGACACCTTTAACTCTAGCCGCCTGGCCGCCGCCATCAACGGCAAGCTGAACCGGCGCAATGCCGCCGCCCAGAAGATCCGGGACACCGAGTCCGTGTACAACCAGCTGGGCGAGCCCCAGAACAAGAAGATCGCCTCCGCCCTGAGCCTGTTCAACAGCGGCGAGCCAGCCCAGCTGCTGGCCGAGCACCTGCTGCCCAACGCCCAGACCTCTGTGACCGAGAGCCTGGCCGCCGCCCTGCGGCAGTACATCCTGGCACACCAGGCCGCCTACGTGCAGAAGATGGAG
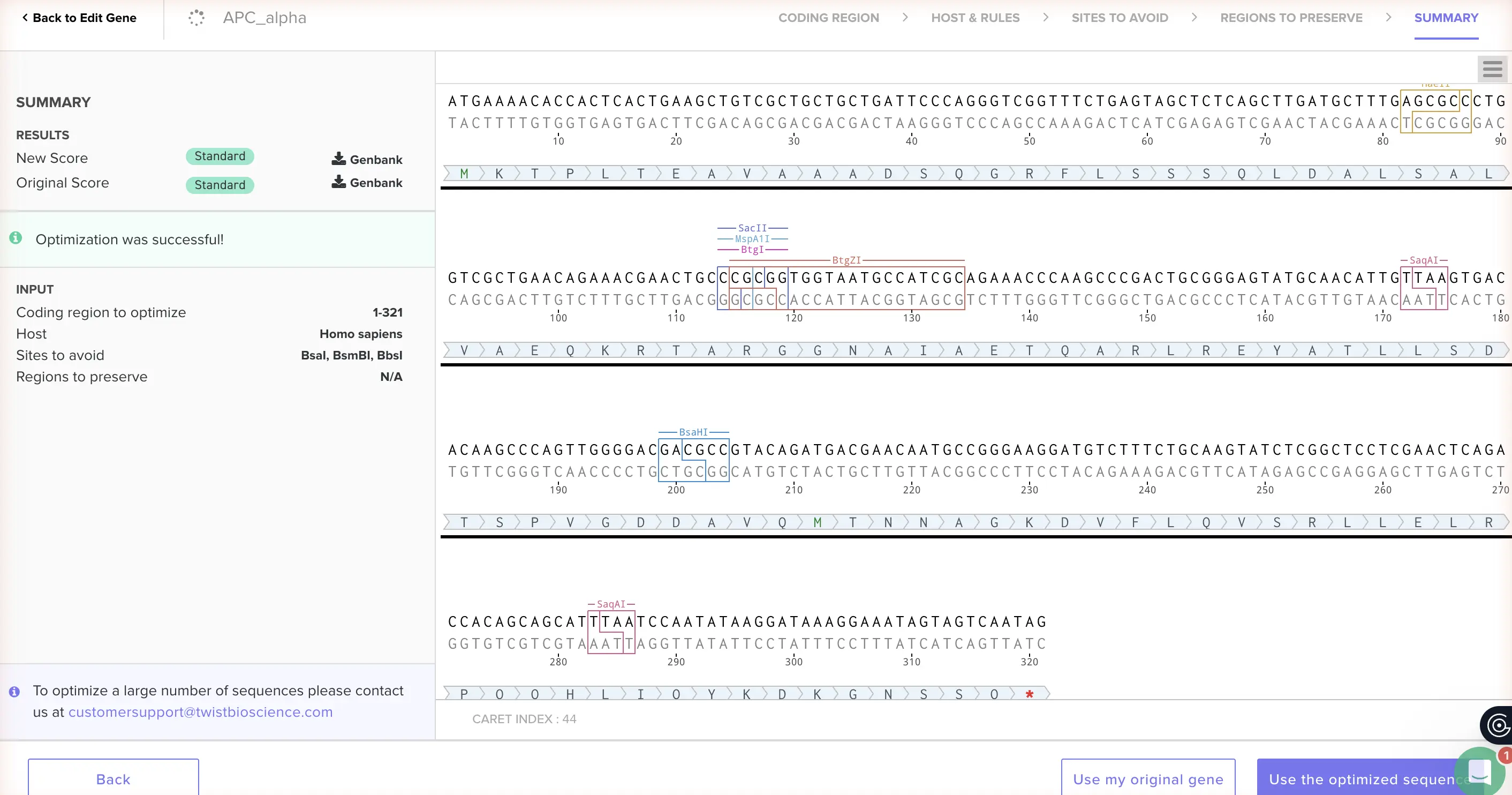
3.4. Your have a ssequence! Now what?
Two main routes: put it in a cell, or skip the cells entirely. Cell-based (in vivo): Clone the optimized sequence into an expression vector (exactly what we’re building in Part 4), transform into E. coli, and let the bacteria do the work. RNA polymerase transcribes the gene into mRNA; ribosomes translate the mRNA into the amino acid chain; the chain folds into APC. Harvest by lysing the cells and purifying via the His-tag. One catch with APC specifically: the protein needs its chromophore (phycocyanobilin) to fluoresce, which requires co-expressing the biosynthetic enzymes HO1 and PcyA — otherwise you get a colorless apoprotein. Cell-free (in vitro): Add the DNA (or mRNA directly) to a cell extract containing ribosomes, tRNA, RNA polymerase, and energy sources — transcription and translation happen in a tube. Faster to set up, works for proteins that are toxic to bacteria, and useful for rapid prototyping. Less scalable for large quantities, but often the better choice early in a project.
3.5. How does it work in nature/biological systems
The information flows one way: DNA gets transcribed into mRNA (every T becomes a U, otherwise same sequence as the coding strand), and the ribosome reads the mRNA in triplet codons, adding one amino acid per codon. AUG = Met = start. Three codons signal stop. For APC alpha: 144 codons, 432 nucleotides, one 144-amino acid protein that folds and binds its chromophore to become a functional light-harvesting unit. One of the more elegant tricks in molecular biology: a single stretch of DNA can encode multiple different proteins. In eukaryotes, alternative splicing swaps exons in and out. In phages like MS2, overlapping reading frames squeeze two completely different proteins out of the same nucleotides just by shifting where the ribosome starts reading. Evolution is efficient.
Part 4: Prepare a Twist DNA Synthesis Order
4.1 Accounts Created accounts at benchling.com and twist-bioscience.com. Benchling for design and annotation; Twist for ordering the synthesized construct.
4.2 Expression Cassette Built the cassette in Benchling as a new linear DNA sequence, annotating each element as I pasted it in. The APC coding sequence replaces sfGFP. Structure: Promoter (BBa_J23106) TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC Constitutive promoter — drives transcription constantly, no inducer needed. RBS (BBa_B0034 + spacers) CATTAAAGAGGAGAAAGGTACC Positions the ribosome for efficient translation initiation. Start Codon ATG Begin. Coding Sequence (APC alpha, human-optimized) [446 bp — see Part 3.3] The codon-optimized APC alpha subunit. 7x His Tag CATCACCATCACCATCATCAC Purification handle. Lets you pull the protein out of cell lysate with nickel-affinity chromatography. Stop Codon TAA End. Terminator (BBa_B0015) CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA Tells RNA polymerase to stop. Prevents transcriptional read-through into whatever’s downstream. Verified the full cassette, confirmed all annotations are in place, and downloaded the FASTA file.
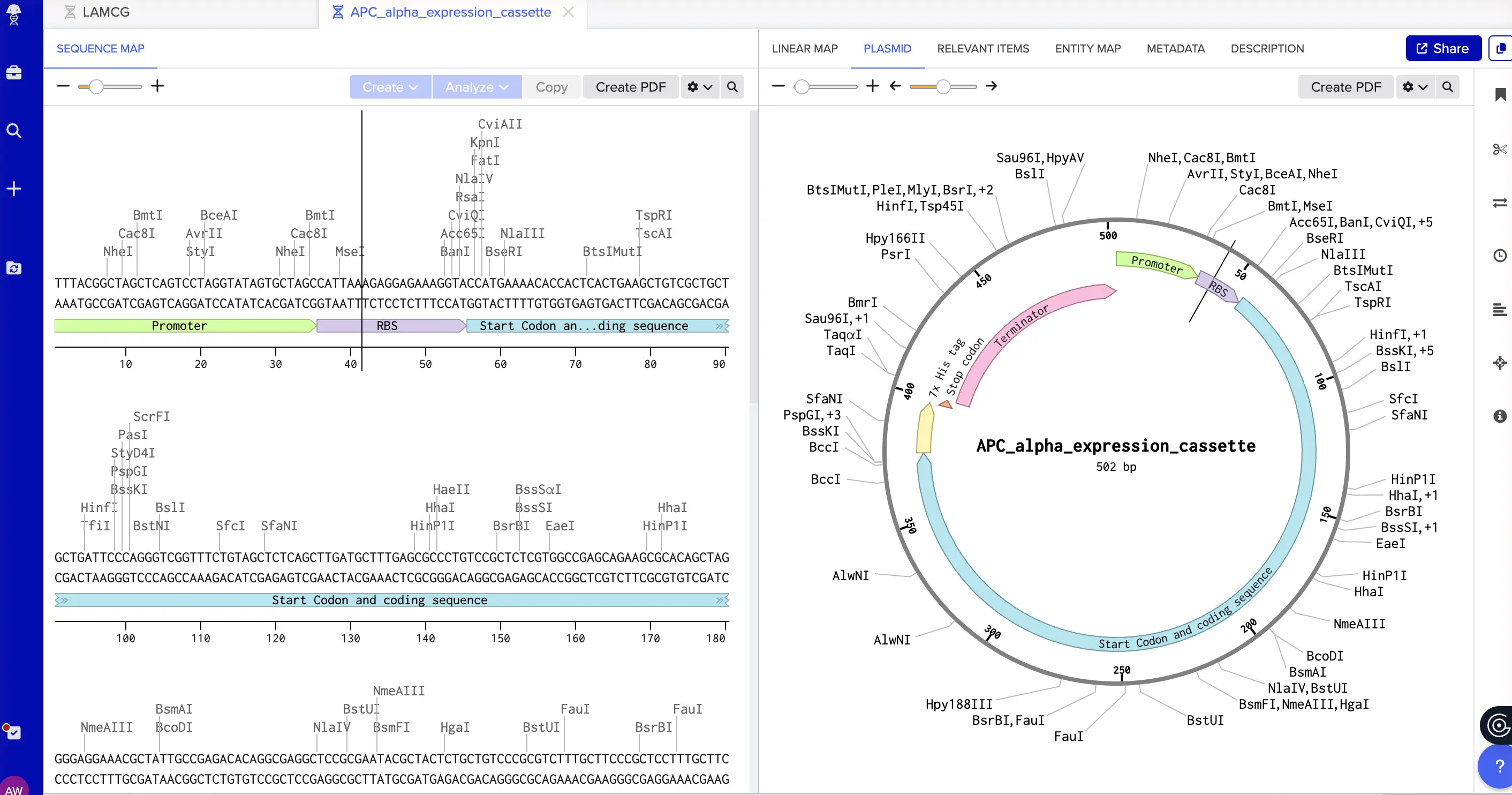
4.3–4.6 Twist Configuration On Twist: Genes → Clonal Genes. Clonal genes arrive as circular plasmid DNA ready for direct transformation into E. coli, which historically saves 1–2 weeks versus gene fragments that need an additional assembly step. Worth choosing unless you have a specific reason not to. Uploaded the FASTA via Nucleotide Sequence upload. For the backbone, I selected pTwist Amp High Copy: ampicillin resistance for selection, high-copy ColE1 origin for good plasmid yield, and a compatible cloning site. Downloaded the full construct as a GenBank file, then imported it back into Benchling. That’s the complete plasmid — pTwist-Amp-APC-alpha — ready for transformation.
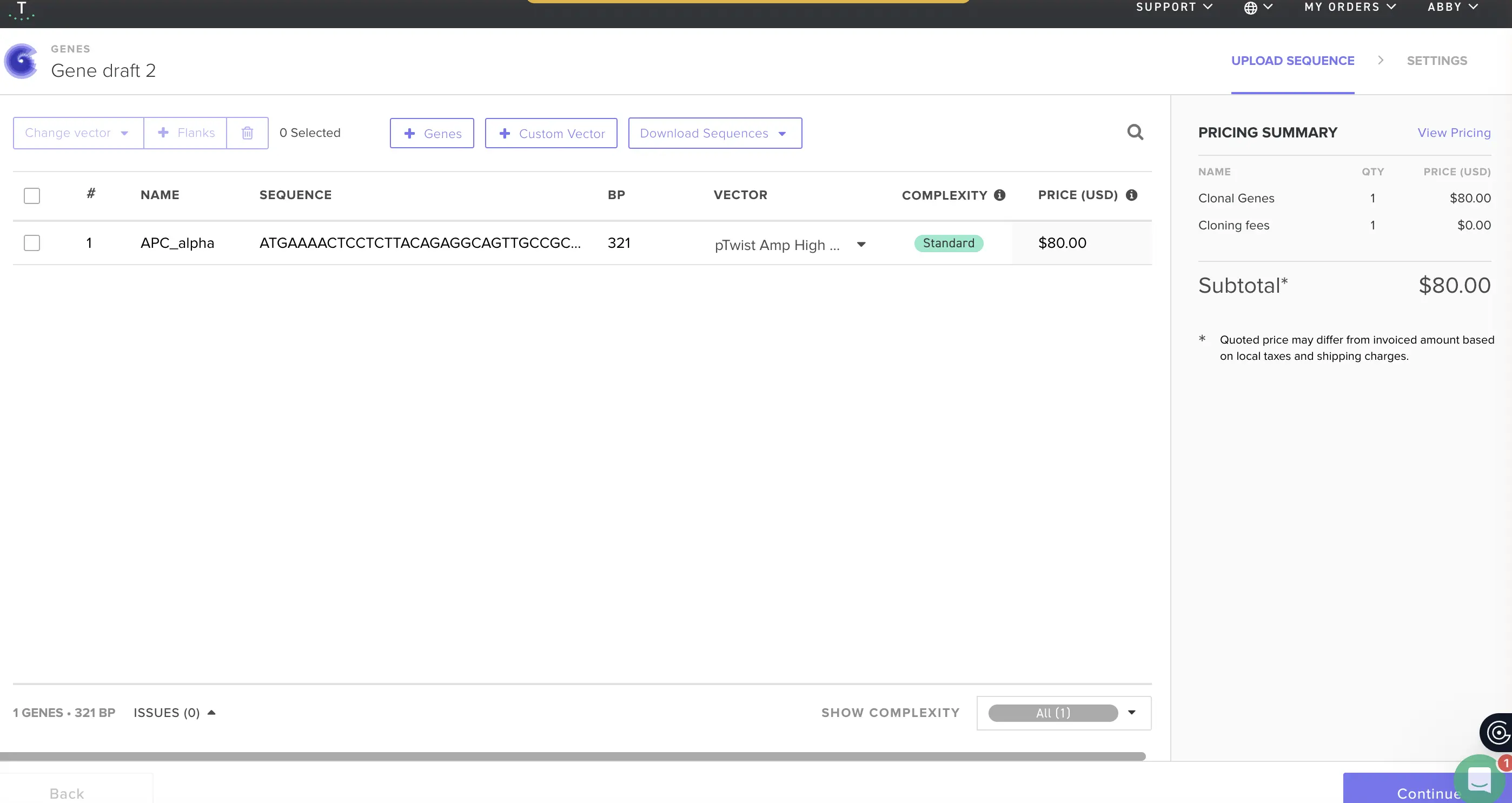
Part 5: DNA Read/Write/Edit
5.1. DNA Read (i) What DNA would you want to sequence and why?
Agricultural soil microbiomes — specifically comparing microbial community composition and functional gene content between conventional monoculture fields and regenerative/polyculture farms. The soil microbiome drives nitrogen cycling, carbon sequestration, disease suppression, and plant-growth promotion, but most of these organisms can’t be cultured in a lab. Metagenomics lets you read the full genetic blueprint of the entire community without culturing anything. Applied to agriculture, it could identify microbial indicators of soil health, track how farming practices shift communities over time, and pinpoint which organisms are responsible for beneficial traits worth protecting or enhancing.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Oxford Nanopore (MinION). Long reads (10–50 kb average, ultra-long reads >100 kb) are essential for metagenomic assembly — short reads from second-gen platforms struggle to resolve closely related strains or span repetitive regions. The MinION is also portable enough to bring to the field, enabling real-time soil monitoring without shipping samples to a sequencing center. Generation: Third-generation. Sequences single native molecules in real time, no PCR amplification required, and can directly detect base modifications like methylation.
Sample prep: • DNA extraction from soil (bead-beating + column purification to remove humic acids that inhibit downstream enzymes) • Optional size selection to enrich for long fragments • End repair + dA-tailing • Ligation of ONT sequencing adapters (which carry the motor protein that threads DNA through the pore) • Load onto flow cell
Base calling: A voltage drives ionic current through protein nanopores in a synthetic membrane. As each DNA strand is threaded through by the motor protein, different bases block the current in characteristic ways. The resulting current trace is converted to sequence by a neural network (Dorado/Guppy). Output: FASTQ files with long reads and per-base quality scores, assembled into contigs for taxonomic and functional annotation.
5.2. DNA Write (i) What DNA would you want to sythesize and why?
A synthetic nitrogen fixation cassette based on the nif gene cluster from Azotobacter vinelandii, for expression in plant chloroplasts. Nitrogen fertilizer is one of the largest agricultural inputs globally and a major source of greenhouse gas emissions and water pollution. Legumes solve this problem by hosting nitrogen-fixing rhizobia in root nodules — if staple crops like wheat or maize could do the same, it would be a massive deal for food security and sustainability. Chloroplasts are a logical target because they’re descended from cyanobacteria, have their own gene expression machinery, and maintain a low-oxygen environment that nitrogenase needs. For a proof-of-concept, I’d start with the core nitrogenase structural genes: nifH (~900 bp), nifD (~1,600 bp), and nifK (~1,500 bp), codon-optimized for Arabidopsis chloroplast expression, plus chloroplast transit peptides and a co-expressed oxygen-protection system (bacterial hemoglobin VHb from Vitreoscilla, which scavenges oxygen to protect the notoriously O₂-sensitive nitrogenase).
(ii) What technology or technologies would you use to perform this DNA sythesis and why?
Twist uses silicon microarray chips to synthesize thousands of oligonucleotides in parallel at low cost per base, then assembles them into full-length genes. It’s the right tool for multi-kilobase constructs where accuracy matters. Essential steps: • Design + codon optimization (removing restriction sites, repeats, problematic secondary structures) • Phosphoramidite chemistry: sequential base addition via coupling, capping, oxidation, deprotection cycles • Synthesis of overlapping ~200 bp oligo fragments covering each gene • Assembly PCR or Gibson Assembly to join fragments into full-length genes • Sequence verification by Sanger sequencing • Delivery as sequence-verified clonal genes
Limitations: practically capped at ~10 kb per construct with high accuracy, so the full 24 kb nif cluster needs to be split across multiple orders and assembled. Error rates (~1 per 1,000 bp) mean sequence verification isn’t optional. Repetitive sequences and extreme GC content remain genuinely hard. Turnaround is 1–3 weeks, which slows down design-test cycles — though costs have dropped enough that this is no longer the bottleneck it once was.
5.3. DNA Edit (i) What DNA would you want to edit and why?
TaGW2 in wheat — a negative regulator of grain size and weight. Loss-of-function mutations produce significantly larger, heavier grains without requiring additional inputs. The same gene in rice (OsGW2) has natural loss-of-function alleles associated with increased grain width, so this is well-validated biology. Wheat is globally responsible for ~20% of human calories; a 10% increase in grain weight per plant compounds into a meaningful food security gain. The catch: wheat is hexaploid (three ancestral genomes, six copies of every chromosome). You have to knock out all three TaGW2 homeologs simultaneously to see the full phenotype. Traditional breeding would take decades of backcrossing to stack three traits. CRISPR does it in one transformation event.
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR-Cas9 with multiplexed gRNAs targeting conserved sequences across the A, B, and D genome copies. The gRNA (~20 bp) base-pairs with the target site; Cas9 cuts both DNA strands; the cell repairs the break via NHEJ, which introduces small indels; indels in a coding exon cause frameshifts that knock out the protein. Three gRNAs, one transformation, all three homeologs disrupted. Steps: • Design 3–4 gRNAs targeting conserved exonic sequences in TaGW2-A, -B, and -D (PAM: NGG) • Deliver as Cas9+gRNA ribonucleoprotein complexes (RNPs) via biolistic bombardment into wheat embryo cells — RNP delivery avoids foreign DNA integration, which simplifies regulatory review • Regenerate plants from transformed tissue • Screen by PCR + sequencing for edits in all three homeologs • Phenotype for grain size, weight, and yield
Limitations: off-target cuts are the main concern, mitigated by careful gRNA design and high-fidelity Cas9 variants. Wheat’s genome is large (~17 Gb) and highly repetitive, making both editing and off-target assessment harder than in model plants. Transformation efficiency in wheat lags behind Arabidopsis. Regulatory status varies significantly by country — some jurisdictions treat CRISPR edits indistinguishable from natural mutations as non-GMO; others require full review regardless.