Week 4 HW: Protein Design- Part 1
Part A: Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat isn’t 100% protein; it’s typically composed of ~20% protein, ~20% fat, and ~60% water. So 500g of meat will approximately contain 100g of protein. Since Daltons is the unit used to measure the weight of small molecules and 1 Dalton is approximately $1.66 \times 10^{-24}$ grams. Since we need to calculate the number of molecules of AAs in our meat, let’s first convert the mass of one AA into grams
$$100\text{ Da} \times (1.66 \times 10^{-24}\text{ g/Da}) = 1.66 \times 10^{-22}\text{ g}$$
Then we divide the total protein mass by the mass per AA
$$\frac{100\text{ g}}{1.66 \times 10^{-22}\text{ g/molecule}} \approx 6.02 \times 10^{23}\text{ molecules}$$
So the total number of AA molecules in 500 g of meat is approximately $6.02 \times 10^{23}$, which is coincidentally (and elegantly) about 1 Mole of amino acids.
- Thus we learn the trick that chemists use when going from molecular weights to weighing scale weights: If a molecule weighs 1 Da then 1 mole of it will weigh 1 gram. In this case since AAs weigh 100 Daltons, 1 Mole of AAs will weigh 100 grams (which is also the amount of protein in 500 grams of meat). Mind is BLOWN!
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Humans dont become cows because we dont incorporate bovine proteins directly. Through digestion, we break down those proteins down into their constituent amino acids. Since the 20 natural amino acids are a universal biological language, our body simply uses them as raw materials to build new proteins based on human DNA sequences.
3. Why are there only 20 natural amino acids?
- There is a theory proposed by Crick that this is a frozen accident; Once life settled on 20, the cost of changing the entire genetic code was too high.
- The other theory says that its the Optimal set; these 20 provide enough variety (acidic, basic, polar, non-polar) to build amost any functional shape. Adding more might have had diminishing returns or caused too many “side reactions”
- As mentioned in the last slide of Joe Jacobsons lecture 20 provides the optimal balance of codon redundancy and diversity.
4. Can you make other non-natural amino acids? Design some new amino acids.
- Every amino acid has the same basic structure: an amino group, a carboxyl group and a side chain (R-group). To design as new one, you typically keep the backbone the same so the ribosomes can still physically link it, but you can change the R-group to do something nature can’t. So lets design a “Metal-Sensing Amino Acid” since I am interested in how metals can be bound to proteins. For this we can use Bipyridine as our modified R-group because it loves to grab onto metal ions. And we can use UAG (stop codon) to code for our new AA. I found this paper titled “Rewiring Protein Synthesis: From Natural to Synthetic Amino Acids” and in it they describe that we need to modify two specific biological parts to make our synthetic AA. Firstly we need to evolve the Aminoacyl-tRNA Synthetase that are essential enzymes that catalyse the attachment of a specific amino acid to its corresponding tRNA. We can take a natural one and mutate its active site to fit our Bipyridine so it ignores the other 20 natural AA. The resulting tRNA is delivered to the ribosome by an elongation factor which we will have to modify to deliver our bulky new AA.
5. Where did amino acids come from before enzymes that make them, and before life started?
- Before enzymes existed, chemistry had to synthesise amino acids through abiotic chemistry fueled by the geological and cosmic energy. The main theories of our time come from:
- Miller-Urey Primordial Soup: This theory suggests that the early atmosphere of earth was a reducing environment rich in methane, ammonia and water vapor. Miller and Urey found that passing electrical sparks through these gases produced several amino acids including glycine and alanine
- Deep-Sea Hydrothermal vents: As we remember watching deep sea thermal vents footage from BBC’s planet earth documentary. These cents provide a constant stream of superheated, mineral rich water and reducing gases like H2 and H2S which provides extreme pressure and temperature gradients which combined with the mineral catalysts could drive the synthesis of organic molecules. This is refferred to as the iron-sulfur world theory.
- Panspermia: One of my favorite theories. Amino acids may not have started on earth at all but were delivered by comets and meteorites. Like a message of life from beyond. Who wrote it? How long ago? Haha. I am gonna write a sci-fi story one day with this premise. The evidence for this theory came from Murchison meteorite which fell in Australia in 1969 and was found to contain over 70 different amino acids.
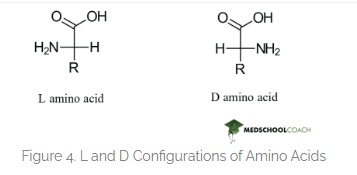
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- This question somehow scares me. Since this relates to Chirality in life. And how fascinatingly all life on this planet is formed by L-amino acids. Contemporary notion is that if we mess around with this and make life from the mirror image which is D-amino acids then we will form helices with a mirrored spin and life that will be incompatible with the enzymes of L-AA oriented life and how catastrophic something like this will be in the wild. But I hold an optimists approach. I feel like this is going to be a huge area of research in synbio and we should approach this problem with good ethics and positivity.
Here is the correct difference between L-D Amino acids.
Here is the hallucinating ChatGPT image that cant draw a correct Amino Acid for the life of me. I have tried many times. But they say its gonna take over the world. It got some parts right.
 prompt: ChatGPT please draw me an image showing L and D aminoacids showing chirality of life and how they form mirror alpha helices
prompt: ChatGPT please draw me an image showing L and D aminoacids showing chirality of life and how they form mirror alpha helices
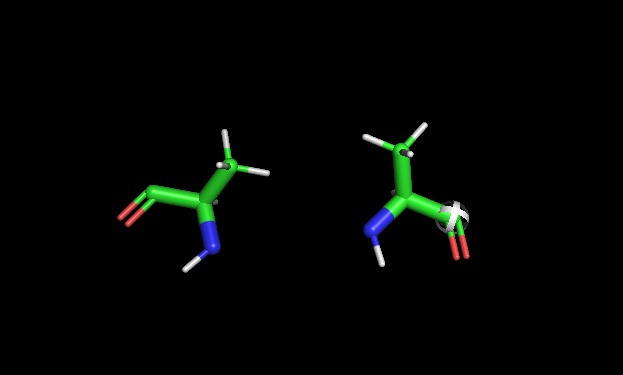
- PyMOL chirality: Later used pymol to show L and D Alanine side by side
7. Can you discover additional helices in proteins?
- Yes, additional helical structures besides the standard alpha delix can be found in proteins. Studies show that other types of helices occus in many proteins, but they are often overlooked or mistaken for small distortions in alpha helices. These helices are especially common in memberane proteins and are found in a significant number of known protein structures. (Vieira-Pires & Morais-Cabral, 2010)
8 Why are most molecular helices right-handed?
- As shown above they are mostly righ-handed because there is more abundance of L-Amino acids. So when coil righ handed helices avoid steric clashes and are more stable and thus more common
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?(also answers Q11 )
The primary sequences of beta strand and sheet prefer alternating hydrophobic and hydrophilic amino acids. This forms exposed hydrogen bond donors and acceptors on both sides that forms the sheet structure and the hydrophobic side chains interlock between the sheets. This is shown in the image below from pyMol
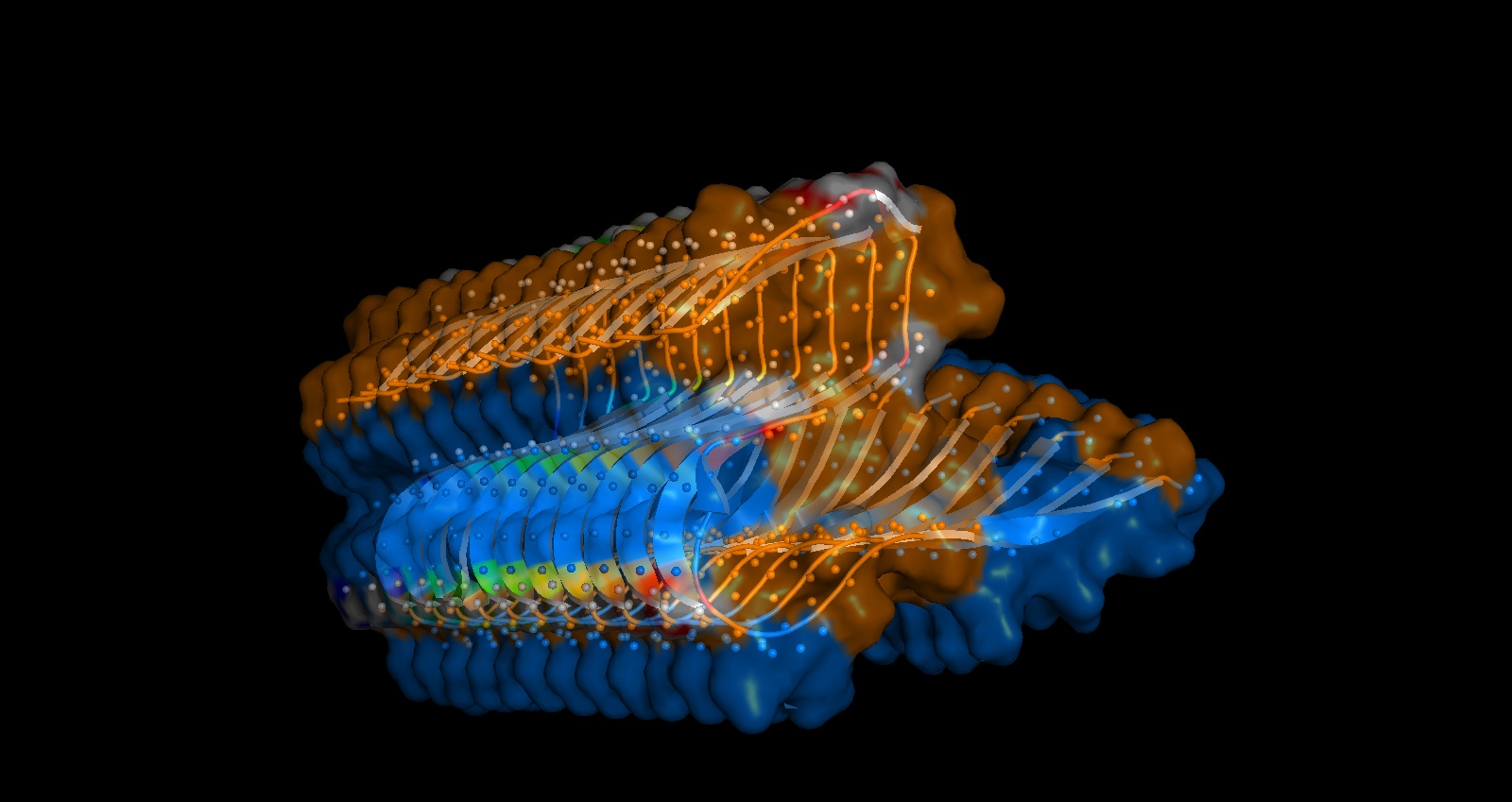 You can shee in the above image that Hydrogen bonds in grey small dots create horizontal structure and Hydrophobic packing on the inside denoted by the orange surface creates the vertical stacking.
You can shee in the above image that Hydrogen bonds in grey small dots create horizontal structure and Hydrophobic packing on the inside denoted by the orange surface creates the vertical stacking.
Part B: Protein Analysis and Visualization:
1. Protein Selection and Rationale
- Protein: I selected 4IS4, the Glutamine Synthetase from the model plant Medicago truncatula. I chose this because GS is the primary engine for nitrogen assimilation in plants. By studying a plant-specific decameric structure, I can better understand how to engineer nitrogen uptake in duckweed—a key goal for my PACT proposal to reduce agricultural fertilizer runoff in Canada.
- Rationale: GS is the “gatekeeper” for nitrogen assimilation. It converts ammonium (found in high concentration in Canadian hog/dairy manure) into amino acids.
- The Goal: To see of we can engineer a version of this protein that works faster or remains stable in the fluctuating temperatures of a canadian lagoon.
2. Amino Acid Sequence:
The length of the protein is: 378 aminoacids. The most common amino acid is: G, which appears 42 times.
- Sequence Homologs: There are thousands of homologs of the protein as this is a highly conserved protein across species.
4. The structure was released on 2014-04-09. The resolution is 2.35 Å so its a very good resolution.
Interactive Model: View the decameric structure of 4IS4 on RCSB PDB
4. 3D molecule visualization software images:
Cartoon Image- front:
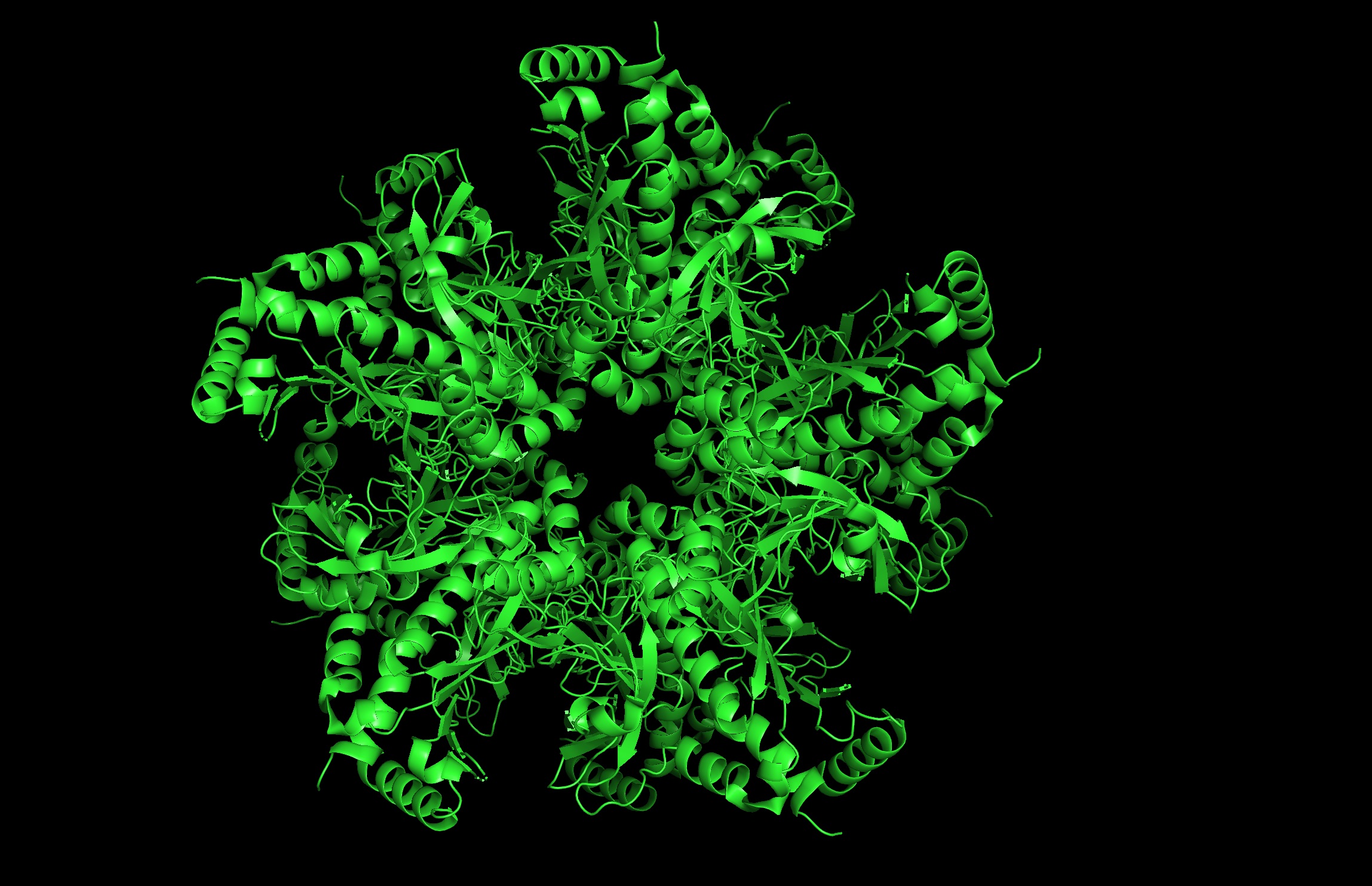 Cartoon Image- side:
Cartoon Image- side:
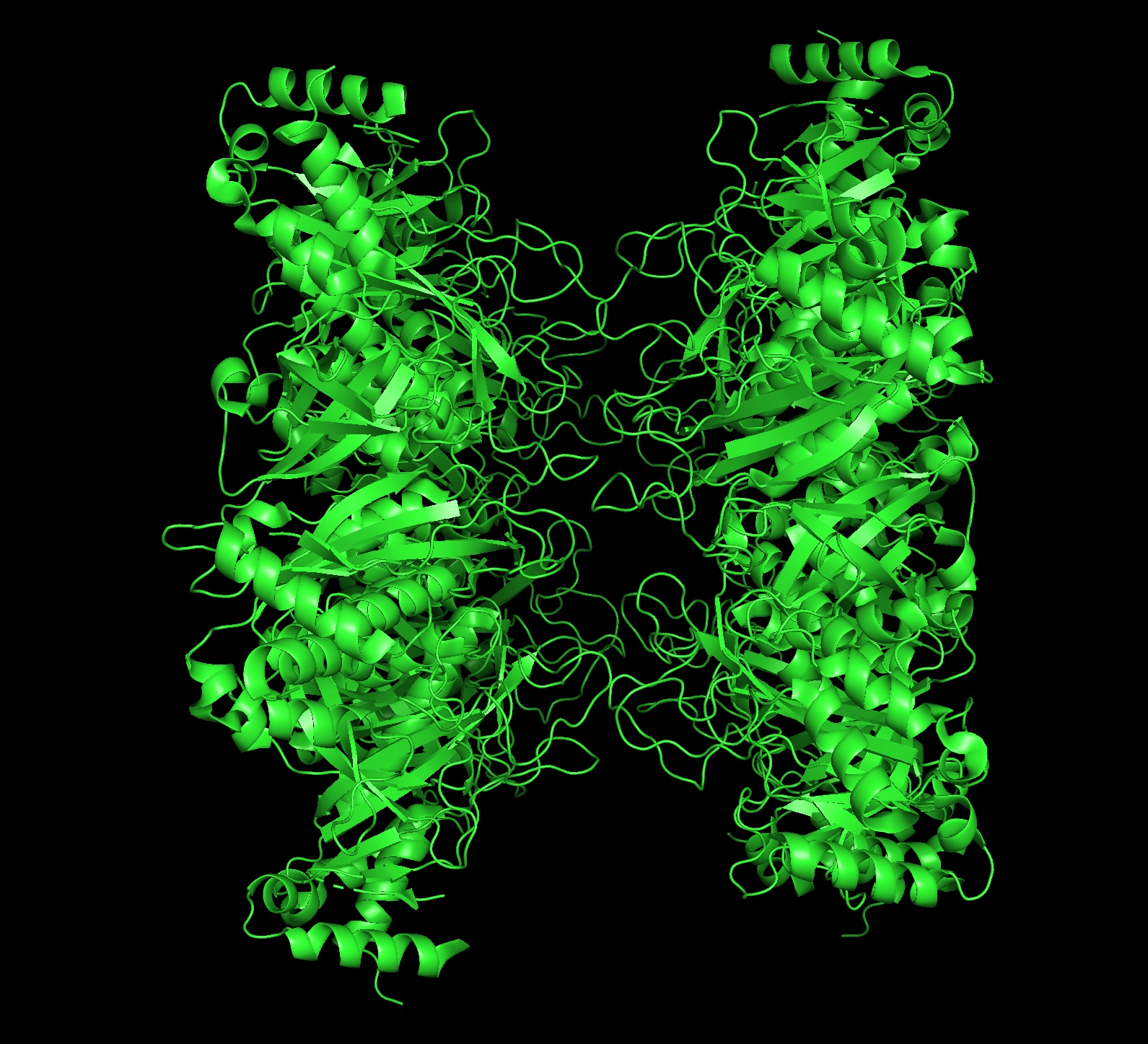 Ribbon Image- Front:
Ribbon Image- Front:
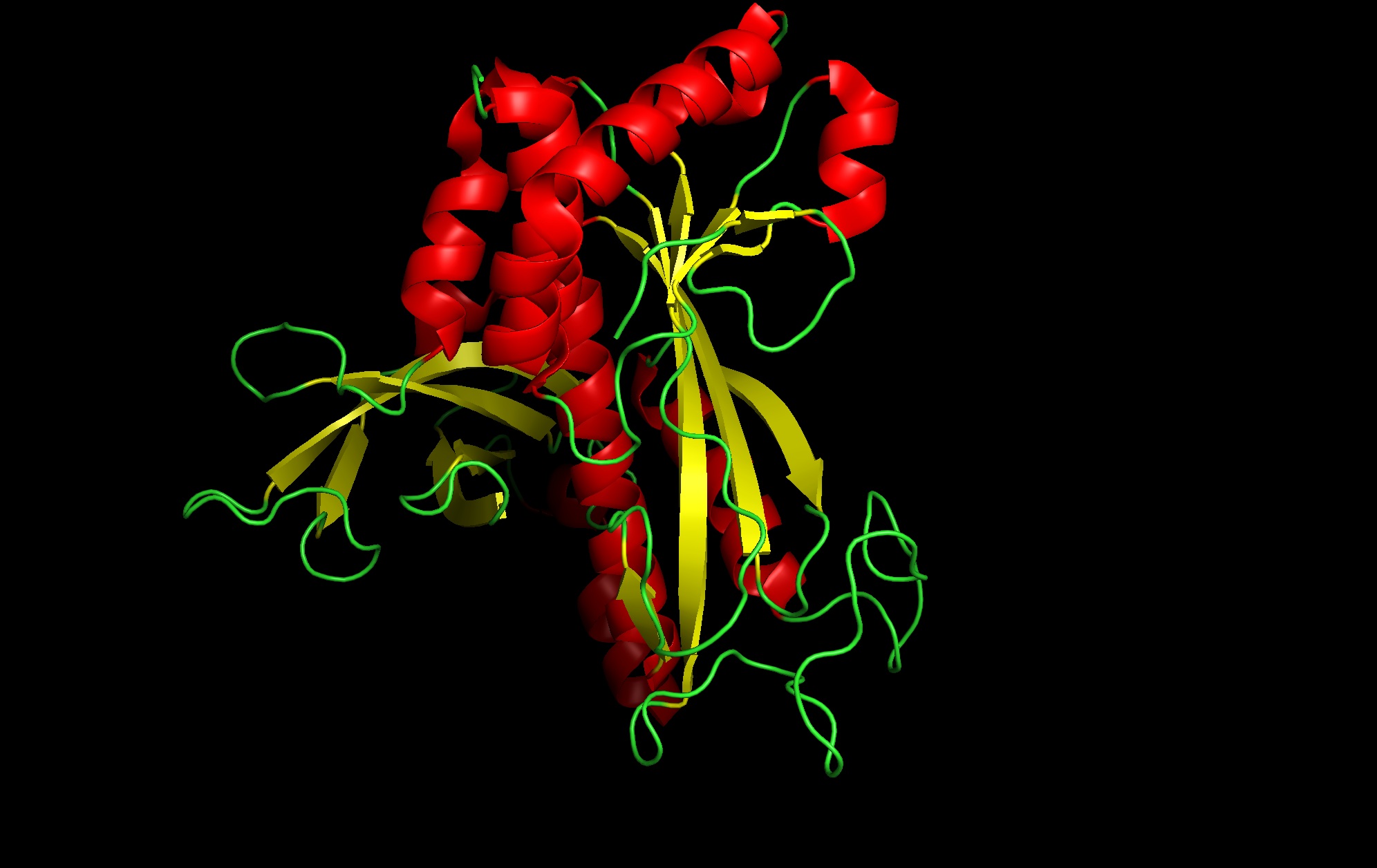
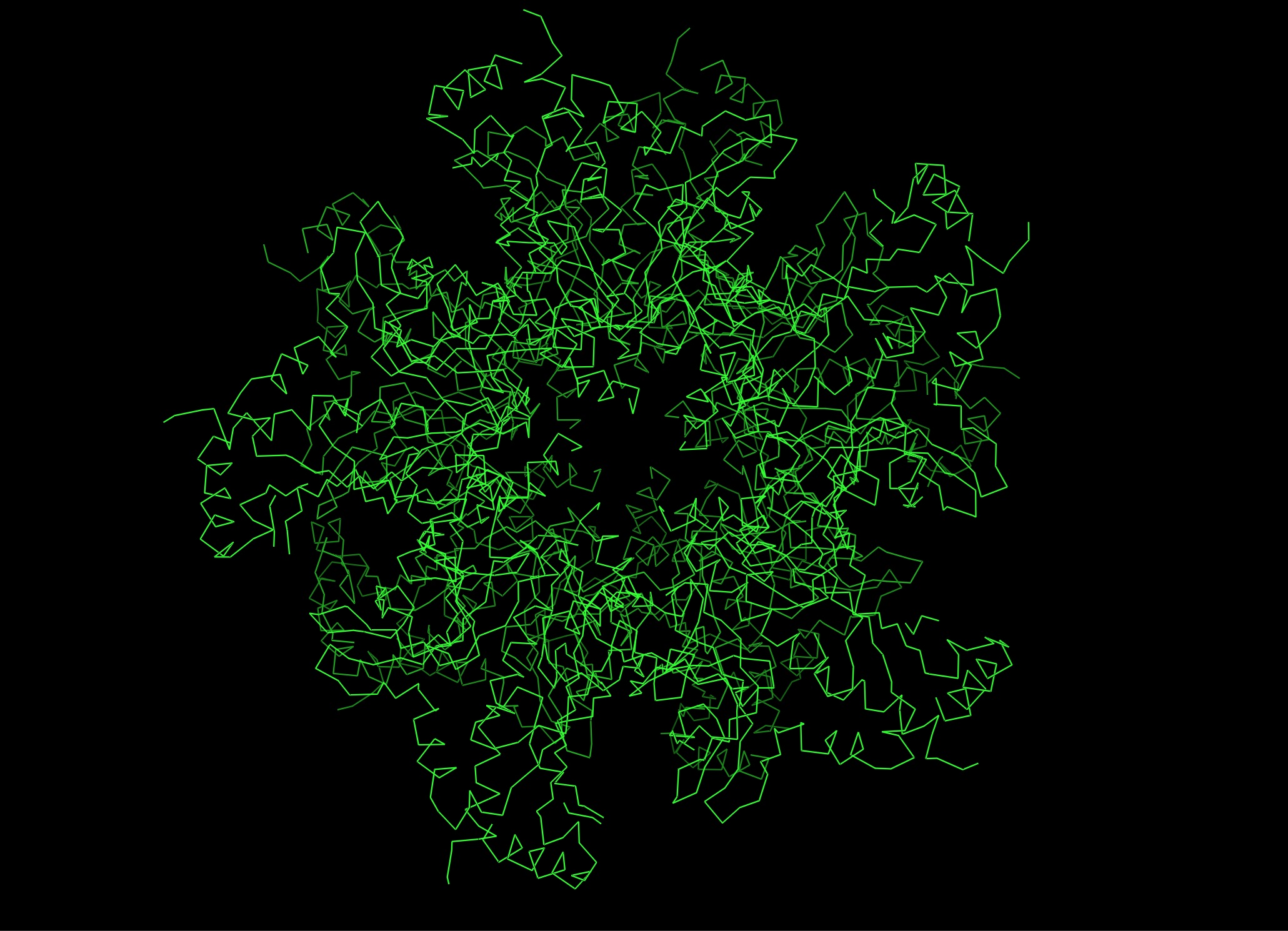 Most- Informative:
Most- Informative:
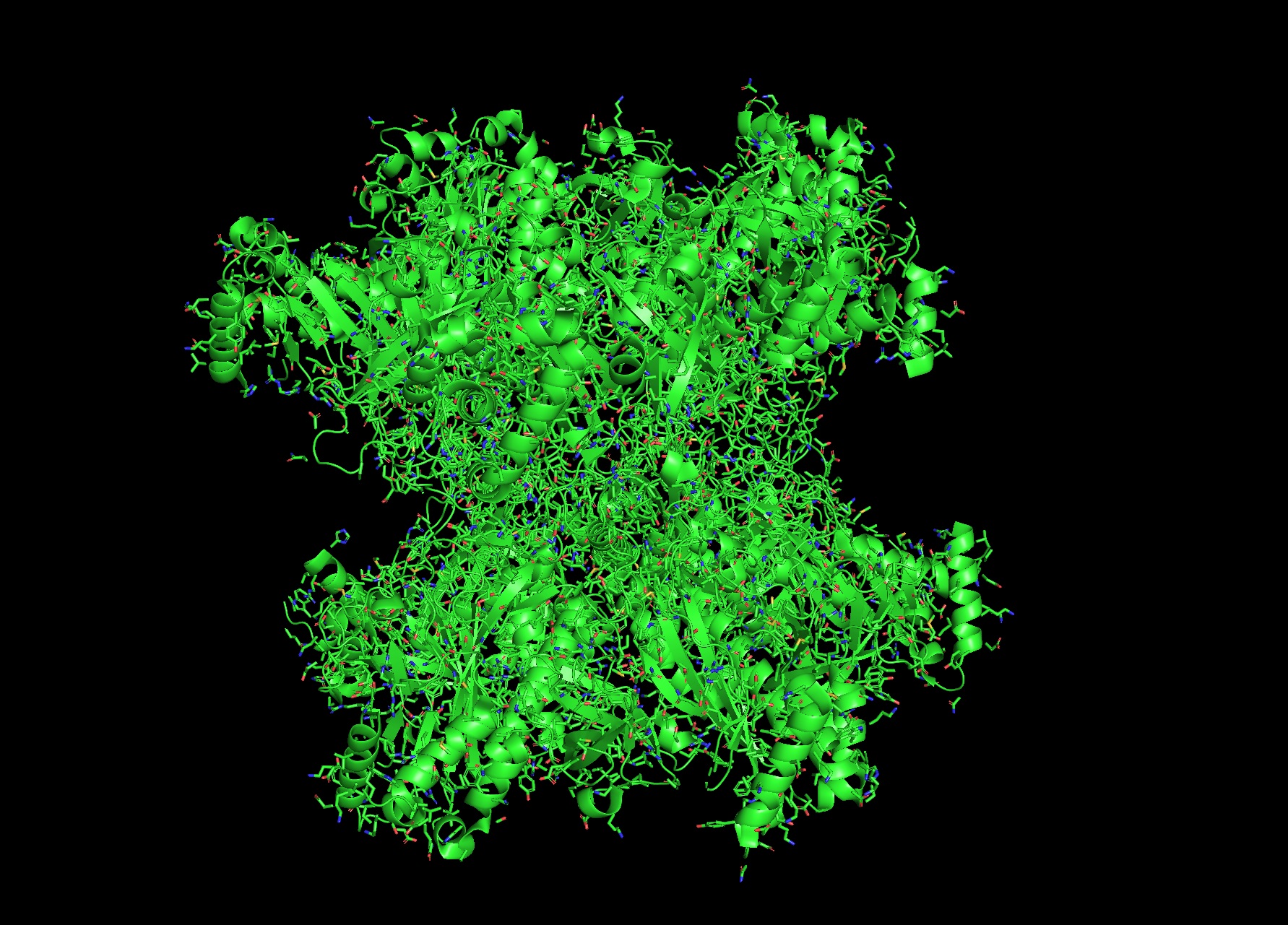
- Color by secondary structure:
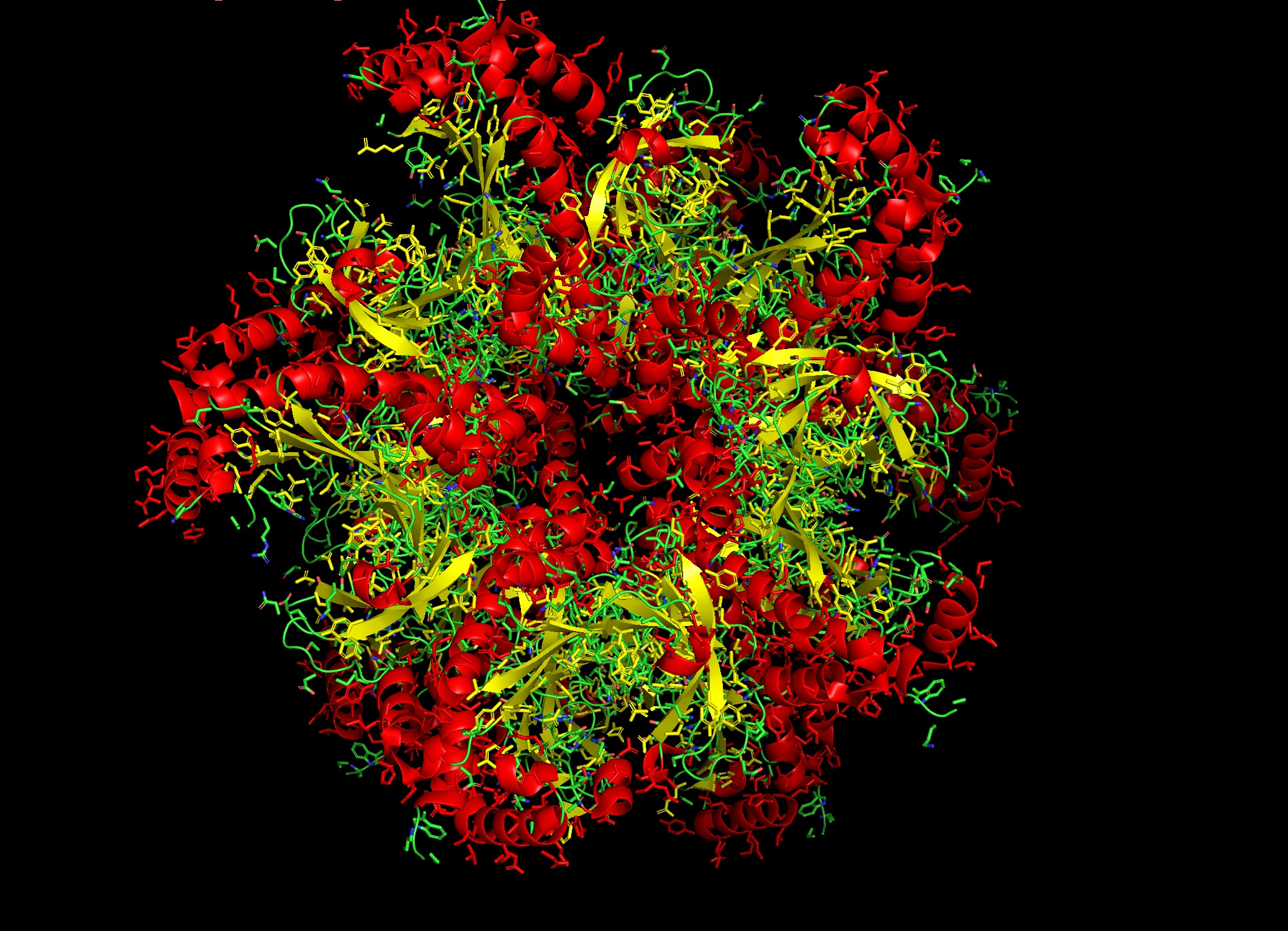
- seems like a pretty equal representation of sheets and helices which is a common feature in enzymes
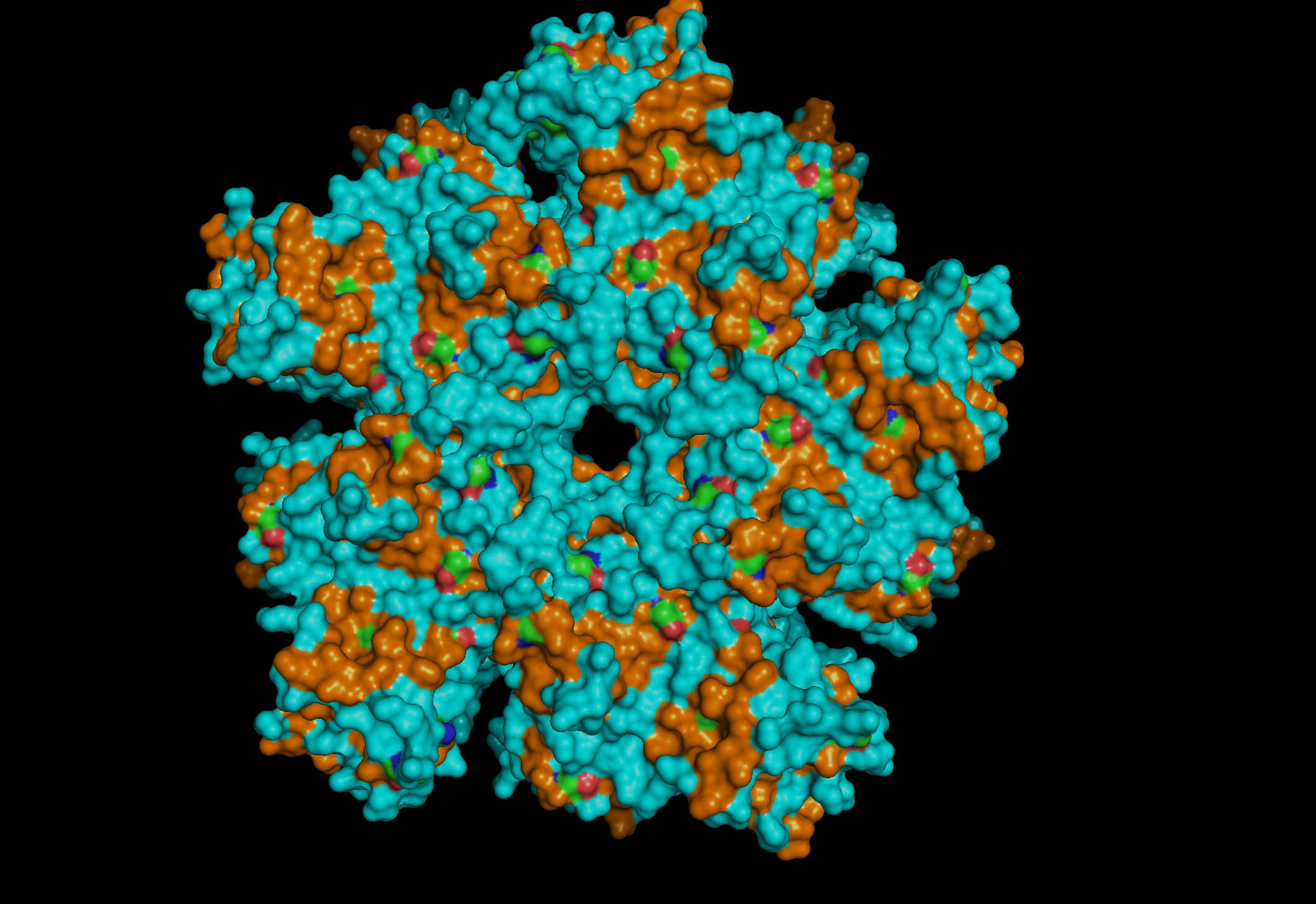
Color by Residue Type: Hydrophobic: Orange; Hydrophilic: Blue
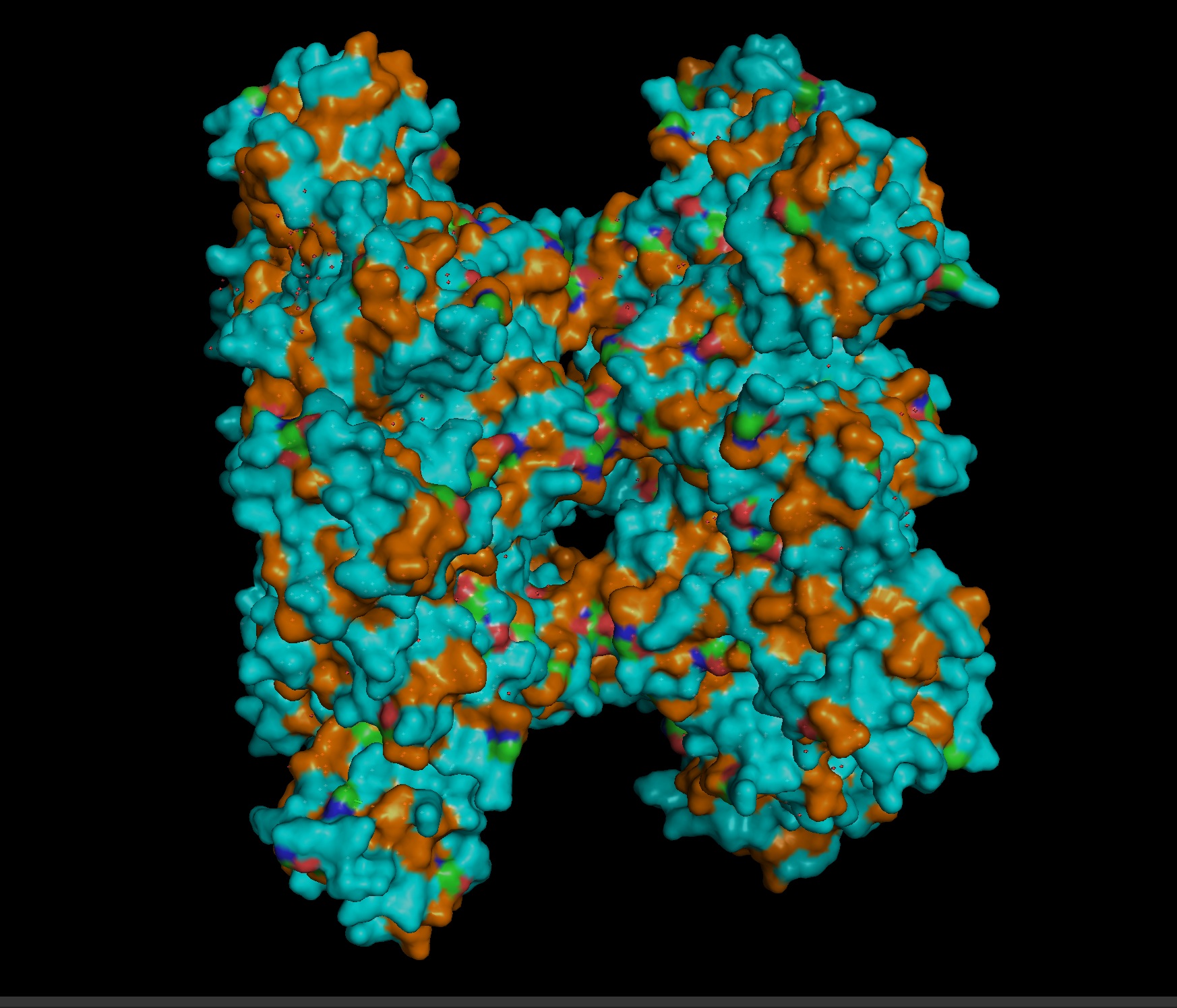
Since this is an enzyme it has tons of pockets
Part C. Using ML-Based Protein Design Tools
- C1. Protein Language Modeling
a. Unsupervised Deep Mutational Scans
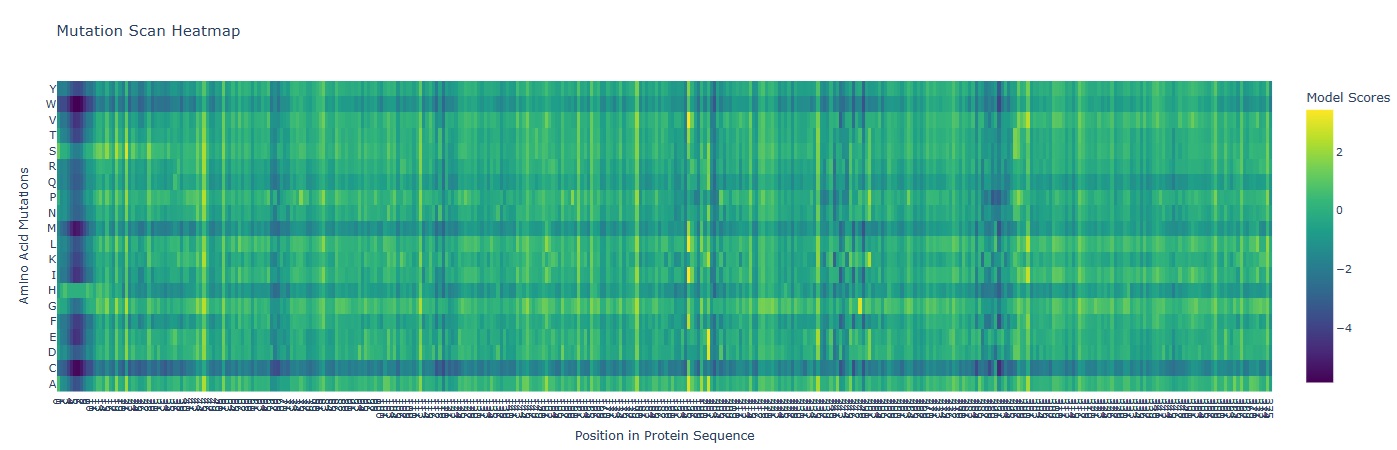 A mutation scan is basically asking “If I mutate every position to every other amino acid, which ones does the model think are tolerated… and which ones are catastrophic?”
In the heatmap the X-axis represents the AA position in the protein (1-378). And the Y-axis represents the 20 possible amino acids.
A mutation scan is basically asking “If I mutate every position to every other amino acid, which ones does the model think are tolerated… and which ones are catastrophic?”
In the heatmap the X-axis represents the AA position in the protein (1-378). And the Y-axis represents the 20 possible amino acids.
- Deep Blue/ Purple Columns: These are “Cold” regions. they represent positions where almost any mutation results in a significantly lower model score. This indicates High Conservation. In glutamine synthetase these are usually residues critical for binding ATP, Glutamate or forming the decameric ring interface.
- Yellow/Green Spots: These are “Neutral” or “Favorable” regions. The model suggests the protein could tolerate these changes without losing its structural integrity.
- Mutation that Stands out: I observed a distinct pattern of high sensitivity around Position 2-12 which probably is the catalytic site of the enzyme.
TBD: (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
b Latent Space Analysis
The Latent Space analysis was cool to visualise but the neighbourhood doesnt give anything useful unfortunately.

C2. Protein Folding
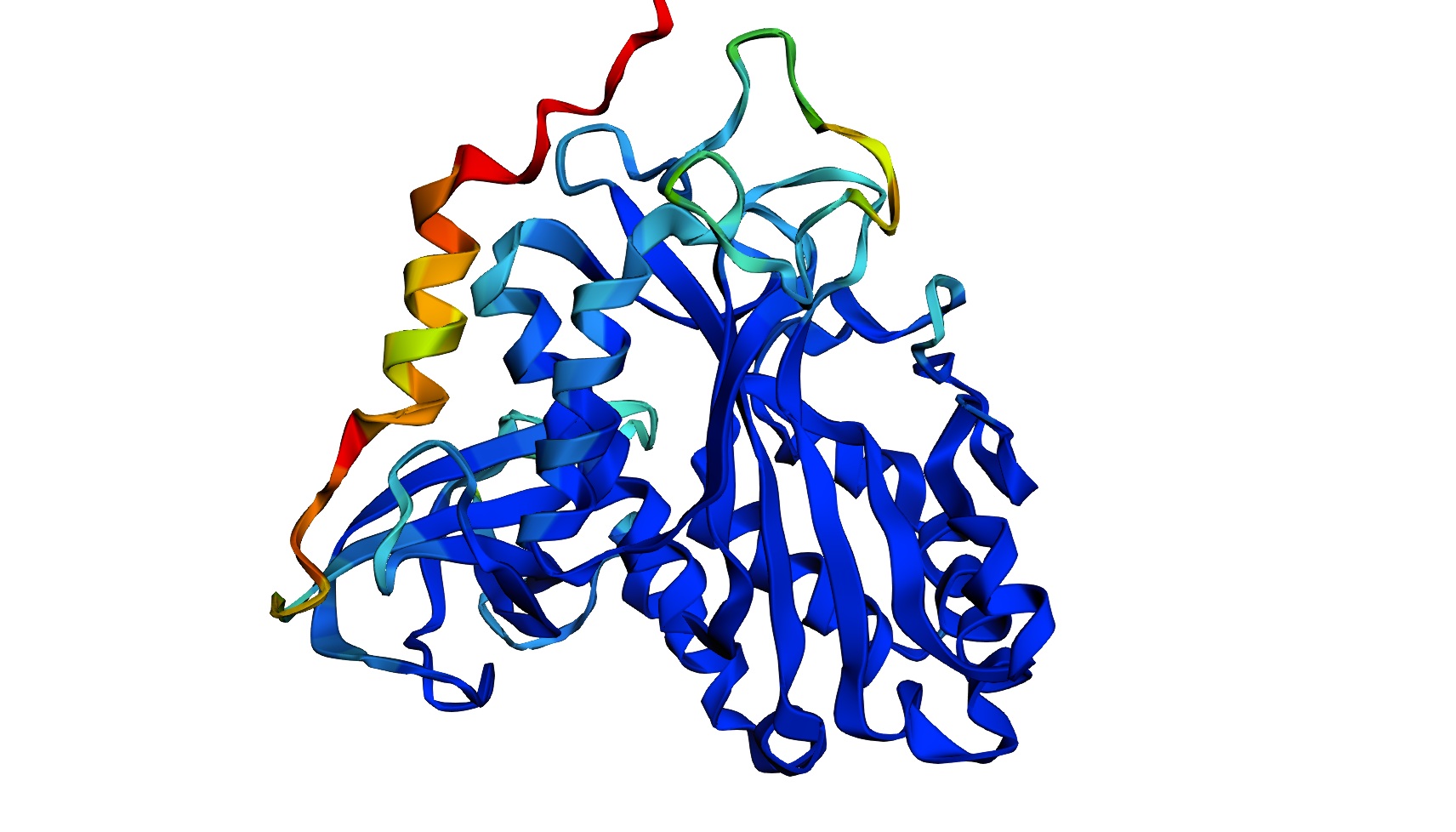
I got a pyMol structure of a monomer of my protein and compared it to the ESM Fold predicted structure and I have to say these snapshots show a lot of similarities of you focus on the number of parallel B-sheets and their location
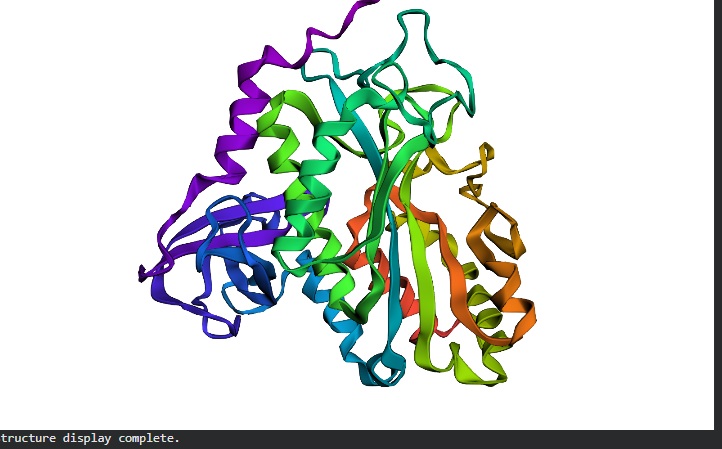
Confidence score: Mostly Blue so ESM has a high confidence in this structure which it should
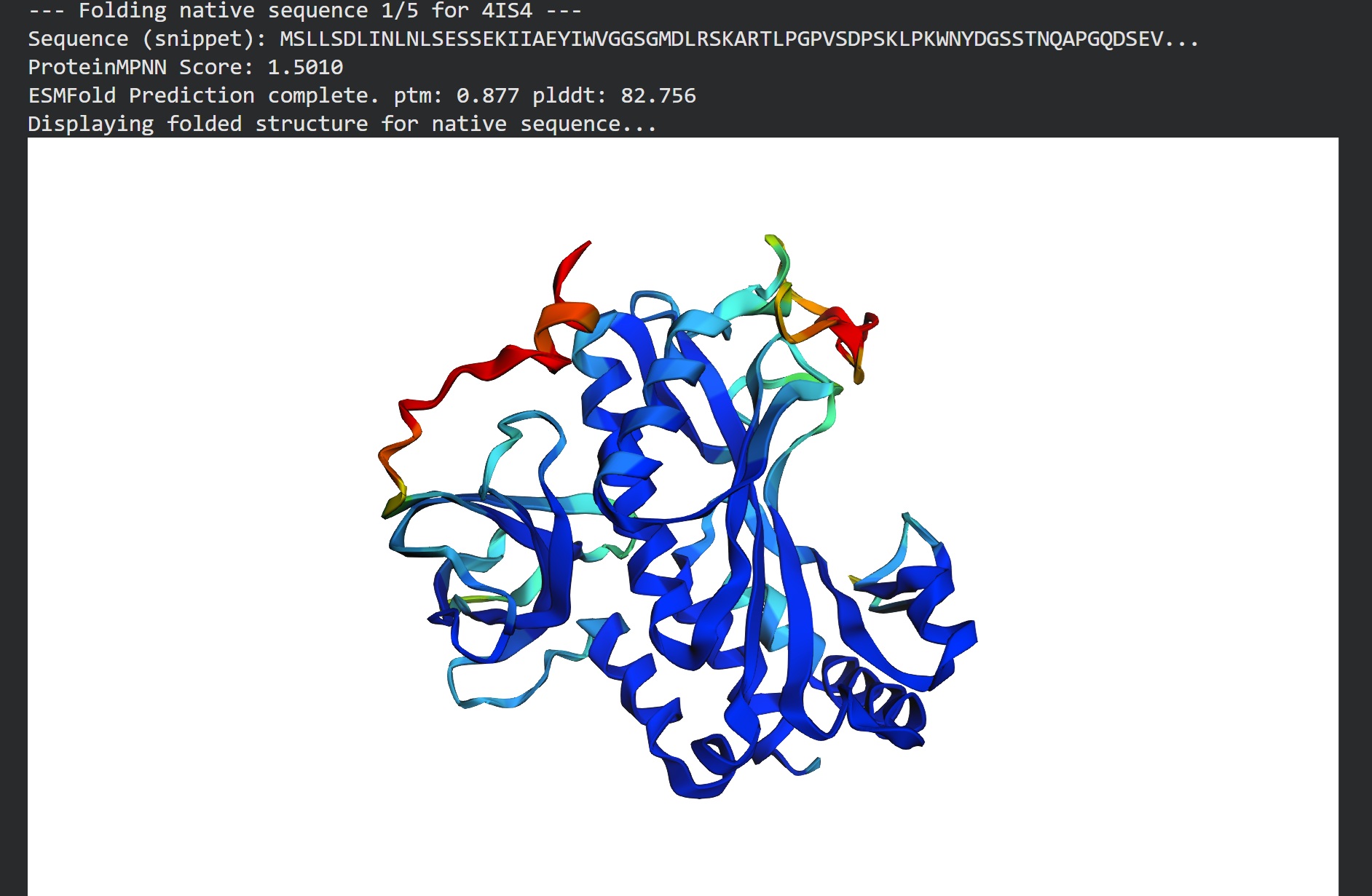
C3. Protein Generation
Inverse folding using protein MPNN
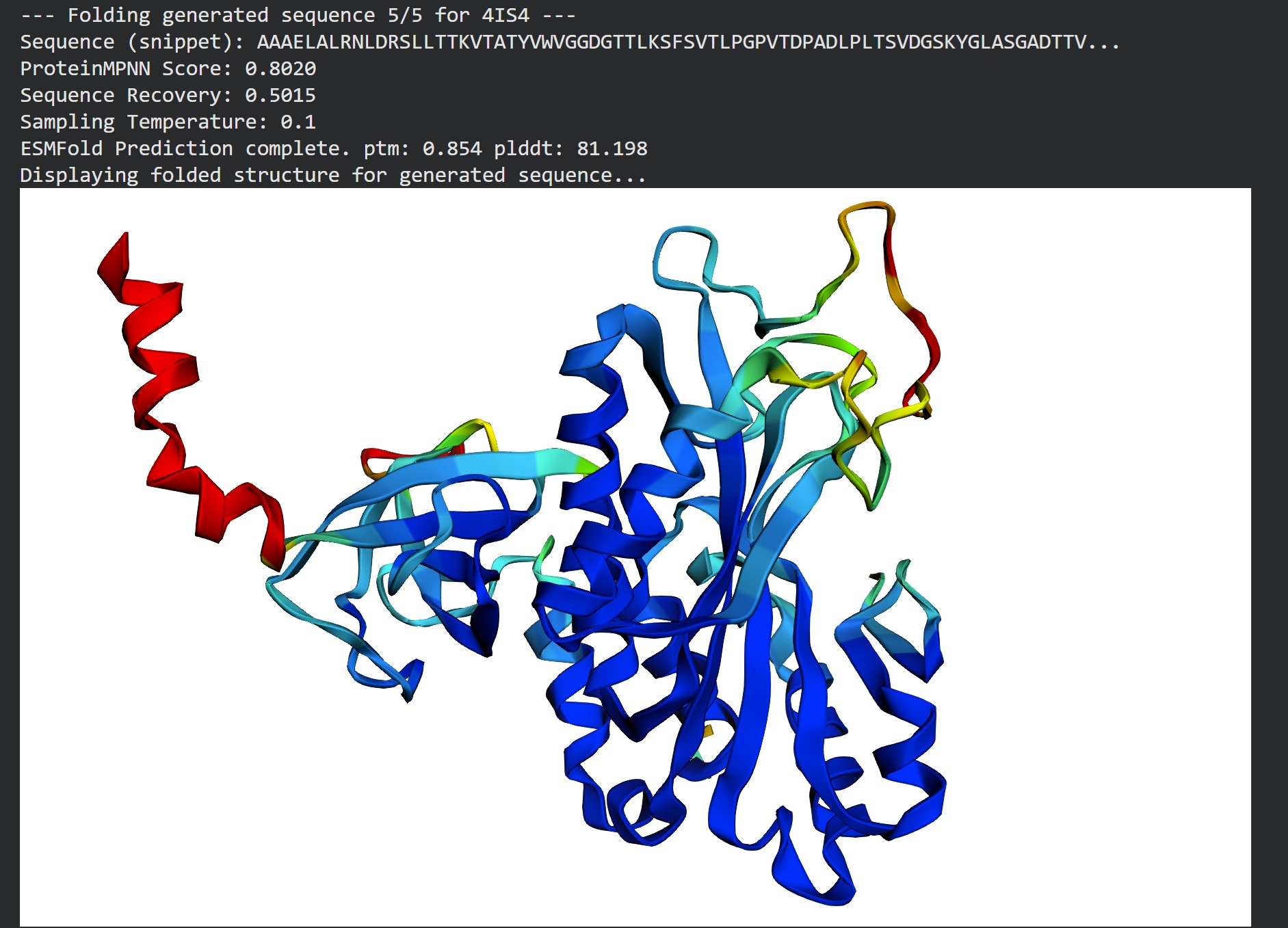
Following were 4 generated sequences along with the 1st being the native sequence
These generated sequences closely resemble our original structure so ProteinMPNN did a great job at finding various sets of sequences giving the same PDB structure we entered.
Generated sequences…
4IS4, score=1.5294, fixed_chains=[], designed_chains=['A'], model_name=v_48_020 MSLLSDLINLNLSESSEKIIAEYIWVGGSGMDLRSKARTLPGPVSDPSKLPKWNYDGSSTNQAPGQDSEVILYPQAIFKDPFRQGNNILVICDVYTPAGEPLPTNKRYNAAKIFSHPDVAAEVPWYGIEQEYTLLQKDTNWPLGWPIGGYPGPQGPYYCGIGADKAYGRDIVDAHYKACLYAGINISGINGEVMPGQWEFQVGPSVGISAGDEIWAARYILERITEIAGVVVSFDPKPIPGDWNGAGAHTNYSTKSMRENGGYEIIKKAIEKLGLRHKEHIAAYXXXXXXXXXXXXXXXXXNTFSWGVANRGASVRVGRDTEKDGKGYFEDRRPSSNMDPYVVTSMIAETTLLWKP
T=0.1, sample=0, score=0.8142, seq_recovery=0.4985 EELKELLENIDLSLLTTKIIATYVWIGGDGTTLKSFSVELPGPITNPADLPLTSVDGSKYNLSSGADTTVILVPRKIFRDPFLKGDNIIVVCSLKTPSGEPLPTDTYAKAAEIFAHPKVAAEQPEFTITIEFTLLDKETGLPLGYPPEGYPGPDNPYRNGTGPDTAFGQEILDETVEACKYAGIPISGSAREVEPGQWSITVGPALGIDAGLHVVAARYILEKIAEKAGVKVSFKDKPYPGPYYSKGAPVEFATKAMRGEGGLALILEAIEKLKKRYSELLAAVXXXXXXXXXXXXXXXXXATPSWSVDTPGSSFQIGAATVEAGRGSFTFLVPKSDADPFIVTGMLAEITLLYED
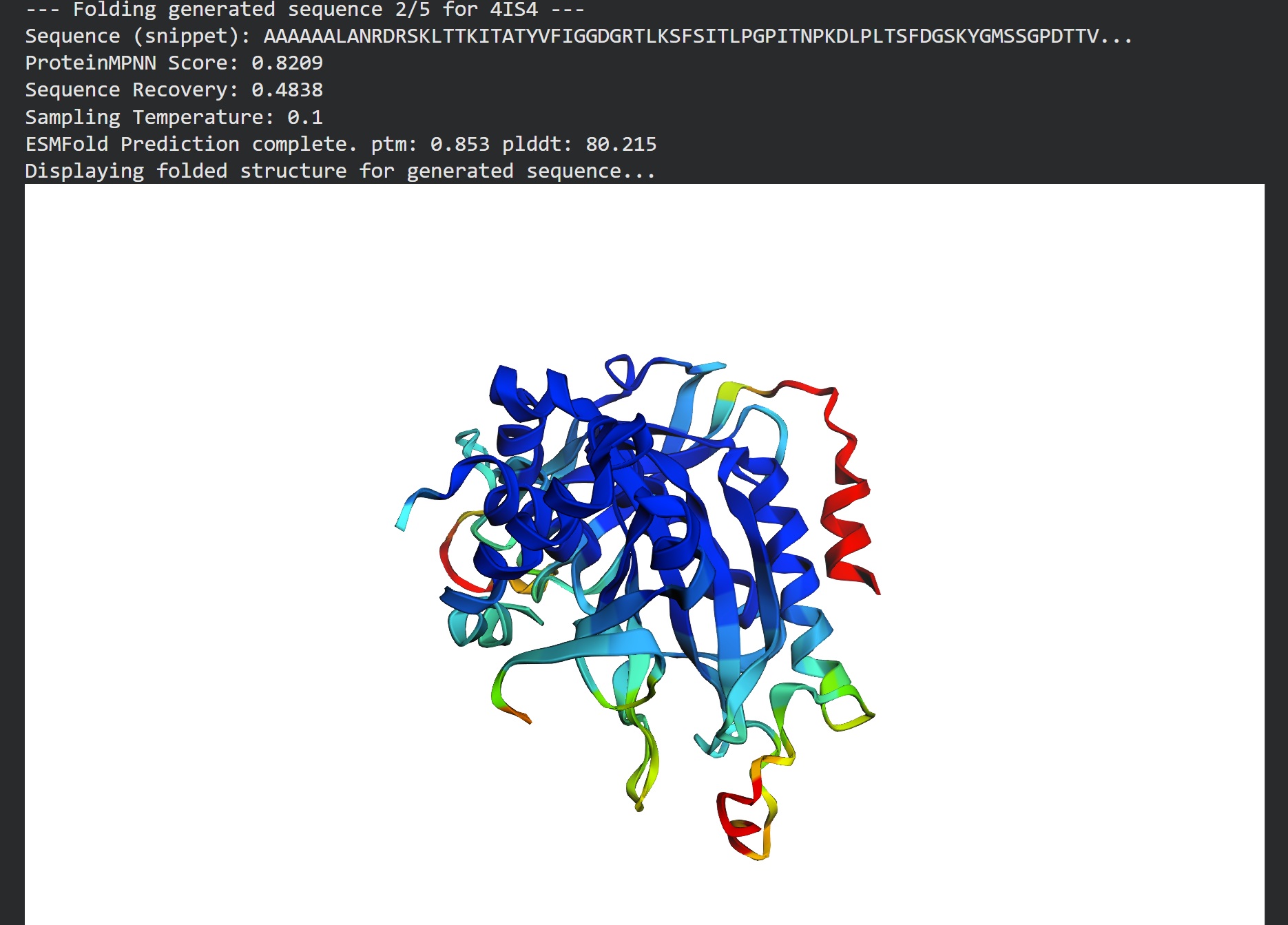
T=0.1, sample=0, score=0.8052, seq_recovery=0.5074 AAAAAALANVDRSLLTTKIKAEYVFIGGDGTTLRSFSVELPGPITDAKDLPLSSFDGSKYNMASGSNTTVILKPEKIFKDPFLGGDNIIVVCSSYTPEGKPLPTDKYAEAKEIFDKPEVKAEQPQFTIRIEFTLLDAETGLPLGYPASGYPGPNGPWRNGTGPNTAFGMDIVNETVAACKYAGIPISGSASEIEPGQWSITVGPALGINAGLNVIAARYILEKIAAKAGVKVSFDDKPVPGPYYSRGAPVGFSTKTMRGAGGLAAILAAIEKLGLRYEELKKAFXXXXXXXXXXXXXXXXXSEFSWSVDTEGSSFRIGAATVAAGKGSFTVLVPPSNANPFVVTSMIAYYTLLYKP
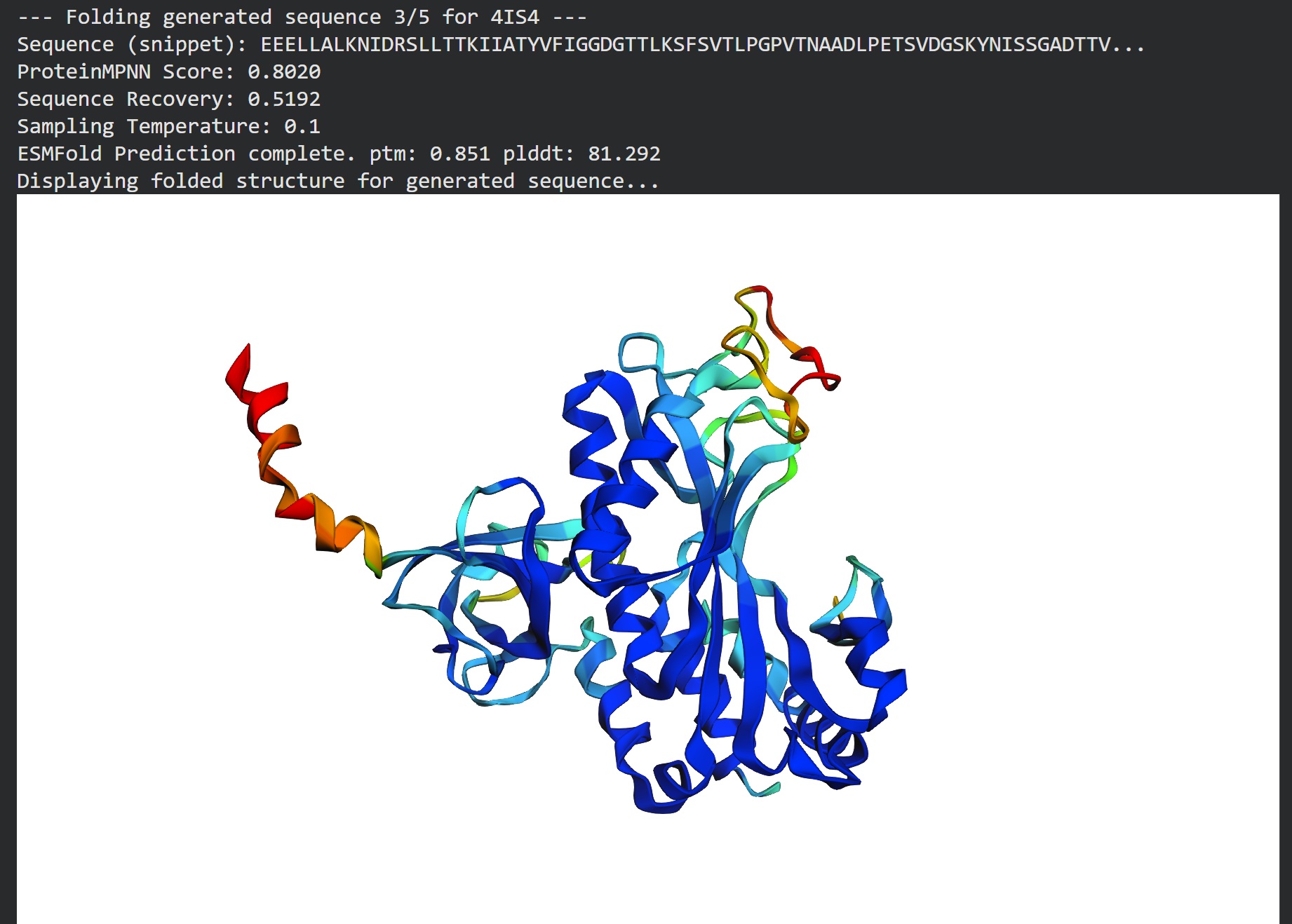
T=0.1, sample=0, score=0.8224, seq_recovery=0.5192 AEALAALENIDLSLLTTKIIAEYVWVGGDGTTLRSFSVELDGPITDPSKLPLTSVDGSKYGLASGPDTTVILVPEAIFKDPFRKGNNIIVVCSLRTPSGEPLPTDKLAEAREIFDKPEVKAQQPEFTVRIEFTLLDKETGKPLGYPPEGYPGPNNPYYNGTGPDTAFGDEIVNEAVEAMKYAGIPISGSEREIEPGQWSYTVGPALGIQAALNVIAARYILEKIAEKHGVNVSFKPKPVPGPWYSRGMPVGYATKAMRGPGGLAKILADIEKLAKRYPELLAAFXXXXXXXXXXXXXXXXXATFSWSVDTPGDSFRIGAETVAAGRGSFTVLLPPSDADPAVVTGMLARVTLLEKP
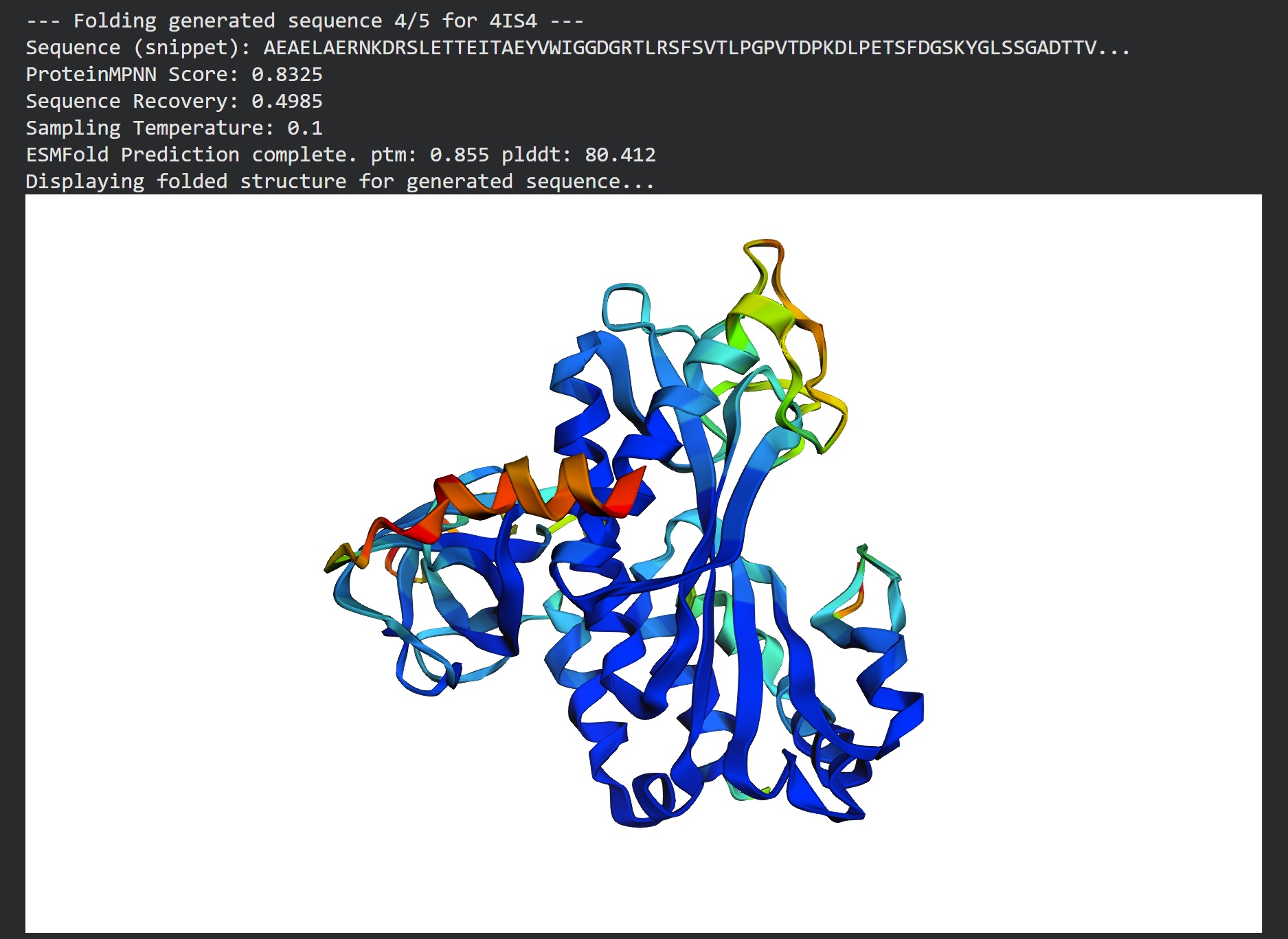
T=0.1, sample=0, score=0.8041, seq_recovery=0.5074 AAEELAKKNVDRSLLTTEITAEYVFIGGDGRTLKSFSVTLPGPVTNPADLPLTSFDGSKYNMASGPDTTVILKPKKIFKDPFLKGNNIIVICSTYTPAGTPLPNNKYAEAEAIFADPAVAAAQPLFTIEIEFTLLDADTGLPLGYPADGYPGPVNGWENGTGPDTAFGKDILDKTVEACKYAGIPISGSAREVEPGQWSFTVGPALGIEAGYHVLAARYILEKIAEEAGVKVSFAPRPVPGPWYTRGAPVSFATAAMRGPGGLAVIDAAIARLAARYPELLAAVXXXXXXXXXXXXXXXXXATPSWSVDTEGASFRVGADTVAAGRGSFTFLVPRSDANPFVVTSLLARVTLLDTP
Part D. Group Brainstorm on Bacteriophage Engineering:
- Make a group: Tammy Sisodiya (London Node); we are looking for collaboration
- Engineering Goal:
- Start with making L-Protein more robust
- Computational tools:
- ESM-2 to perform deep mutational scan to find which mutations the model thinks will improve stability
- Protein MPNN to redesign the sequence around the core structure to find more optimal amino acid arrangements
- ESMFold to check if new sequence still folds correctly
- Proposal:
- Sequence ——> ESM2 Scan ——> ProteinMPNN ——–> Redesign ——> ESMFold Validation
- I chose ESMFold because its faster at testing multiple mutation iterations in silico
- ML Models have limitations; they are great at folding but might not perfectly predict how a protein behaves inside a complex living cytoplasm.
- Reading:
MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ:
Mutational analysis of the MS2 lysis protein L
*Thanks for reading!*