Week 5 HW: Protein Design- Part 2
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM:
1. Human SOD1 AA sequence from UniProt (P00441):
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
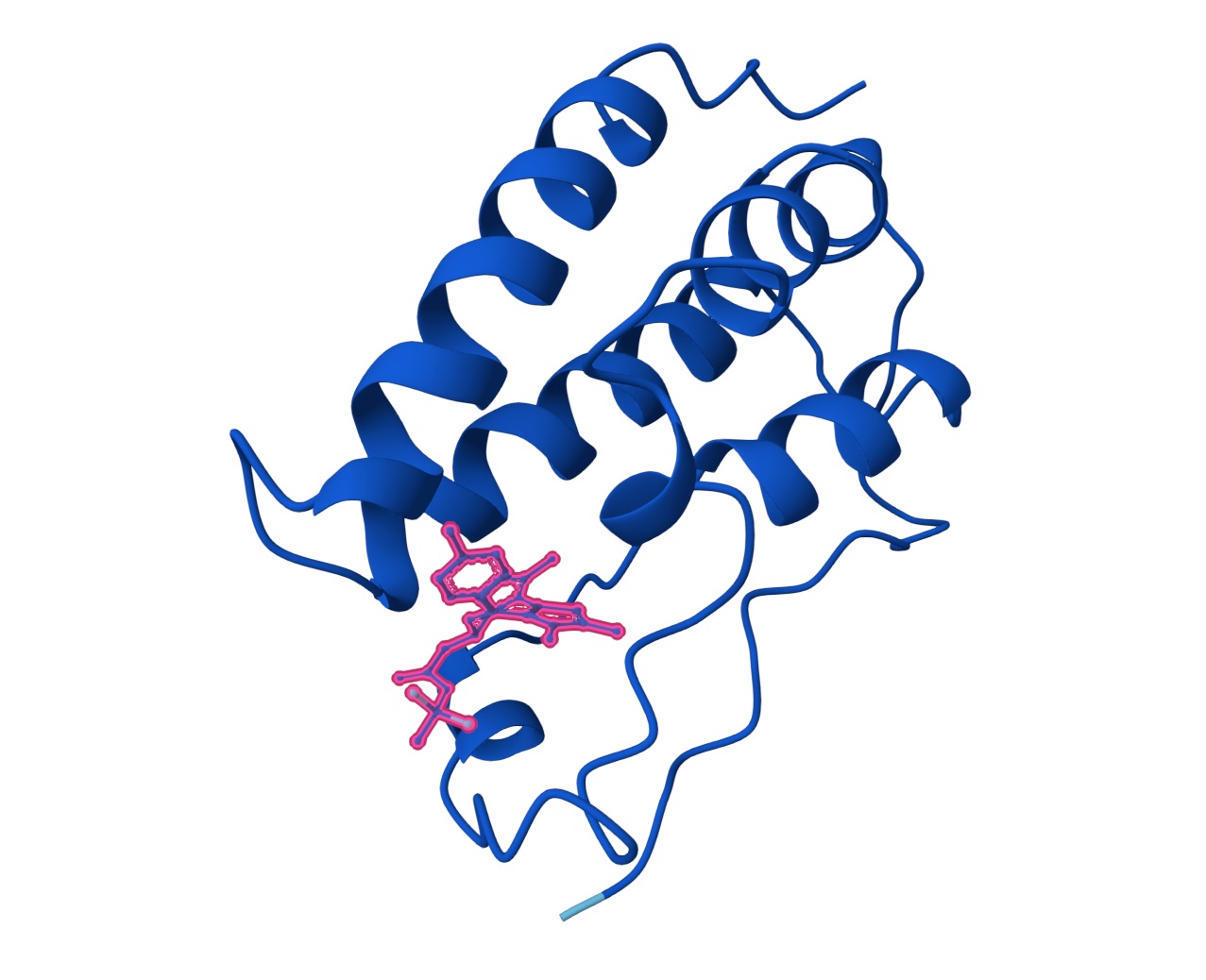
PyMOL image with A @4 is highlighted
Interesting find: I didnt know that all protein synthesis starts with amino acid Methionine(Met) which is cleaved off by Methionine Aminopeptidases as an essential process in a large majority of cells. It also has a condition where only the sequences with these small uncharged AAs as a second AA can be functionally folded instead of being degraded. (Nguyen et.al.2019)
Introducing SOD1 Seq with A4V Mutation (removing Alanine and adding Valine) associated with the most aggressive form of the ALS disease (and removing the initial methionine) :
ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. PepMLM Generation Results
| Candidate | Sequence | Pseudo Perplexity |
|---|---|---|
| Design 1 | WRSPATGARHKK | 13.60 |
| Design 2 | WRVPAVAVRHKK | 12.17 |
| Design 3 | HRYPVVGAEWKK | 15.56 |
| Design 4 | WRYYAAAIAHKK | 13.00 |
| Control | FLYRWLPSRRGG | 22.53 |
Note: Lower perplexity indicates higher model confidence in the sequence’s fit for the SOD1 mutant.
Part 2: Evaluate Binders with AlphaFold3:
AlphaFold3 Evaluation Table
| Candidate | Sequence | ipTM Score (Good Score > 0.80) | pTM Score (Good Score > 0.50) | Avg. pLDDT | Binds at A4V? |
|---|---|---|---|---|---|
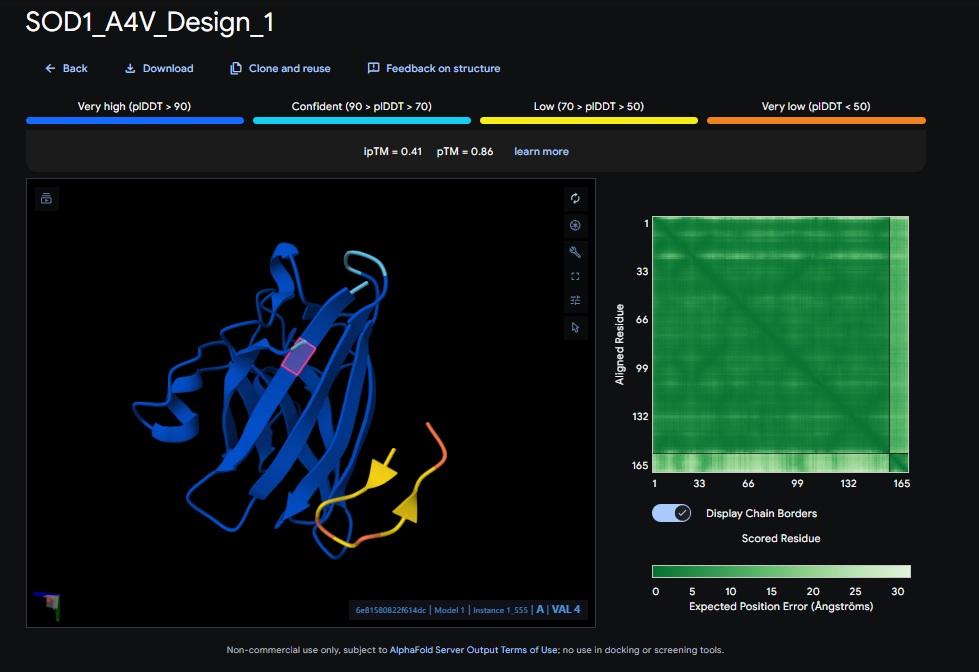
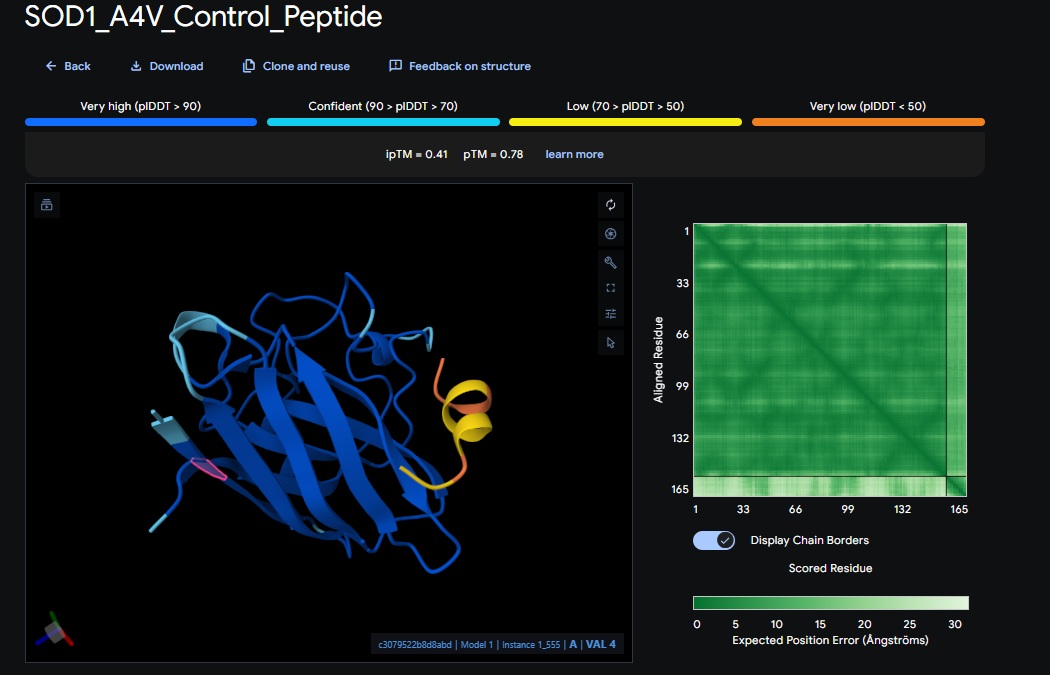
| Design 1 | WRSPATGARHKK | [0.41] | [0.86] | [low] | No |
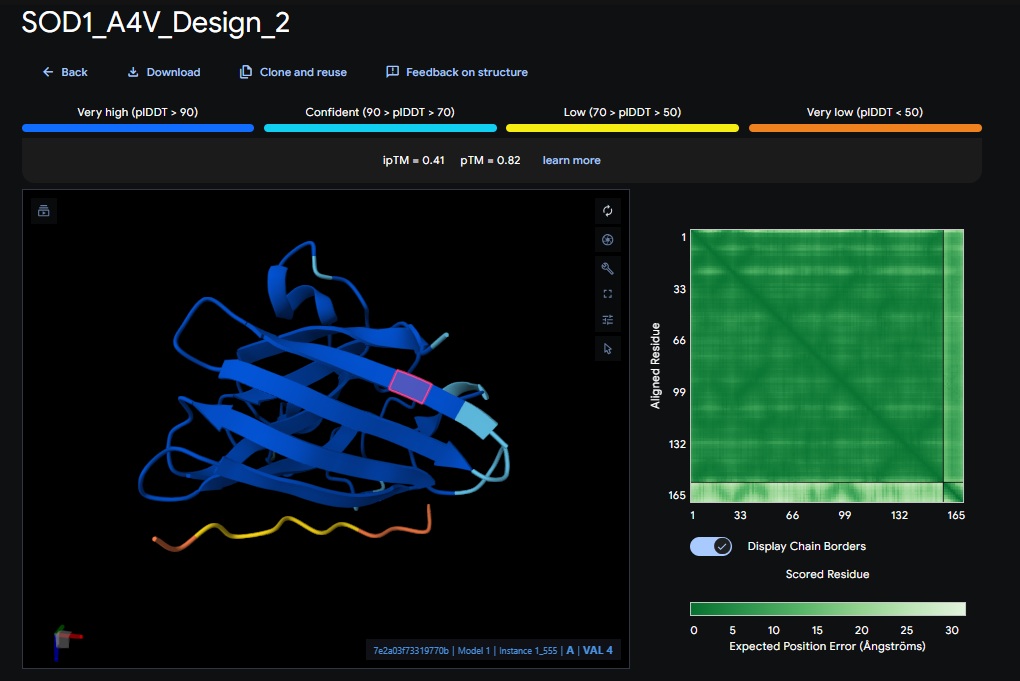
| Design 2 | WRVPAVAVRHKK | [0.41] | [0.82] | [low] | No |
| Design 3 | HRYPVVGAEWKK | [0.25] | [0.87] | [low] | No |
| Design 4 | WRYYAAAIAHKK | [0.34] | [0.72] | [low] | No |
| Control | FLYRWLPSRRGG | [0.41] | [0.78] | [low] | No |
1. Design_1 (WRSPATGARHKK) + SOD1:
2. Design_2 (WRVPAVAVRHKK) + SOD1:
3. Design_3 (HRYPVVGAEWKK) + SOD1:
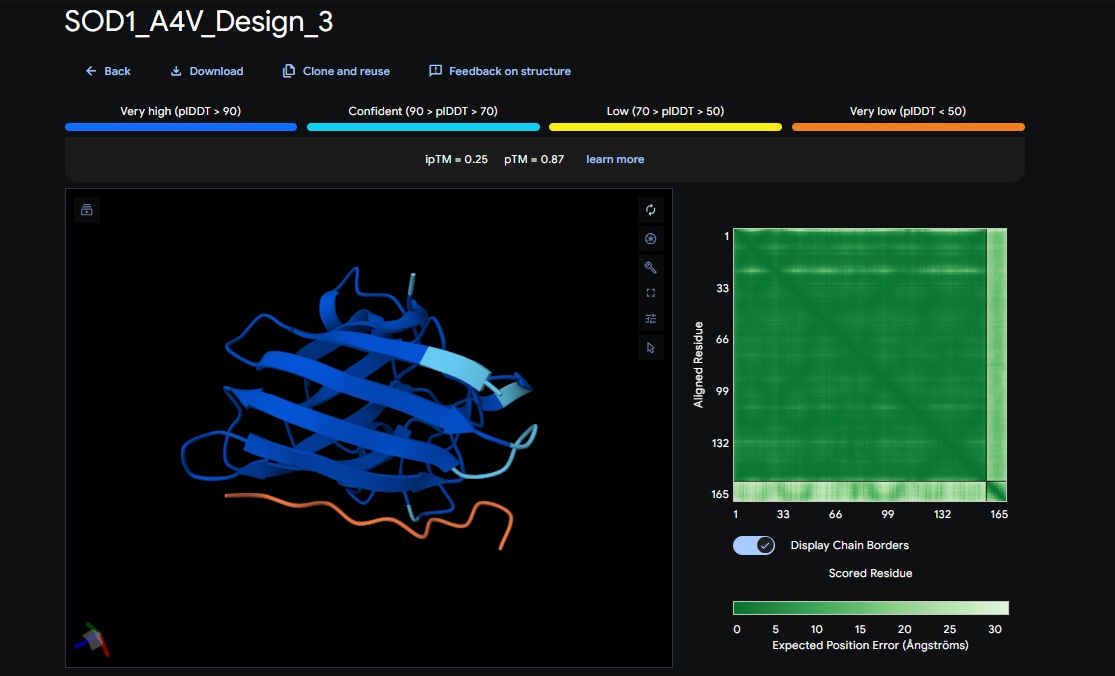
4. Design_4 (WRYYAAAIAHKK) + SOD1:
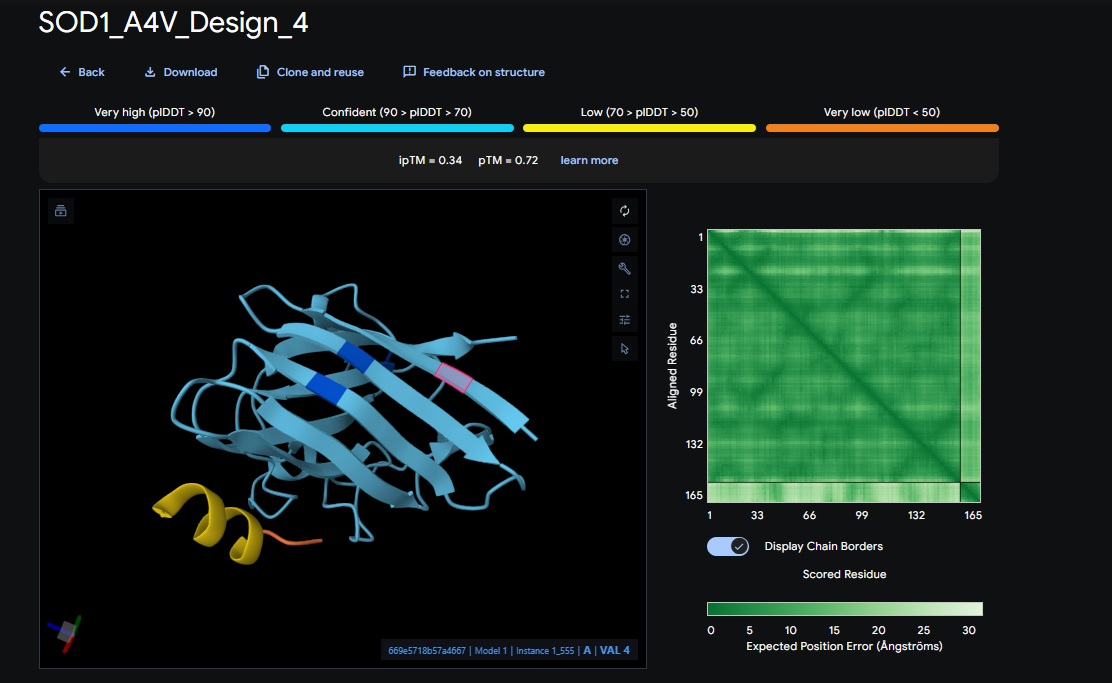
5. Control_Peptide (FLYRWLPSRRGG) + SOD1
At this point I realised that I was only doing the pepMLM and alpha fold with the monomer of the protein so the binder I am trying to design is for the enzymes natural dimer state, So I am going to redo the above sections and see if I get different scores.
2. PepMLM Generation Results for Dimer and 15 AA Peptide length (first 4 designs):
| Candidate | Sequence | Pseudo Perplexity |
|---|---|---|
| Design 1 | WTWVVHVATHKHHLK | 15.02 |
| Design 2 | DTVVHHHATHEHKKK | 14.28 |
| Design 3 | WTVEHHLVTKQEKKK | 10.65 |
| Design 4 | DTWDHHLATKEHKKK | 10.52 |
| Design 5 | WTRVHAVVEKKK | 13.10 |
| Design 6 | ATHVHVAIHHKK | 7.738 |
| Control | FLYRWLPSRRGG | 31.133 |
Note: Lower perplexity indicates higher model confidence in the sequence’s fit for the SOD1 mutant.
Part 2: Evaluate Binders with AlphaFold3:
AlphaFold3 Evaluation Table
| Candidate | Sequence | ipTM Score (Good Score > 0.80) | pTM Score (Good Score > 0.50) | Avg. pLDDT | Binds at A4V? |
|---|---|---|---|---|---|
| Design 1 | WTWVVHVATHKHHLK | [0.81] | [0.86] | [low] | No |
| Design 2 | DTVVHHHATHEHKKK | [0.84] | [0.89] | [low] | No |
| Design 3 | WTVEHHLVTKQEKKK | [0.75] | [0.83] | [low] | Closest Yet |
| Design 4 | DTWDHHLATKEHKKK | [0.67] | [0.79] | [low] | No |
| Design 5 | WTRVHAVVEKKK | [0.82] | [0.87] | [low] | No |
| Design 6 | ATHVHVAIHHKK | [0.88] | [0.91] | [low] | Almost |
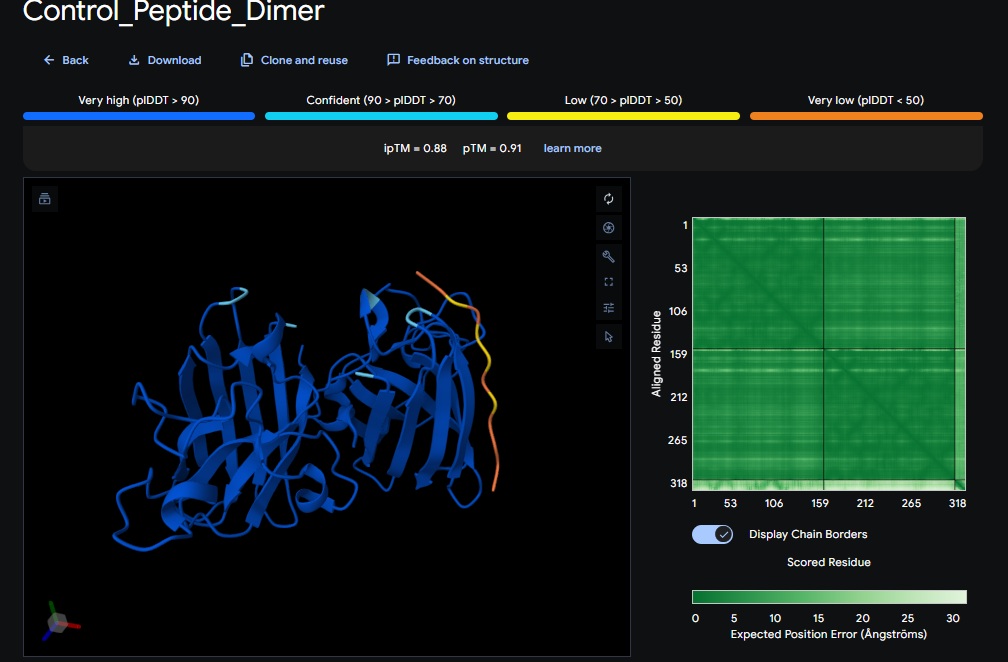
| Control | FLYRWLPSRRGG | [0.88] | [0.91] | [low] | No |
1. Design_1 (WTWVVHVATHKHHLK) + SOD1:
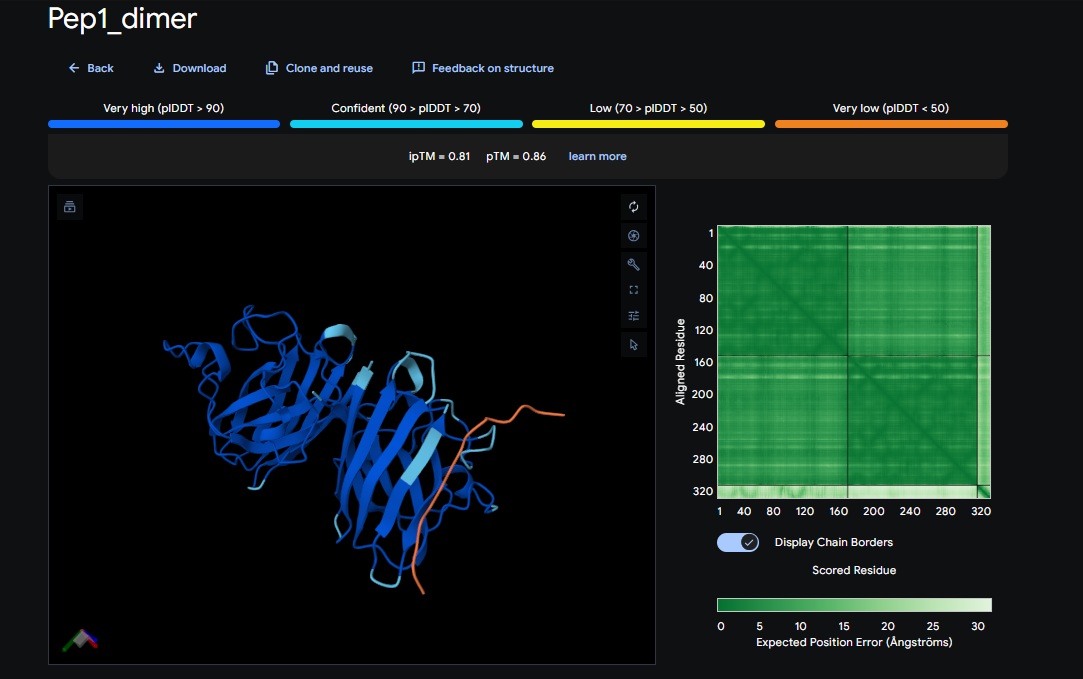
2. Design_2 (DTVVHHHATHEHKKK) + SOD1:
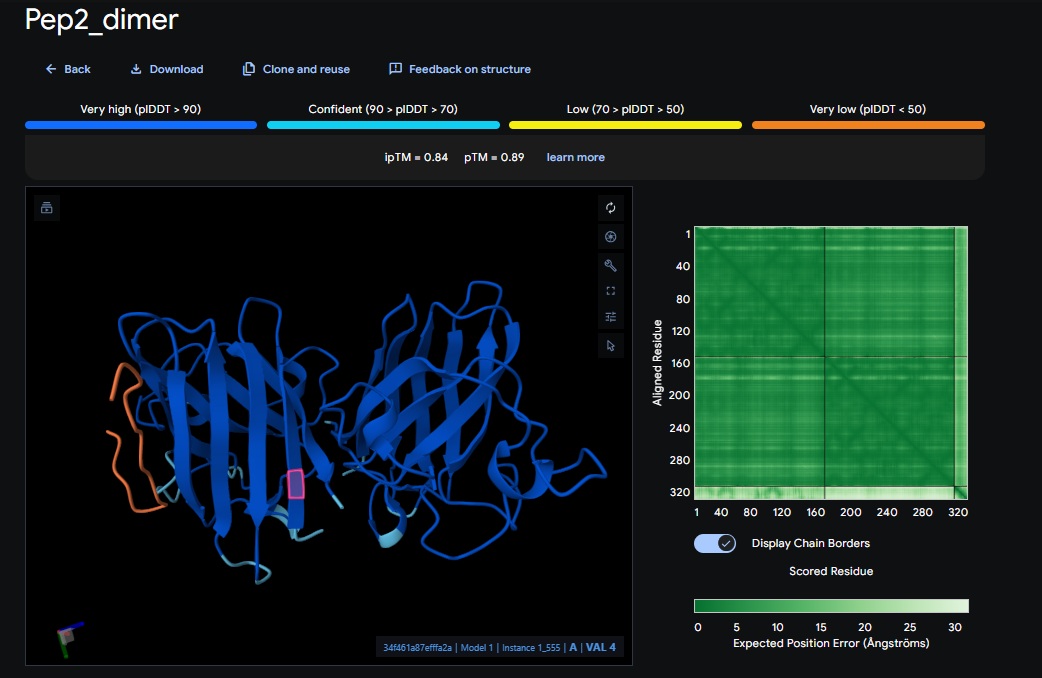
3. Design_3 (WTVEHHLVTKQEKKK) + SOD1:
You can see the peptide burried in the active site for the first time and both ends closest to the A4V Mutation sites of both monomers of the enzyme
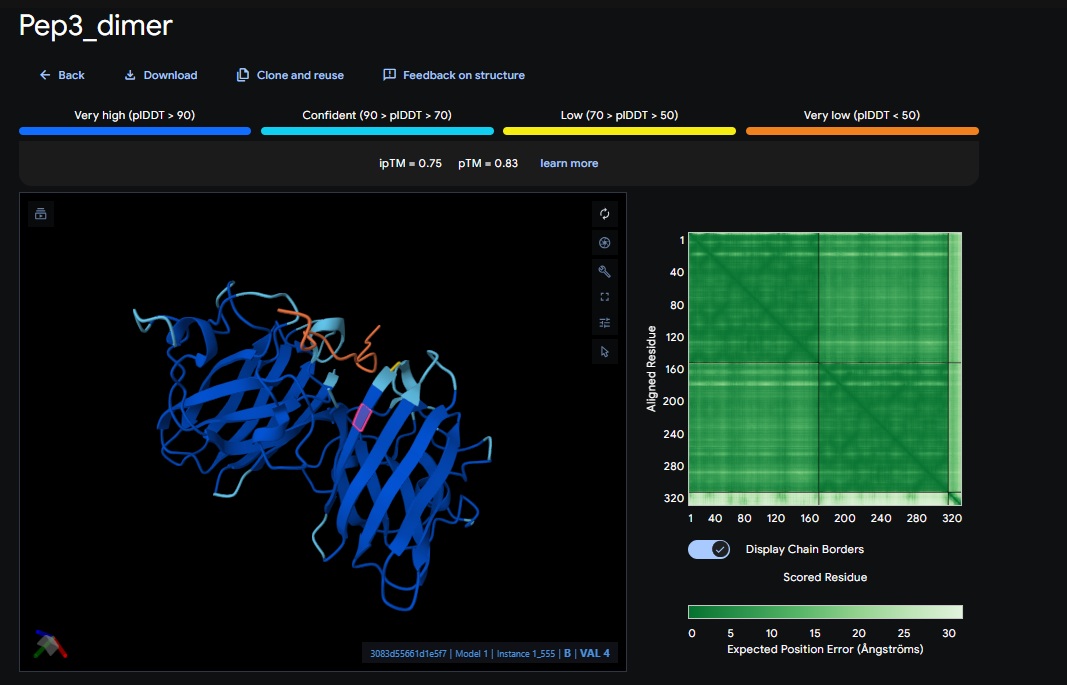
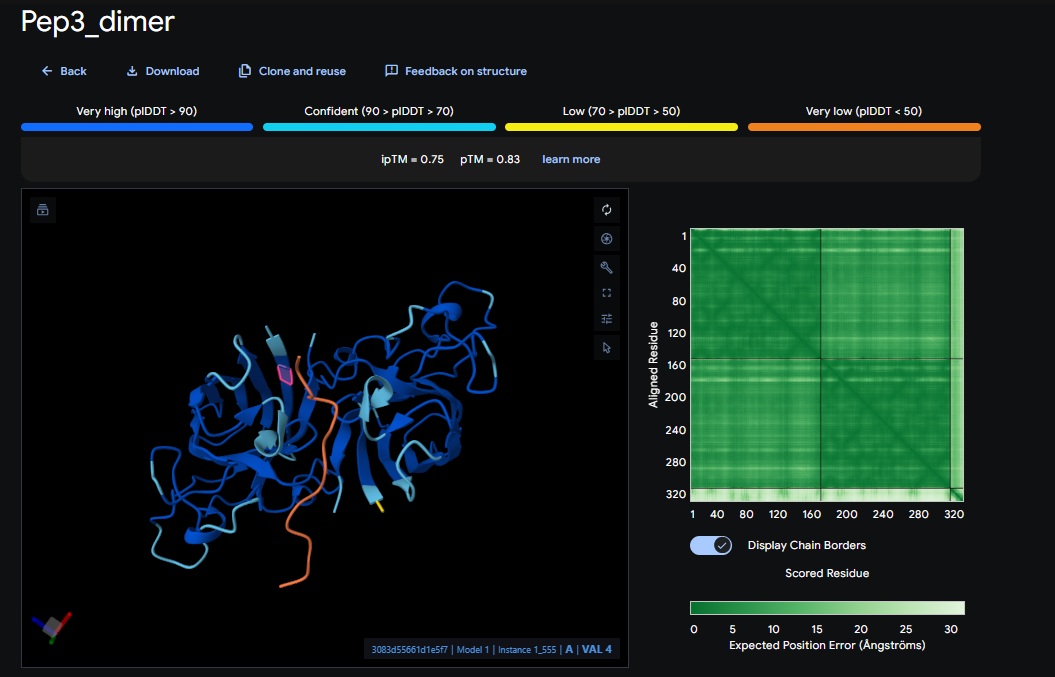
4. Design_4 (DTWDHHLATKEHKKK) + SOD1:
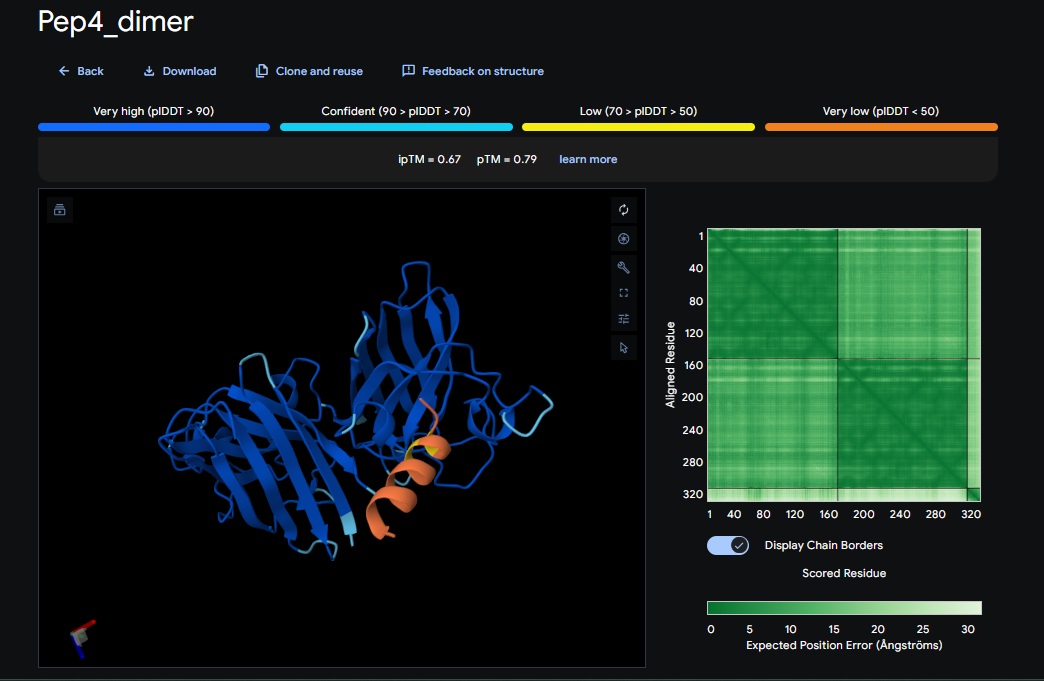
5. Design_5 (WTRVHAVVEKKK) + SOD1:
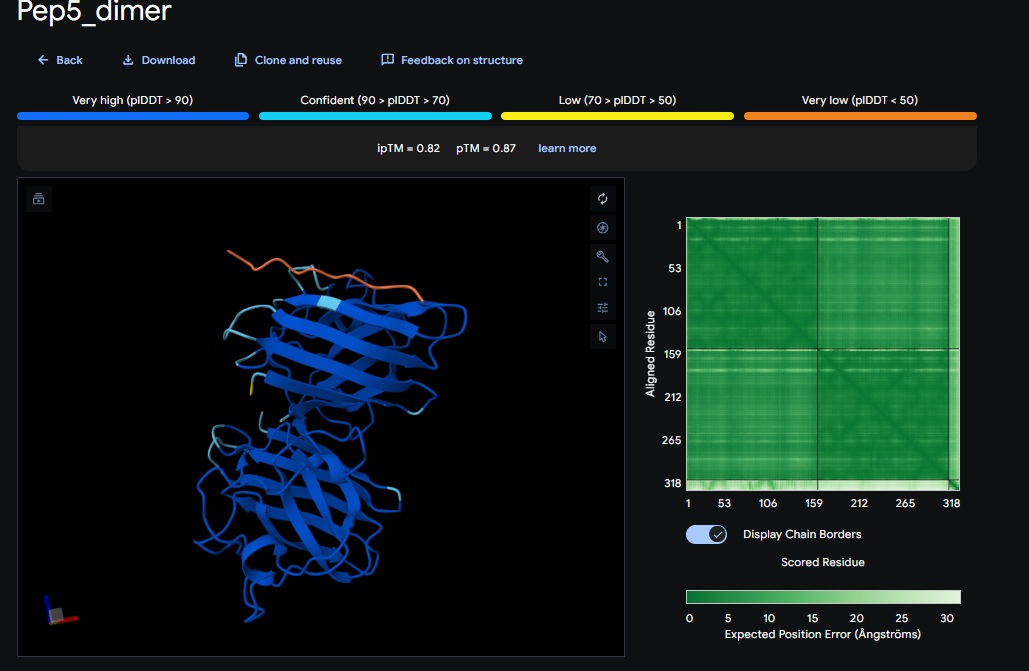
6. Design_6 (ATHVHVAIHHKK) + SOD1:
burried peptide
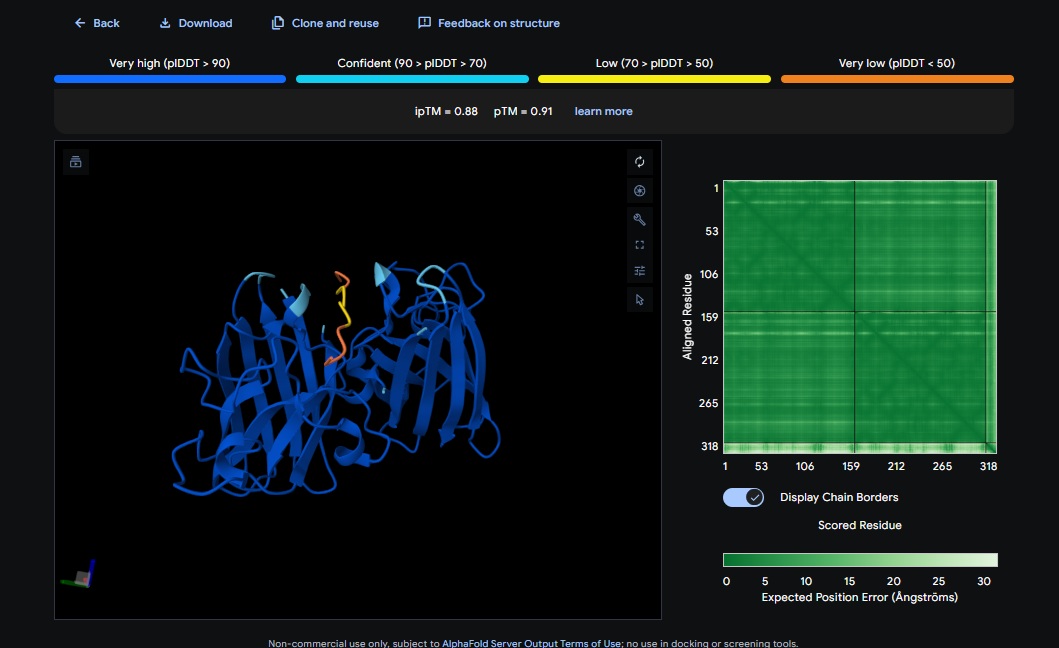
Control_Peptide (FLYRWLPSRRGG) + SOD1
PepMLM and AlphaFold Results Interpretation:
- Significant improvement in Confidence (ipTM): Dimer breakthrough: Switching to a homodimer target in PepMLM was the turning point for better AplphaFold scores.
- Perplexity vs structure correlation:
Visually the best peptides for the dimer were Design 3 (
WTVEHHLVTKQEKKK), Design 6 (ATHVHVAIHHKK) and Design 4 (DTWDHHLATKEHKKK). Interestingly these also had the best perplexity score (10.65, 7.73 and 10.52 respectively) but not necessarily the best TM scores. - The Design 6 peptide with the dimer (
ATHVHVAIHHKK) got the highest ipTM (0.88) and pTM (0.91) scores closest to the control (FLYRWLPSRRGG) which got the same ipTM (0.88) and pTM (0.91) scores. The control didnt get a good perplexity scores and interestingly the control peptide and several designs seem to be on the same spot not near the N-terminus but near one side of the b-sheets. . Both scores dont always seem to align. It seems that perplexity measures how natural the sequence feels to the language model, but ipTM measures the physical docking. - Targeting the A4V Mutation: While the Control Peptide and designs showed high general binding scores, Design 6 and 3 were the only ones that were localized near the N-terminus, which is the actual site of the A4V mutation. This suggests that raw scoring (ipTM) doesn’t always guarantee site-specific binding. Alpha fold likes the control peptide, pepMLM doesnt.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
| Candidate | Sequence | Solubility (Prob) | Hemolysis (Prob) | Binding Affinity (pKd/pKi) | Net Charge (pH 7) | MW (Da) |
|---|---|---|---|---|---|---|
| Design 1 | WTWVVHVATHKHHLK | [1.000 (Soluble)] | [0.035 (non-hemolytic)] | [5.742 (weak)] | [+2.11] | [1879.2] |
| Design 2 | DTVVHHHATHEHKKK | [0.995] | [0.032] | [4.652 ] | [+1.2] | [1804.0] |
| Design 3 | WTVEHHLVTKQEKKK | [1.000] | [0.020] | [4.990 ] | [+1.94] | [1891.2.1] |
| Design 4 | DTWDHHLATKEHKKK | [1.000] | [0.068] | [6.092] | [+1.02] | [1874.1] |
| Design 5 | WTRVHAVVEKKK | [1.000] | [0.023] | [6.168] | [+2.85] | [1480.8] |
| Design 6 | ATHVHVAIHHKK | [1.000] | [0.019] | [4.846] | [+2.14] | [1377.6] |
| Control | FLYRWLPSRRGG | [1.000] | [0.047] | [6.098] | [2.76] | [1507.7] |
After comparing all the results seems to me that the best binders (binder 3 and 6) with respect to the perplexity scores and AlphaFold dont necessarily have the best binding affinity. The best affinity seems to be of Design 5 which actually had a good AlphaFold scores as well (ipTM :0.82) but not far more than the other peptides. They all seem to have good solubility and are non-hemolytic.
Based on all the calculations above I will choose Design 6 (ATHVHVAIHHKK) with the best perplexity score, best AlphaFold score and good therapeutic properties.
Part 4: Generate Optimized Peptides with moPPIt:
| Candidate | Binder | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| Design 1 | GEKVCYKLKCMH | 0.959324 | 0.750000 | 9.463325 | 0.597243 |
| Design 2 | CQDWYKSYRKYR | 0.942937 | 0.916667 | 7.473063 | 0.467137 |
| Design 3 | RQYDTYYEKCVS | 0.948101 | 0.916667 | 8.280322 | 0.467137 |
All three peptide binder designs from moPPIt-v3 just knocked the binder design out of the park compared to the PepMLM binders. In no time it created stable binders at the right site (as confirmed with alphafold results below) with great affinity (highest @ 9.4 vs 4.8 from our best design above). All three are very soluble, non-hemolytic and decent binding to our target motif residues in the SOD1 protein (I chose motif residues 2-6 and 143-154).
Design 1:
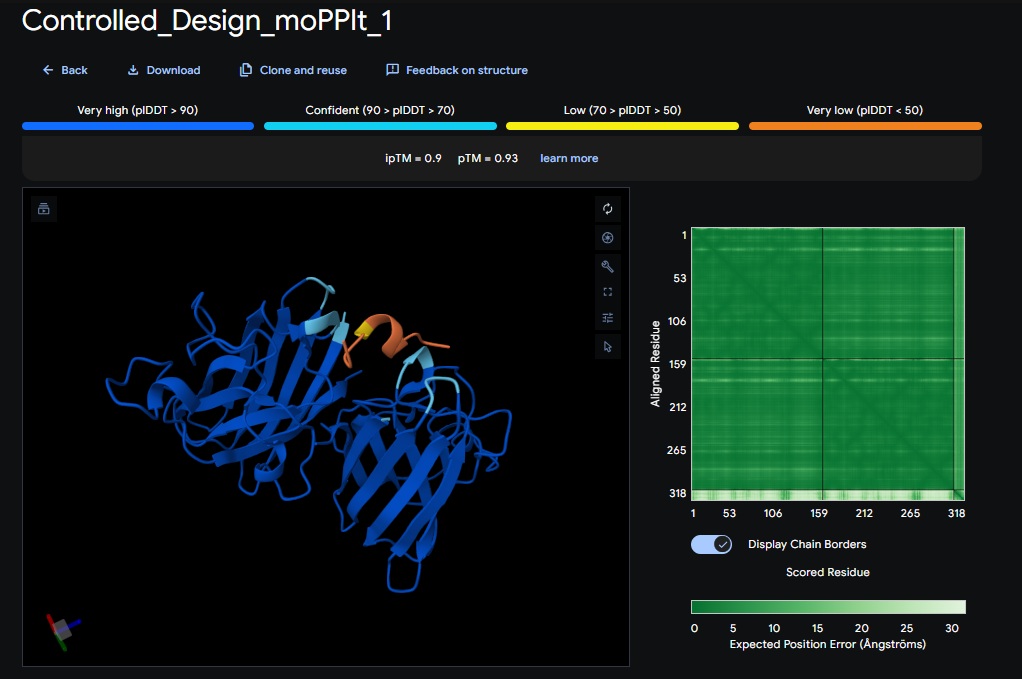
Design 2:
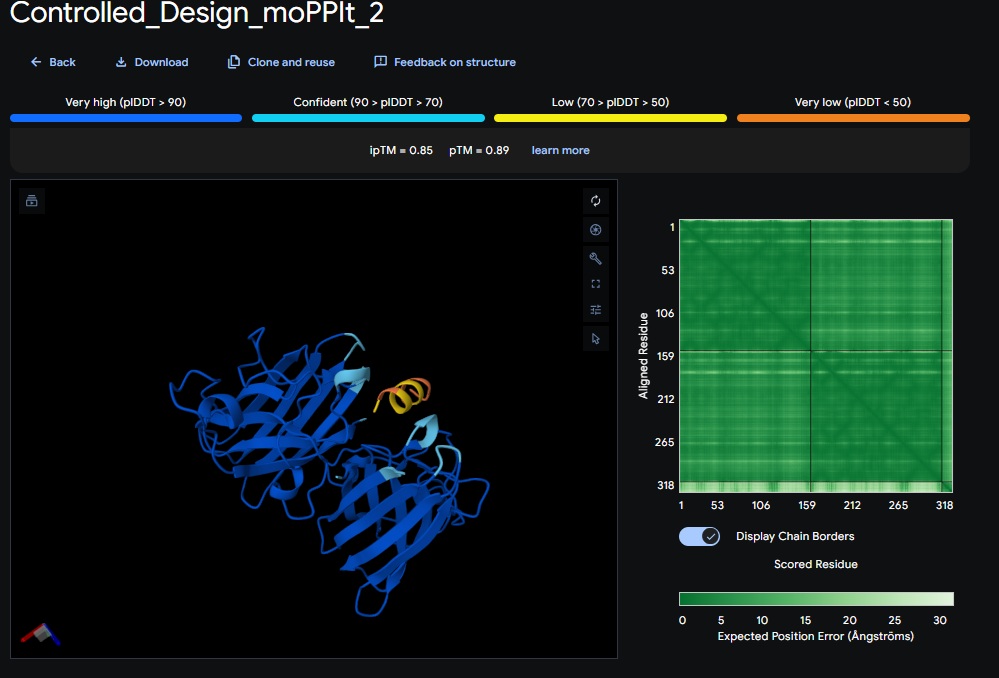
Design 3:
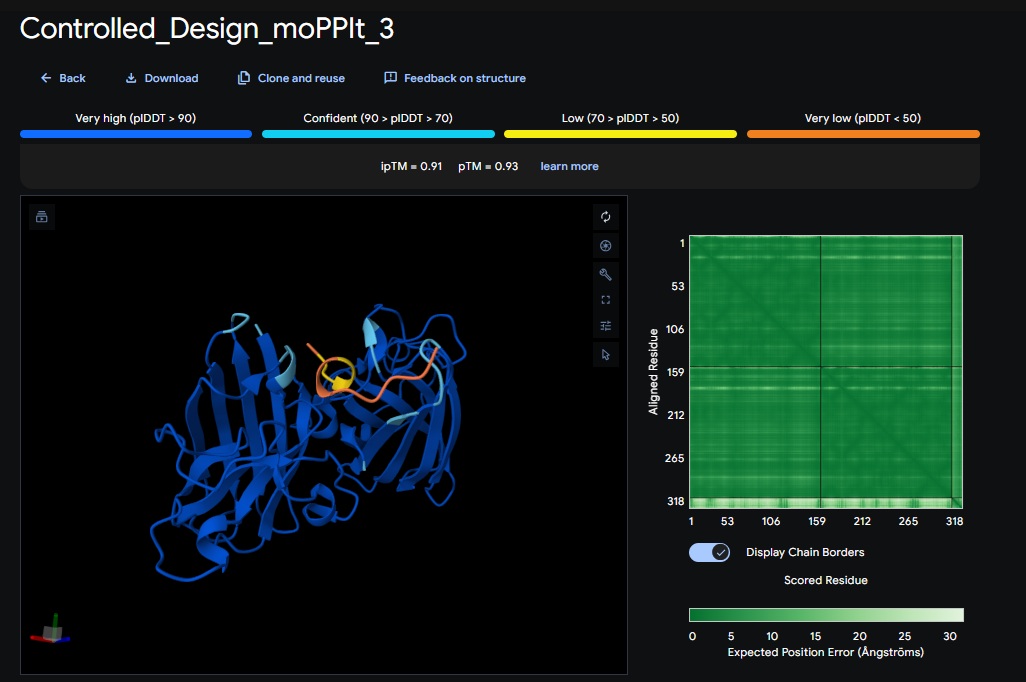
Part C: Final Project: L-Protein Mutants
Objective: Improve the stability and auto-folding of the lysis protein of a MS2-phage
Wild-Type L-protein: Here is what we are working with
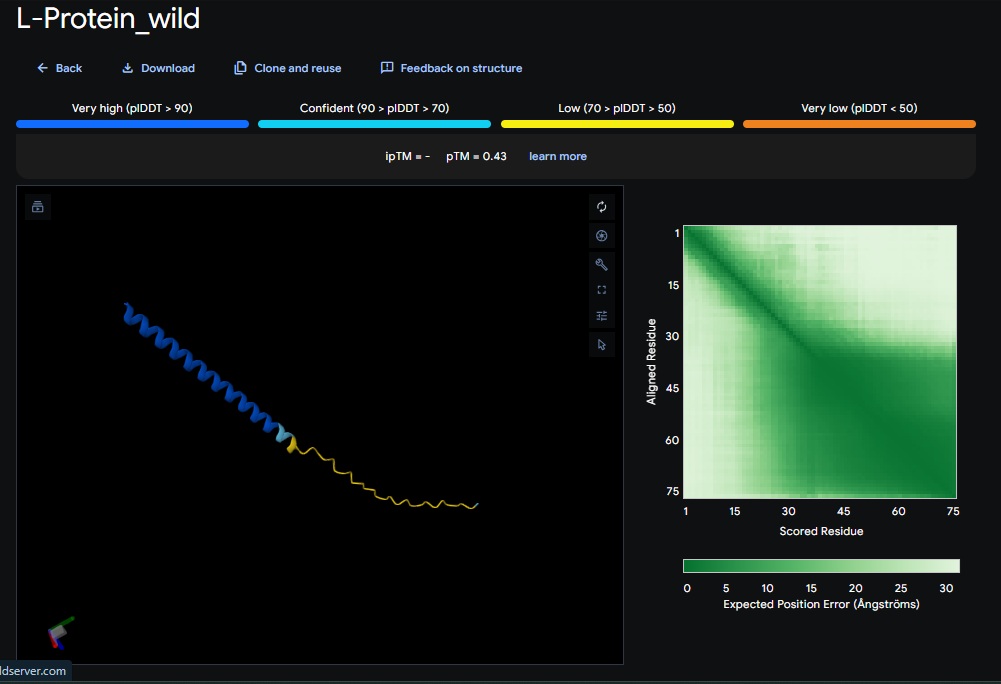
Key Summary of findings: The mutational analysis of MS2 L identifies the highly conserved LS motif (Leu 48 - Serine49) as the functional “trigger” for lysis. Experimental data also shows that even trace amounts of functional protein can cause lysis, but the protein is currently limited by its dependency on the DNA J host chaperone which the bacteria uses by P330Q mutation to get resistant(Chamakura et al., 2017).
De Novo L-Protein Strategy:
To bypass the DNAJ dependency, I am using moPPIt to redesign the L-protein as a self-folding antimicrobial agent.
Targeting the N-terminus: I am replacing the disordered, basic N-terminal domain (residues 1-35) with a stable, de novo- designed alpha-helix. This removes the regulatory domain that normally requires DNAj for displacement.
Maintaining Lysis activity: I am preserving the conserved hydrophobic C-terminal domain and the LS motif to ensure that the protein retains its ability to integrate into the host membrane and trigger autolysis.
Stability optimization: I am using AlphaFold2 to validate that the new sequence achieves a high confidence score in isolation. A high score suggests the protein will fold independently and rapidly, reducing the time window for the host to mount a resistance response.
Tool Pipeline Used:
moPPIt (for multi-objective optimization) ——-> AlphaFold2 (for structural validation of autofolding potential) ——–> PeptiVerse (check for high solubility and binding affinity)
Method:
- target protein to modify on moPPIt :
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT - binder length to be generated by moPPIt: 75 (original size of the L-protein)
- Objective and weights in moPPIt:
- Hemolysis: We choose nontoxicity as score 1 at first eventhough we are creating a lysis protein. We can change this to 0 if we get low affinity.
- Solubility: We choose 1 because we need good solubility. This will hopefully fix the unstable N-terminal.
- Affinity: We choose 1 because we are looking for high affinity in the c-terminal portion.
- Motif: Keep the generated sequence anchored to the functional lysis residues (i.e. transmembrane region: 41-75, LS-Motif: 48-49).
Results:
L-Protein Variants designed my moPPit:
| index | Variant | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| 1 | YSDRFAITIVVEERAHSGVLKDYIAVVNKPWRNLCETKFFCSDTNACIIMLMMKYQPKGMGHVCWMYSHTTMVSN | 0.9257961213588716 | 0.5733333230018616 | 9.356831550598145 | 0.6821509003639221 |
| 2 | SDPWDMEGIGRLSAPAAVLEYACDHKHHLWVLPRNPFQHRGPHSWLNQKTEIKEDENIEGCRMWELVTVPPDATA | 0.8822786584496498 | 0.5600000023841858 | 9.074358940124512 | 0.6412587761878967 |
| 3 | WQWKHGSLQWDIYDGVPAYESMCVHDYGTCVTQSMNVEPWSVWFMLHVSNVADCDNGGLDNAWLEEERRFKDYSS | 0.8875014930963516 | 0.6266666650772095 | 9.25324535369873 | 0.6841824054718018 |
| 4 | TVDQSFEVPSLCEIDFHTGTSPPHRWSHARTGNCNGSYIFLYEDAMTLKKTWEMEGVEESPVPNSHTFTTDGAYF | 0.904684230685234 | 0.653333306312561 | 8.863005638122559 | 0.7156356573104858 |
| 5 | YIYWCQLRYEKDGLACVNCCVVCVALHWFDDVRGKMEPFSPPQPHPYIPQLCYEDMDCLAYMGPRVLFHAGGEMN | 0.9292040765285492 | 0.5466666221618652 | 9.797521591186523 | 0.6883391737937927 |
- Variant 1 AlphaFold2 structure:
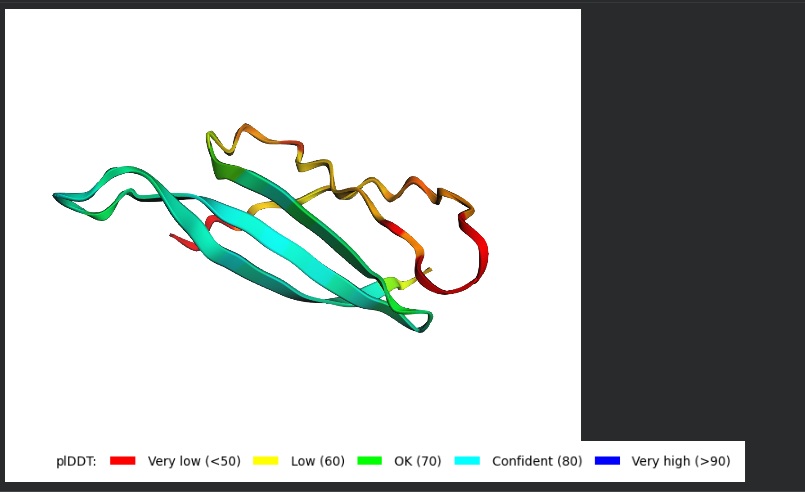
Interestingly the AlphaFold3 version that we were using for Pranams Assignment (which is using diffusion models instead of geometry transformers in AF2) gives a slighlty better image
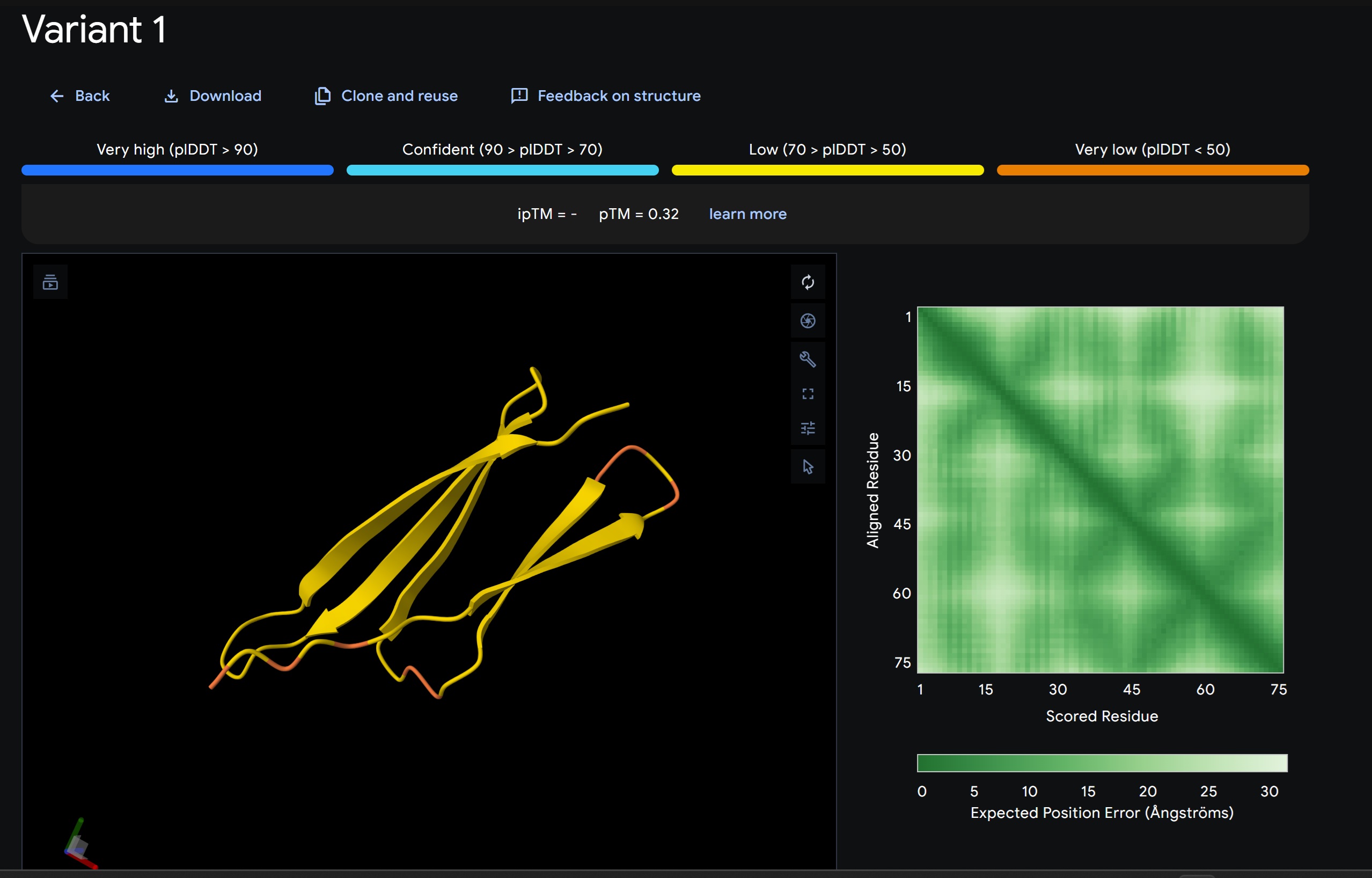
Variant 2:
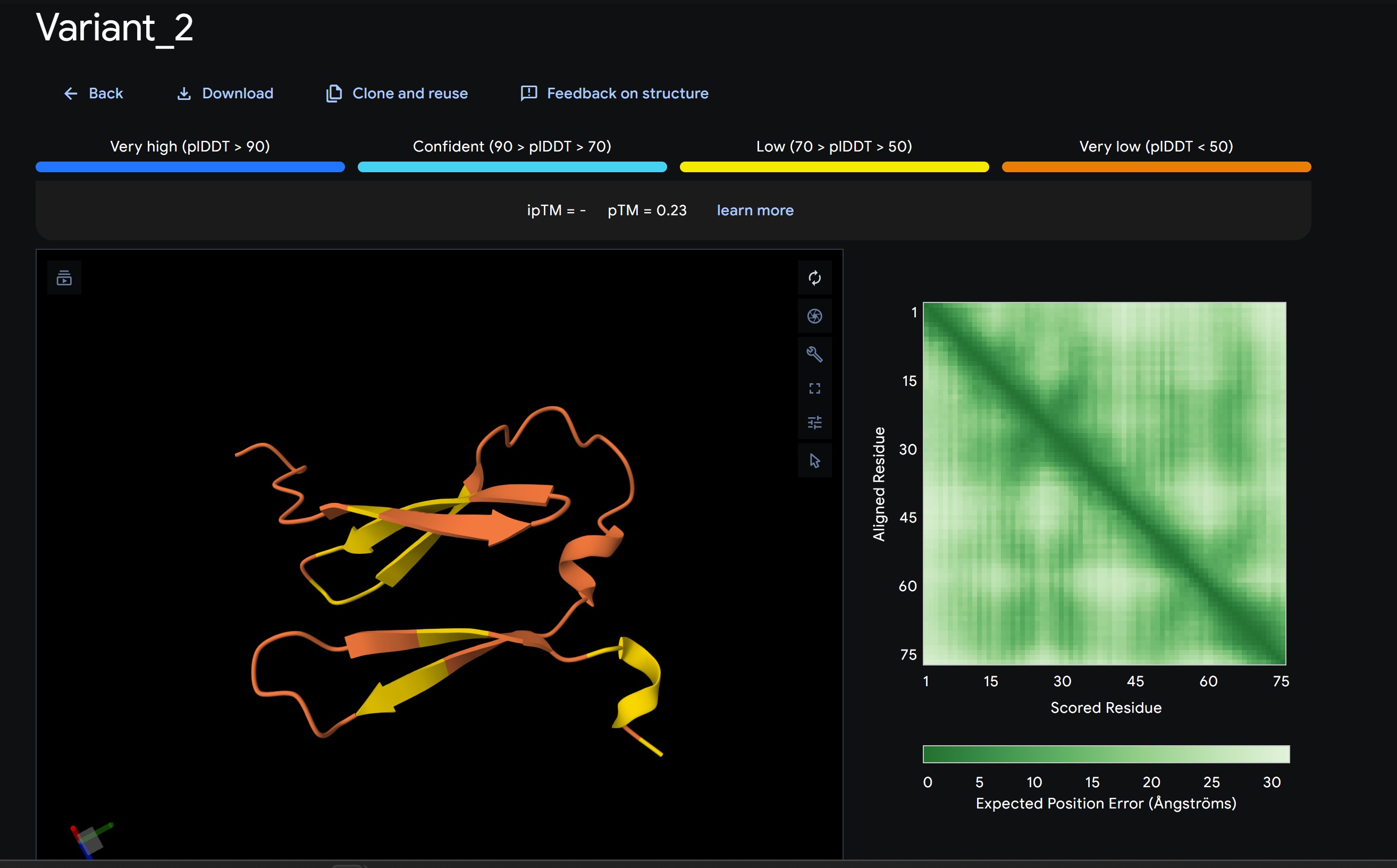
Variant 3:
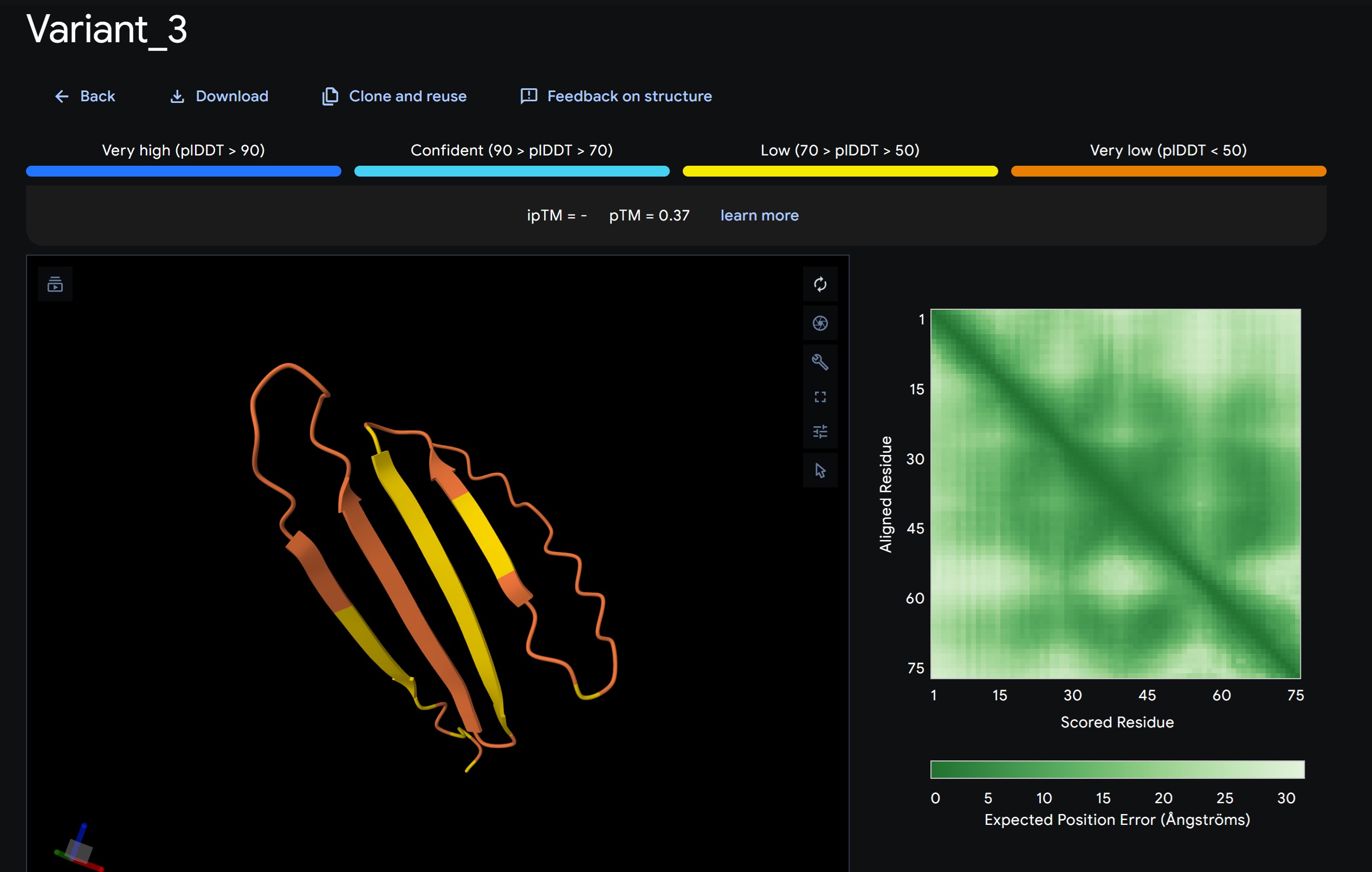
Variant 4:
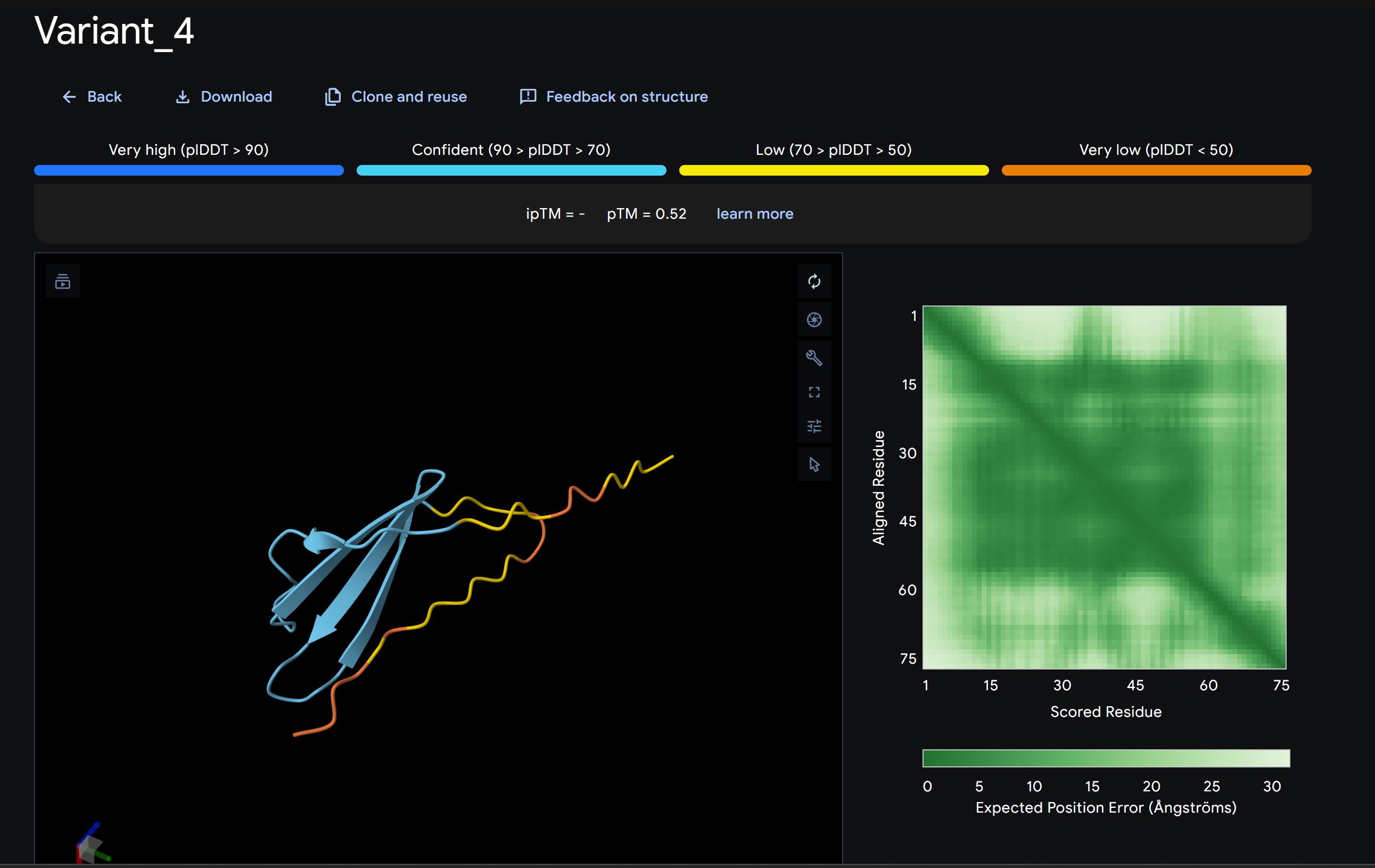
Variant 5:
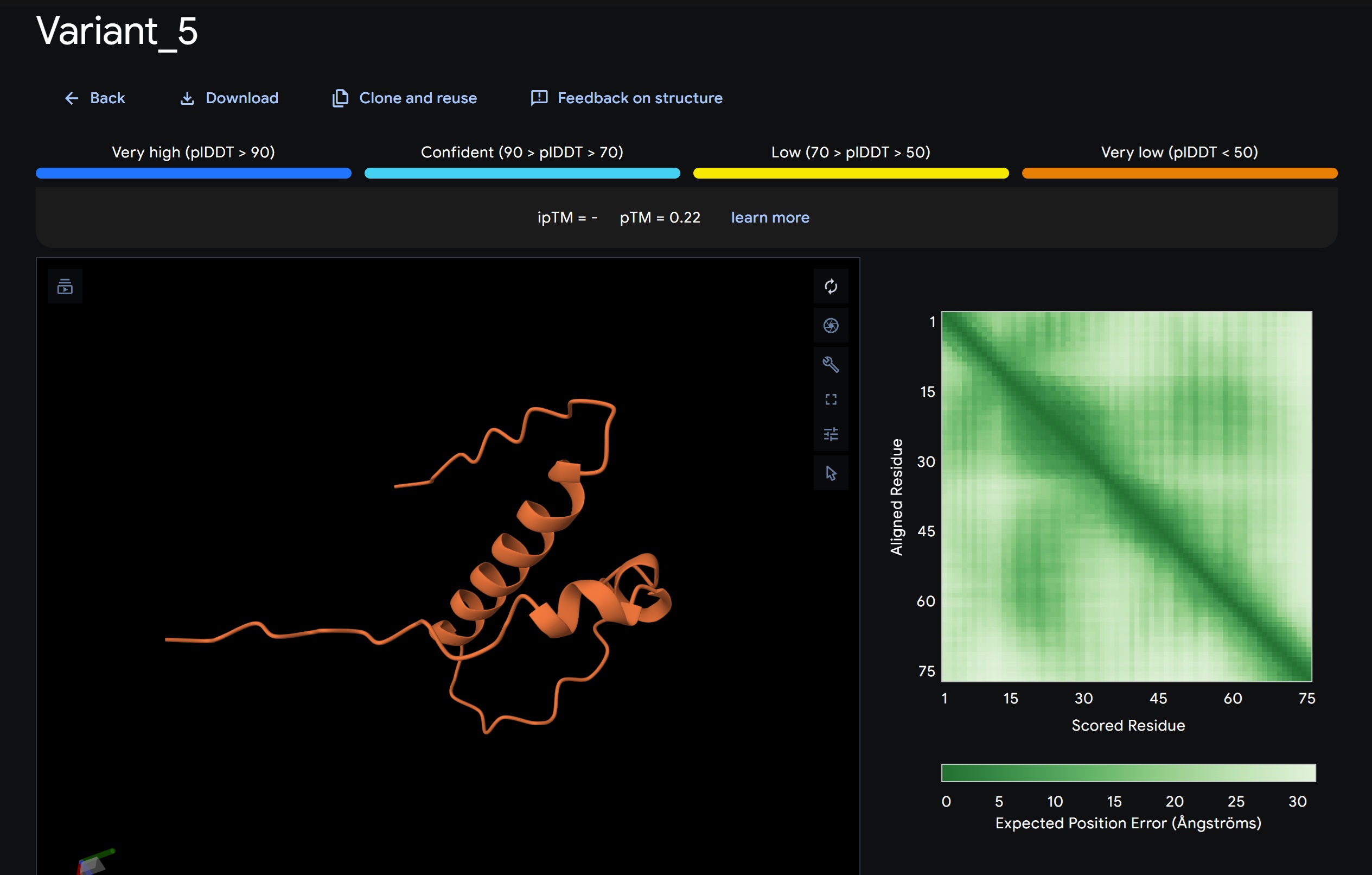
Discussion:
My lead candidate, Variant 1, was generated with a high Affinity score (9.35) and Motif score (0.68). Validation via AlphaFold3 shows the emergence of beta-sheet secondary structures in the redesigned N-terminus. While the overall pTM score is 0.32, the localized folding in the region previously dependent on DnaJ suggests a more rigid, self-stabilizing scaffold. This supports the strategy of using de novo helical/sheet designs to bypass the need for host chaperones like DnaJ (P330Q). By comparing these variants, it is evident that moPPIt can navigate the trade-offs between solubility (for DnaJ independence) and membrane affinity (for lysis efficiency). Variant 4 emerges as the primary choice for synthesis due to its superior motif preservation and balanced biophysical properties.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele):
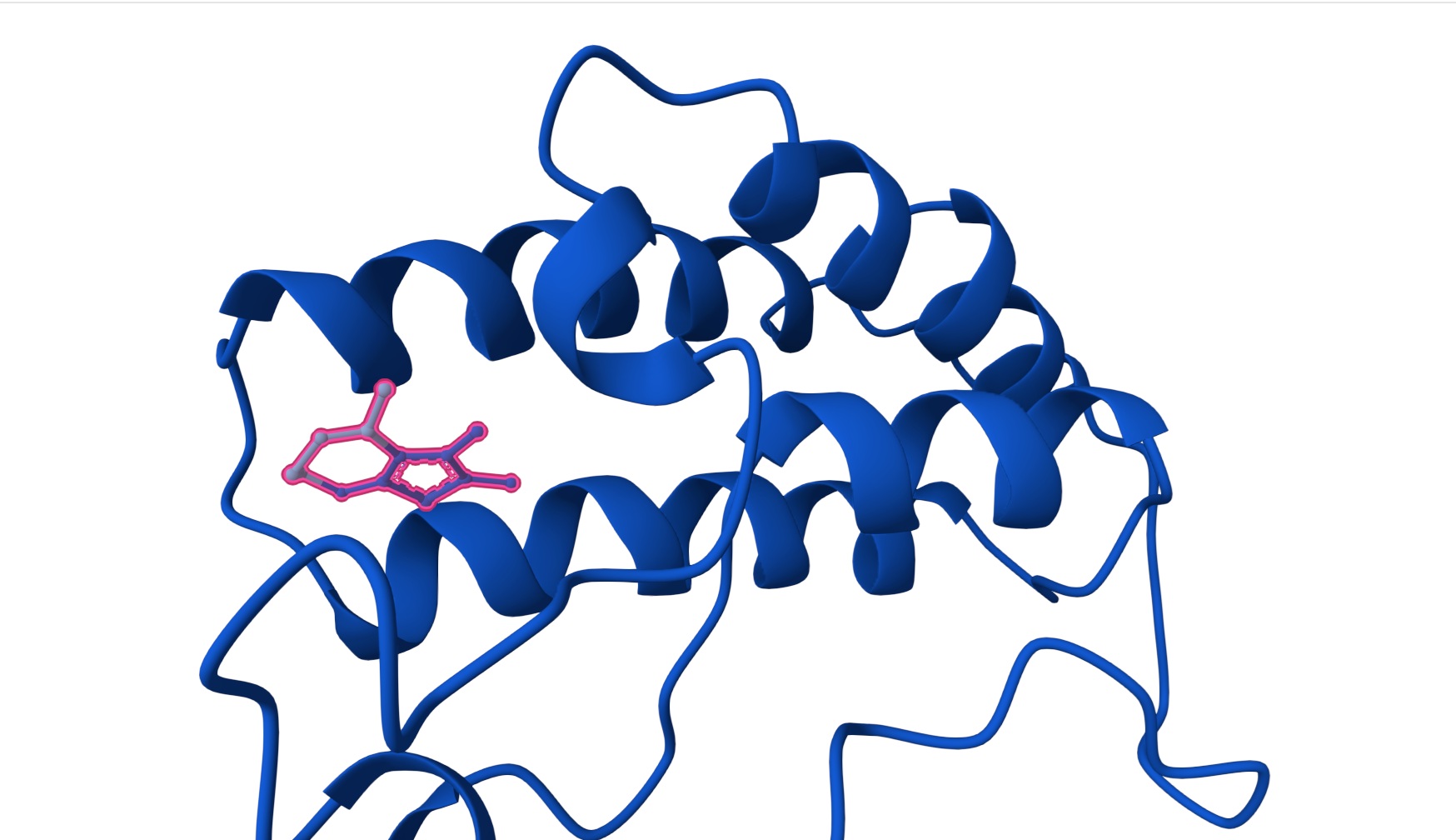
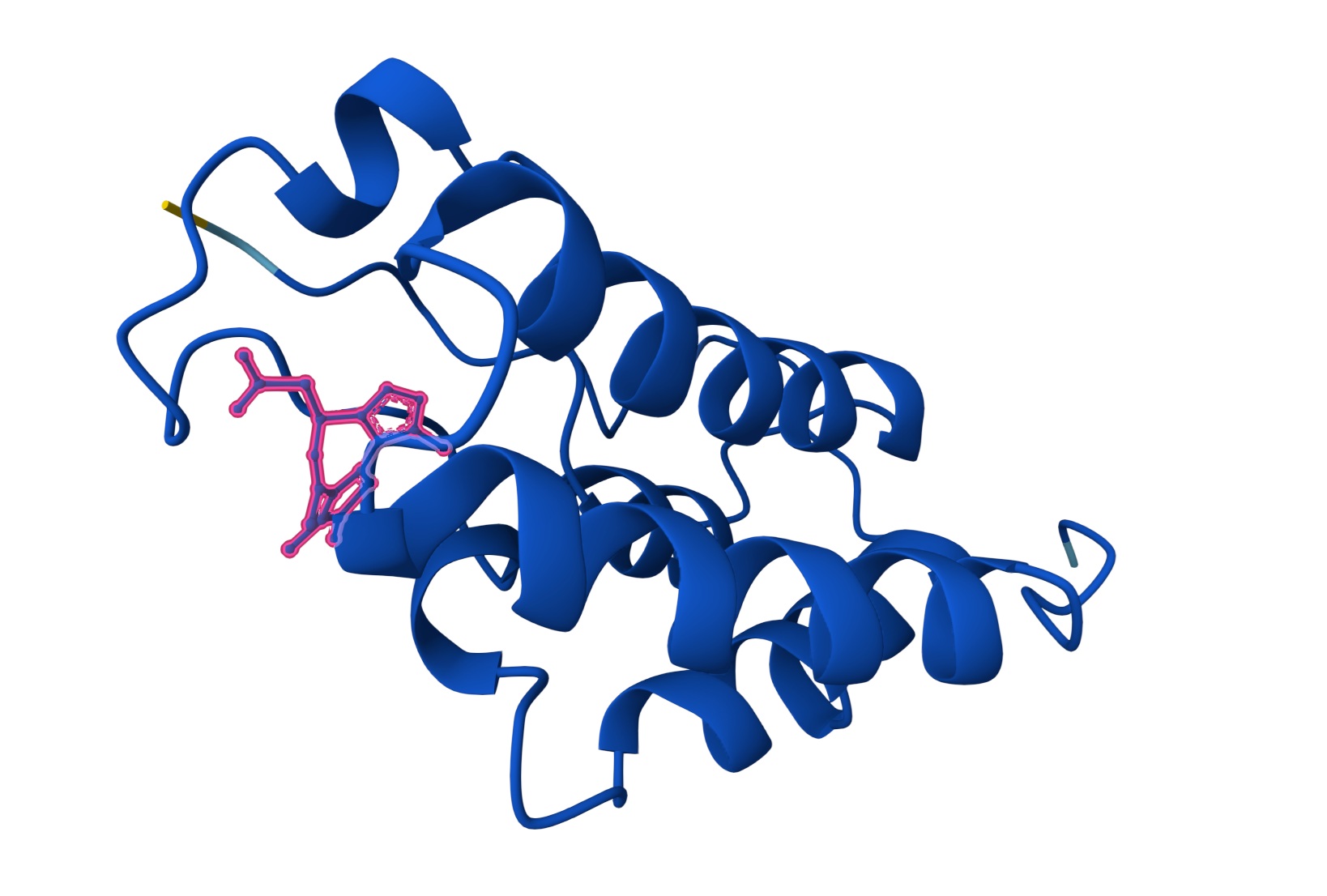
| Compound | SMILES | Binding Confidence | Optimization Score | Structure Confidence |
|---|---|---|---|---|
| Hit | CC1C2C(=C(SC=2NCCN=1)C)C | [0.44] | [0.22] | [0.97] |
| Lead | O=C(C[C@@H]1N=C(C)C2C(=C(SC=2N2C1=NN=C2C)C)C)O | [0.74] | [0.26] | [0.98] |
| JQ1 | O=C(C[C@H]1C2=NN=C(N2C3=C(C(C4=CC=C(C=C4)Cl)=N1)C(C)=C(S3)C)C)OC(C)(C)C | [0.96] | [0.44] | [0.98] |
Hit:
Lead:
Candidate JQ1: