I am a Biotech graduate exploring how synthetic biology can be used to solve real structural problems and how it can be used to bring the new age of materials that moves us away from petrochemicals.
I have worked in industrial bioprocessing and now I want to learn through this HTGAA commmunity how we can design systems that cooperate with life instead of extracting from it.
Many of the problems that motivate me are personal. Treatments that are ineffective, Polluted water in my birth country India and my new home in Canada, wasted nutrients, polluting materials, and technologies that are geographically bound.
I am here to build, fail early, document carefully and most importantly learn alongside people who believe in biology as an engineering platform and who want to implement it safely to increase good while minimizing harm.
I propose a smart biopolymer hydrogel patch that is designed to treat recalcitrant cutaneous HPV.
The Problem: Traditional treatment like Cidofir are effective but limited by poor penetration and systemic toxicity risks.
The Solution: A patch that acts as a local manufacturing unit, producing therapeutics only when specific triggers are met.
Personal Motivation
My commitment to this project stems from watching close friends and family struggle with recalcitrant cutaneous HPV for decades. Despite enduring numerous rounds of painful cryotherapy and laser treatments, their condition remains unchanged. I am struck by the stagnation in treatment options for such a common and stubborn virus. Through this course, I want to bridge that gap by developing a localized, non-invasive solution that offers hope to those who have exhausted traditional medical paths
Learning Sandbox Goals
Protein Design: I am really interested in learning tools like Alphafold and rosetta to design peptides that can specifically degrade the HPV-specific oncoproteins.
Designing genetic circuits that use logic to ensure precision delivery.
I love the idea of Living Materials that act like mini computers embedded into the biopolymer to produce the antiviral peptide on demand.
2. Governance Policy Goals:
From reading the resource material and this weeks presentations I think to ensure a technology like this to be developed ethically, here I propose three primary policy goals:
Safety & Security:
Prevent the accidental release of potent antivirals or engineered microbes out of the living material to unintended places.
Treating patient safety as paramount. So the first trials will always be done under medical supervision.
Making the product as inert as possible when not in use and easily biodegradable.
Equity and Autonomy:
In the longer run, the patch needs to be affordable and self-administration capable. This will reduce expensive clinic visits which currently limits access to HPV care.
Facing the shame associated with such conditions head on and help patients feel understood and cared for. Making information public is the first step towards that.
Constructive Use:
Prevent the genetic circuit delivery system to be used by bad players to deliver harmful compounds.
As with the fields half pipe of doom these technologies must be steered away from bad actors. But since learning to tame fire humans have managed to live with dangerous but useful things. So keeping an optimists perspective will help be a step ahead of evil.
3. Proposed Governance Action:
(Three options)
i: Cell-free Mandate:
This requires all biological machinery on peoples skin to exist in a cell free state to avoid mishap.
Use cell free systems that come alive under the right condition.
There should be a clear On and Off state trigger and indication that is mandatory to perform in order to use the polymer.
ii: Open Source Design Ledger:
Create a local registry of peptides used in the genetic circuits.
Allow for a broader collaboration platform that allows various participants in the building of such smart biomaterials.
iii: Tiered access control:
Limits access to high potency therapeutics to certified labs while keeping biopolymer open-source
Learn from existing methodologies and governance practices that deal with sensitive and potentially harmful information and build on top of them.
Current actors interested in this will be researchers who already work with antivirals but might not be native to synthetic biology tools. I would like to share MVPs with them to understand there safety concerns and whether they think my design has any flaws. Also learning about local biosafety laws during the design of the MVP will be paramount. Also health care providers and industry manufacturers of such antivirals would be good collaboration partners.
Details
4. 📊 Governance Scoring Rubric
(Scale: 1 is Best)
Does the option:
Cell-free Mandate
Open Source Design Ledger
Tiered access control
🛡️ Enhance Biosecurity
• By preventing incidents
1 🥇
3
1 🥇
• By helping respond
1 🥇
3
2
🔬 Foster Lab Safety
• By preventing incident
1 🥇
3
1 🥇
• By helping respond
1 🥇
3
2
🌿 Protect the environment
• By preventing incidents
1 🥇
3
3
• By helping respond
2
3
3
⚙️ Other considerations
• Minimizing costs/burdens
3
1 🥇
2
• Feasibility?
2
2
2
• Not impede research
1 🥇
1 🥇
2
• Promote constructive apps
1 🥇
1 🥇
1 🥇
5. Prioritization:
Thinking about the scores achieved by the proposed governance actions against this rubric I have found that the cell-free mandate should be prioritized. My learnings from this weeks class is that we should prioritize safety by design during every step of the DBTL cycle. This consecutively will allow broader open source collaboration on the biopolymer itself since harm is reduced from step 1. Throughtout the week I came up with a number of biopolymer and syn bio ideas ideas and did the governance rubric on each of them. These ideas included making a tennis string out of proteins, keratin based insulation material that has mold prevention immobilized enzymes, recreating breast milk to replace inadequate infant formula, using synbio to clearn the worlds waters through living synbio mats, programmable cambium. This weeks reframing taught me that this technology is still at its nascent stage and needs careful administration into crucial gaps in human requirements. Every thing we do changes the perception of syn bio to people. So we dont just want to present it as a novelty but an essential tool for the future of life. We must not only think about doing things that are possible but do them while keeping saftey, environment and access a priority. As much as we would like to replace petrochemicals with biopolymers straightaway there use is currently highly specialised. Also using it to solve a perosonally inspired project will make sure that I keep safety and access paramount in my mind. And through every stage of the project, design the safety before hand and not treat it as an impediment just to jump regulatory hoops. The designers of the biology must also be the designers of safety around that biology. Also I would say the trade off of making tiered access could cause some friction but having cell-free will make sharing this information open source more robust and possible.
Thanks to this new framework of thinking I will try to incorporate my bold claims here into practice in my project.
Week 2 Lecture Prep: Q&A
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Answer:As mentioned in the slides it appears that the error rate of DNA Polymerase is $10^{6}$ meaning there will be approximately 1 error every $10^{6}$ base additions. Based on a quick search the length of the human genome seems to be around 3 billion pairs $3\times10^{9}$ bp. So based on that there should be 3000 errors per cell division. So to avoid this DNA polymerase has a built in proof reading that corrects some of these errors. Further research shows that there are additional repair mechanisms that brings the final error to one per $10^{10}$ bases. Later in a slide we also learn about the MutS Repair System found in all DNA.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Answer: DNA nucleotide code codes for amino acids in triplet codons. Since there are four neuclotides it makes it 64 codons (4x4x4) that code for 20 amino acids. This gives each amino acid around 3 possible codon options. Since an average human protein is around 400 aa long that gives each protein $3^{400}$ possibilities of codon options for every protein. That is equal to $10^{190}$ which is more than the atoms in the universe which is $10^{80}$ . This is due to the combination of reasons. One of them being the amount of GC neuclotide pairs which make stronger bonds than AT pair. So nature needs to balance the amount of these GC pairs. Too few and the resulting structure is unstable and gets degraded and too high means excessive folding making the DNA/RNA innaccessible for the polymerases and the ribosomes. In the final slide fabrication complexity mathematically explains that biology lives at the half max of complexity where a polymer of N monomeric building blocks of Q different types has Q acheiving the half max at 20 for a polymer of length 500. Which is the same number as number of aa (20) for an average human protein size (500). So nature lives right at this balance of complexity and redundancy.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
Answer: Solid-phase phospohoramidite DNA synthesis invented in 1981 by Caruthers which happens one neuclotide at a time in the 3’ to 5’ direction reverse of how nature (DNA polymerase) does it. Dr Prosts slides and Prof Jacobsons slides show some cool mechanism of removing something called the DMT group in certain neuclotide and then flooding the system with neucleiotides and thus synthesizing many parallel oligos at once. Quite remorkable . While the chemistry is the same as in 1981 its is now scaled using silicon-based microchips (like those used by Twist Bioscience) to synthesize upto 1 Million unique oligos at once much more efficiently, significantly reducing associated energy consumption and costs.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Answer: In Phosphoramidite synthesis small inefficiencies compound so with the biological error correction mechanism even industry standard coupling efficiency of 99% becomes catastrophic over hundreds of cycles so even a 200-mer has a overall yield of $0.99^{200}$ which is approximately 13% accuracy. Plus it probably becomes very messy to control because I would assume that the momomers would wanna fold incorrectly.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Answer: Error rates would lead to multiple mutations per molecule at that scale leading to very few correct full length sequences and will also take a long time. Thus the slides recommend assembling of smaller gene fragments (around 5kb according to the slides) to reduce error and increase control.
Homework Question from George Church:
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Answer: The 10 essential amino acids (once we cant produce ourselves) include 9 core EAAs namely Histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan and valine. Arginine is considered essential for specific species. I find it intreseting that life didnt progrzm itself to be fully self sufficient by producing them all. But then maybe thats a higher expenditure on the cellular machinery. Maybe biology evolved to produce the things only if it wasnt abundant in the environment. The Jurrassic park reference of the lysine contingency seems ridiculous since lysine is one of the 10 amino acids animals dont produce themselves sufficiently yet we are able to get them from eating meat and plants. Thus you couldnt keep the dinasours from surviving without suppliying them with lysine because just like us they had the option of getting lysine from eating other herbivores and plants or the Tourists. The slide also shows NSAA (Non-standard amino acids) that can pave the path for us to design synthetic life dependent upon different sets of amino acids. How would that change things I wonder!
Chat GPT 5.2 generated image with prompt: “Futuristic medical illustration of a smart biopolymer patch…”
*Thanks for reading!*
Week 2 HW :DNA Read, Write and Edit
Part 1: Benchling & In-silico Gel Art
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion
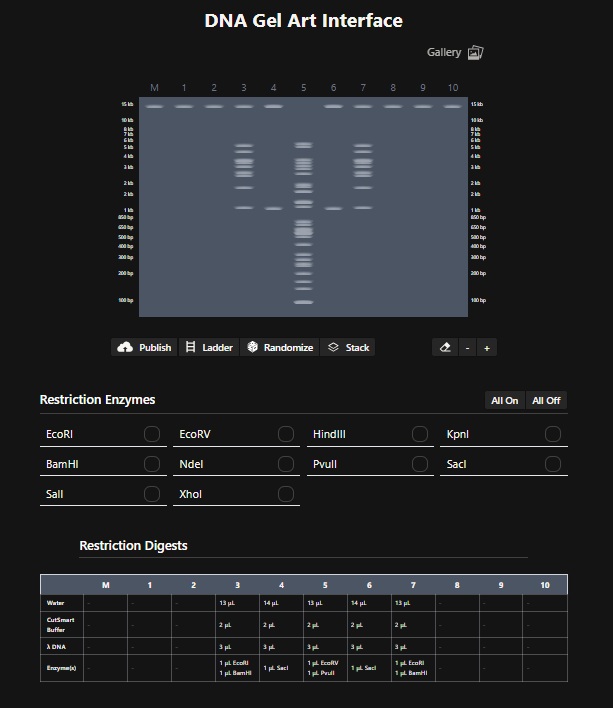
Design using Ronan’s website: Cactus Digest. I found this tool very useful. For the longest time I was just trying to get a good pattern randomly. Then i realised I can edit individual columns. With some more digging it became clear that I need to find restriction enzymes that slice the DNA in the bands that appear like something on the digest. So for the lane 5 I used EcoRV and BanHI to get the nice trunk of the Cactus design. For lane 4 and 6 i used SacI since it only makes two bands. And for lane 3 and & I used EcoRI and BamHI to make the 2 stems of the cactus. A little throwback to where I am from (Rajasthan).
Create a pattern/image in Benchling- I then recreated ronans website design by performing the digest on the lamda phage genome downloaded from GenBank in Benchling using the digest enzymes that ronans website described. The following is a screenshot of the Virtual Digest in Benchling
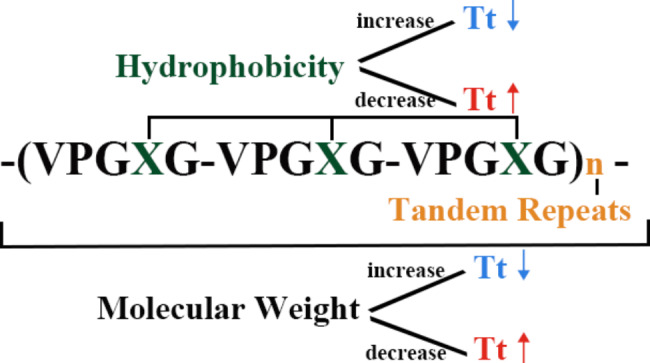
Since I am working on building a smart hydrogel I did some literature review and found that Elastin Like Polypeptides (ELPs) are good candidates. ELPs are recombinant protein polymers inspired by the natural protein Elastin, found in the extra cellular matrix, which is responsible for the flexibility of our bodies. ELPs are thermally responsive biopolymers and capable of having customizable properties. ELPs undergo something called an inverse temperature transition. This means that when heated, normal polymers dissolve but ELPs do the opposite. So ELPs stay soluble below a certain temperature and above a transition temperature (Tt) they undergo hydrophobic collapse and aggregate. ELPs interestingly are just repeat pentapeptides with amino acidv(AA) sequence Val-Pro-Gly-X-Gly (VPGXG)n where X can be any canonical amino acid except proline. This interchangebaility of X gives ELPs their customizability and such engineered ELPs exhibit excellent potential as customizable platforms for drug delivery and tissue repair.
structure of ELPs and its impact on its own Tt (Guo Y, Liu S, Jing D, Liu N, Luo X. The construction of elastin-like polypeptides and their applications in drug delivery system and tissue repair. J Nanobiotechnology. 2023 Nov 11;21(1):418. doi: 10.1186/s12951-023-02184-8. PMID: 37951928; PMCID: PMC10638729.)
For this weeks assignment I designed a short ELP consisting of only 10 repeats with X= Serine to creat a moderately hydrophilic scaffold:. So the AA sequence will be:
VPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSG
3.2 Reverse Translate: Protein (AA) sequence to DNA (NA) sequence:
For this part I used reverse translation tool online for this as suggested in the homework instructions and got this as a result
“>reverse translation of Untitled to a 150 base sequence of most likely codons.
gtgccgggcagcggcgtgccgggcagcggcgtgccgggcagcggcgtgccgggcagcggc gtgccgggcagcggcgtgccgggcagcggcgtgccgggcagcggcgtgccgggcagcggc gtgccgggcagcggcgtgccgggcagcggc
3.3 Codon optimization:
Why optimize?
Now the problem here is that its a highly repetitive DNA with repeats of GTG CCG GGC AGC GGC repeated 10 times. Its correct biologically but as mentioned in Dr Prousts lecture this is what you want to avoid for synthesis stability. So we need to figure out a way to make the DNA sequence less repetative eventhough the AA are repetative. We can do this because each amino acid has more than one codon that can code for it. So lets find the options for each of our AAs.
Here they are:
Valine: GTT, GTC, GTA, GTG
Proline: CCT, CCC, CCA, CCG
Glycine: GGT, GGC, GGA, GGG
Serine: AGT, AGC, TCT, TCC, TCA, TCG
Now we need to find a way to use these options to create a new DNA sequence thats less repetative and more synthesis friendly.
Codon optimization on benchling
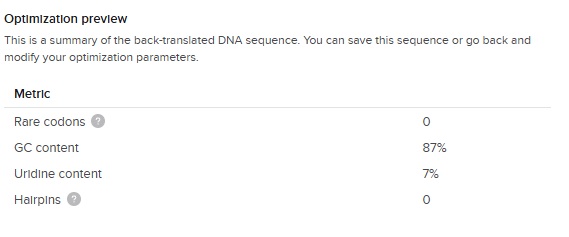
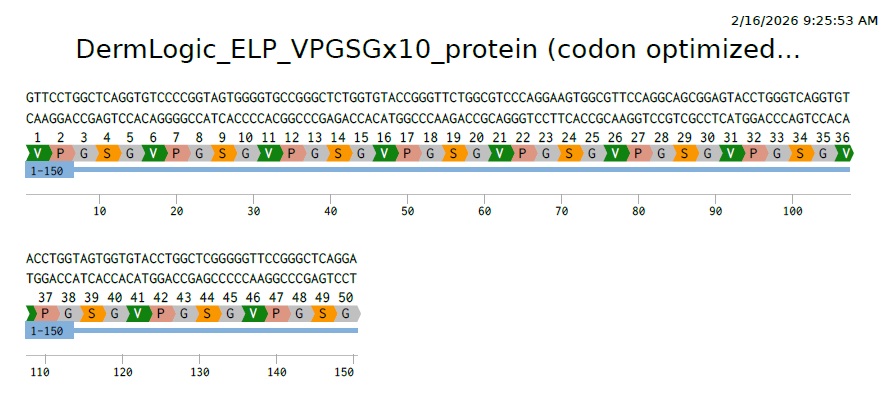
On Benchling I uploaded the 10 repeats of my AA sequence (VPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSGVPGSG). Then selecting the whole sequence choose back translate. Choosing E.Coli (O157:H7 EDL933). Then Match codon usage to target organism method.
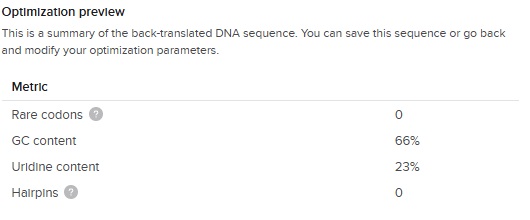
The output of the codon optimization gave the following optimization preview results:
The GC content (66 %) is slightly higher than the E.Coli average (around 50%) but great considering our first DNA sequence was 87 % !
So this is our final optimized 150 bp sequence. It translates to VPGSG 10 times. It has clearly become less repetative.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Production Technology:
Ideally, I would like to produce this ELP using a cell-free protein synthesis (CFPS) system. Cell-free system will allow me to rapidly prototype different ELP lengths without the time-consuming steps of cloning and cell-culture. However, since I am learning the foundational workflows of synthetic biology and tools, I have chosen to codon optimize and design the sequence in E.Coli.
Why this works:
Since the common cell free systems (such as TX-TL) are actually made from e.coli extracts, optimizing the codon for e.coli is a robust strategy that will allow this DNA to be used in both living cells and cell-free synthesis.
Mechanism (Central Dogma):
To produce the protein the DNA is transcribed by RNA Polymerase (recognizing the J23106 constitutive promoter in my design in Part5 below) into messenger RNA. This single stranded mRNA will then be recognized by the ribozomes present in e.coli at the ribozome binding site and thus translating the codon-optimized sequence we made earlier. The ribozome will read the codons three neucleotides at a time and then recruit tRNA with matching amino acids like Valine-Proline etc (that we want and we optimized in benchling for e.coli so they have these tRNAs present and there is no rare codons) and links them together until reaching a stop codon in the mRNA (the T1 terminator) and releasing the ELP polymer. Quite magical how this is identical in all living cells.
3.5 How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
Despite the universality there are important variations between various organism to control this process of DNA code becoming protein.
In eukaryotes A single gene can code for multiple proteins primarily through Alternative Splicing. During this process, the pre-mRNA (containing both coding and non-coding regions) is produced from transcription. Then something called a spliceosome splices the introns and stitches together the exons. The alteration crucially comes from the spliceosome which doesn’t always stitch together in the same order. It can skip certain exons or include others depending on the cells need. This results in distinct mRNA transcripts from the same DNA template which are then translated to unique protein isoforms with different structures and thus functions.
This is what it would look like to have a gene that codes for our ELP polymer with introns and exons in Eukaryotes.
Prokaryotes like E.coli do not use splicing, they often use Polycistronic mRNA (Operons). In this system, a single promoter drives the transcription of multiple distinct genes arranged in a row. The resulting long mRNA strand contains multiple Ribosome Binding Sites (RBS), allowing ribosomes to translate each gene into a seperate protein independently. This allows bacteria to turn on entire metabolic pathways with a single switch, rather than cutting and pasting RNA like eukaryotes do.
I personally am fascinated with the eukaryotic spliceosome and wonder what it would be like to engineer something as complicated and sofisticated.
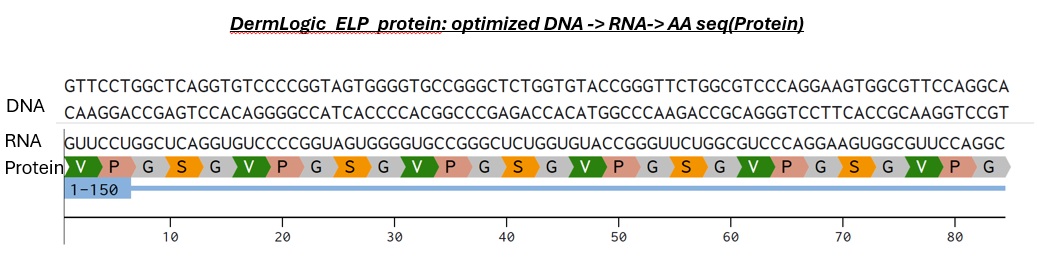
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!!
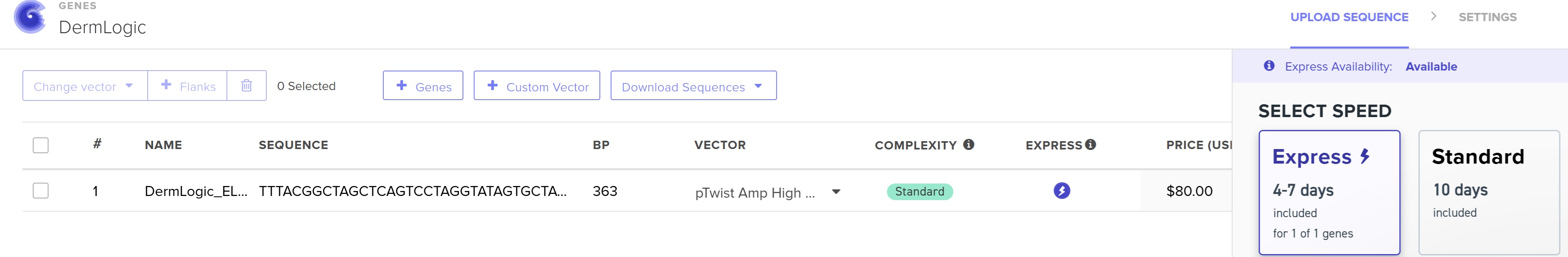
4.3 - 4.6: Final Plasmid from Twist with our ELP insert.
Also our complexity score in twist came out to be standard that means our codon optimization worked and the plasmid is constructable even with our initial repeat codon sequence with high GC content.
Part 5: DNA Read/Write/Edit:
5.1 DNA Read:
(i) What to sequence:
I would like to read the DNA of a Beaver’s gut microbiome and see how it manages to survive eating just trees. I love going camping in the beautiful parks of Ontario such as Algonquin park and always see tons of beavers on my trips. And I am fascinated at the uniqueness of beavers and how they are really architects of their environment much like us humans. And a big part of that is their unique microbiome that lets them survive on something so abundant (trees) and their amazing skills to live under water in the winters. ALso their abilities to hibernate and how thats beneficial to them. Sequencing them could reveal novel enzymes for degrading tough plant materials, or even traits that support their survival under ice during canadian winters.
(ii) DNA Sequencing approach:
Since the beaver gut probably contains thousands of unknown bacteria and fungi working together to break down wood, I would need to sequence the entire soup of DNA. So Metagenomic sequencing would be a good approach using the Oxford Nanopore Tech (ONT). Nanopore sequencers like MiniON are portable which would be ideal to take with me to algonquin park and sample fresh sample from the field!
This is very third generation. To be more precise it is Single Molecule Real-TIme (SMRT) sequencing. Since Nanopore unlike 2nd gen methods (that required PCR amplification) can read native, single molecule DNA directly in real time without amplification. This allows for extremely long reads critical for assembling complex metagenomes.
Wet Steps
I will need a field ready extraction kit like once from Zymo Research to lyse the hardy fungal/bacterial cells.
I will also need to ligate motor proteins and sequencing adapters to the ends of DNA fragments which will lead the DNA into the nanopore.
How it Decodes
The Nanopore is a protein pore through which current is passed and each combination of neucleotides in the DNA inside the pore creates a unique squiggle that Neural Netorks translate into a nucelotide seq.
Output
Primary output is usually a FASTQ file which is the sequence data with quality scores. For the gut soup project we will have to take the collection of millions of reads and compare it with the Metagenome-Assembeled Genomes to identify our wood-degrading microbes of interest.
5.2 DNA Write:
(i) What to synthesize:
Thinking about the DermLogic concept all week I think beyond simple ELPs, I would like to synthesize a “Living Bandage” gene circuit. This could be a genetic circuit designed using skin bacteria like S.epidermis that can sense inflammation markers and respond by synthesizing and secreting healing hydrogel matrix directly into the wound.
(ii) I like the concept of De Novo DNA synthesis using polymerase nucleotide conjugates shared by Prof Joe Jacobson. Its a fast and non-toxic compared to the traditional Phosphoramidite Chemistry. Also its capable of long fragment synthesis which is the future of potentially synthesizing several kilobases without need for extensive assembly. But for the gene fragments ad vectors needed for this type of live bandage I would use the Oligo synthesis using Phosphoramidite chemisry used by companies like Twist Biosciences.
5.3 DNA Edit:
(i) What to edit:
I would like to edit the connective tissue cells (fibroblasts) in older adults to restore their ability to produce functional elastin. Since reading about the role of elastin in the human body I was wondering if we could somehow mimic the natural elastin in the extra cellular matrix of patients with lack of elastin causing various dieseases and locally edit the ECM to reverse some of the elastin related aging in older adults. Such ELPs could someday help an old person heal their weak broken bones that are still “fixed” by doctors using screws and foreign metal parts and instead hope for a future where we could perhaps edit the local stem cells to secrete high performance ELPs that regenerate the tissue naturally, restoring the elasticity and strength.
(ii) Technologies
Design:
Since we are dealing with humans we wouldnt want to use standard CRISPR-Cas9 to cut ds DNA but rather use something advanced in CRISPR technologies like Prime editing. It uses a nickase (a broken Cas9 that can only cut one strand) fused to a reverse transcriptase to find a specific site using pegRNA (guide RNA for the system) in the fibroblast genome and directly write a corrected sequence for elastin without causing any dangerous ds breaks.
Inputs:
Firstly we will need the fusion protein delivered as a plasmid or mRNA encoding the nCas-9RT fusion.
Thenn we will make the custom guide pegRNA containing the target spacer (DNA site in the fibroblast genome for elastin).
Lipid nanoparticle delivery system can be useful for delivery into the fibroblast cell.
Limitations:
Its less efficient (20-50% successful cells edited) than standard CRISPR-Cas9 (80%)
Designing pegRNA is harder than standard gRNA
The Problem:
Manually pipetting cell-free reactions is slow, inconsistent across different researchers, and difficult to scale for field use
Their Solution:
The Authors used the Opentrons OT-2 to automate the assembly of flouride riboswitch biosensors
The Novelty:
They didn’t just automate the liquid moving, they optimized the robot’s physical parameters (like blowout height and dispense speed) to handle the viscosity of cell-free extracts. This allowed them to produce 384 consistent, functional sensors in 30 minutes- something that would take a researcher much longer and with higher risk of error.
The Result:
They proved that low-cost robotics can produce “shelf-stable” diagnostics that perform nearly as well as those made by expert humans, making it possible to manufacture sensors for environmental toxins anywhere in the world.
Question 2: What do you intend to do with automation tools for your final project?
Project Context: My project, DermLogic, invovles creating a smart biopolymer hydrogel patch that utilizes cell-free genetic circuits to detect and treat cutaneous HPV.
Automation Plan:
I intend to use the Opentons OT-2 to standardize and scale the production of these “Living” patches. I envision that the primary challenge with DermLogic will be ensuring the precise ratio of cell-free extracts, DNA logic gates, and biopolymer precursors (hydrogel) to maintain therapeutic consistency. Here are some lessons taken from the above paper:
Viscosity-Optimized Liquid Handling:
Following the methodology in Brown et al. (2024), I will program specific robotic parameters to handle the high viscosity of the hydrogel-extract mixture. This includes tuning the aspirate/dispense rates and implementing touch-tip/blowout sequences to prevent the “bubbling” that often occurs during manual pipetting of cell-free systems.
High-Throughput Logic Testing:
I will use automation to screen an array of genetic circuits designed to detect HPV oncoproteins. The robot will facilitate a “Master Mix” approach, where the base cell-free machinery is distributed across 96-well plate, followed by the addition of unique DNA constructs. This allows me to test hundreds of logic-gate variants in a single run to see which provides the sharpest “ON” trigger in the presence of HPV markers.
Automated Lyophilization Prep:
To meet governance goals of a “cell-free mandate” for safety proposed in week 1’s assignment, i will use the Opentrons to dispense the final reactions into patch molds before they are frozen and lyophilized. Automation ensures that every patch has a uniform concentration of the therapeutic peptide, which is critical for clinical safety and efficacy.
Pseudocode for DermLogic Patch Assembly:
forwellinpatch_molds:p20.pick_up_tip()p20.aspirate(10,hydrogel_master_mix,rate=0.5)# Slow aspirate for viscosityp20.dispense(10,well,rate=0.5)p20.touch_tip(v_offset=-2)# Ensure clean release from tipp20.drop_tip()
Final Project Ideas Slide
Week 4 HW: Protein Design- Part 1
Part A: Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat isn’t 100% protein; it’s typically composed of ~20% protein, ~20% fat, and ~60% water. So 500g of meat will approximately contain 100g of protein. Since Daltons is the unit used to measure the weight of small molecules and 1 Dalton is approximately $1.66 \times 10^{-24}$ grams.
Since we need to calculate the number of molecules of AAs in our meat, let’s first convert the mass of one AA into grams
So the total number of AA molecules in 500 g of meat is approximately $6.02 \times 10^{23}$, which is coincidentally (and elegantly) about 1 Mole of amino acids.
Thus we learn the trick that chemists use when going from molecular weights to weighing scale weights:
If a molecule weighs 1 Da then 1 mole of it will weigh 1 gram. In this case since AAs weigh 100 Daltons, 1 Mole of AAs will weigh 100 grams (which is also the amount of protein in 500 grams of meat). Mind is BLOWN!
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans dont become cows because we dont incorporate bovine proteins directly. Through digestion, we break down those proteins down into their constituent amino acids. Since the 20 natural amino acids are a universal biological language, our body simply uses them as raw materials to build new proteins based on human DNA sequences.
3. Why are there only 20 natural amino acids?
There is a theory proposed by Crick that this is a frozen accident; Once life settled on 20, the cost of changing the entire genetic code was too high.
The other theory says that its the Optimal set; these 20 provide enough variety (acidic, basic, polar, non-polar) to build amost any functional shape. Adding more might have had diminishing returns or caused too many “side reactions”
As mentioned in the last slide of Joe Jacobsons lecture 20 provides the optimal balance of codon redundancy and diversity.
4. Can you make other non-natural amino acids? Design some new amino acids.
Every amino acid has the same basic structure: an amino group, a carboxyl group and a side chain (R-group). To design as new one, you typically keep the backbone the same so the ribosomes can still physically link it, but you can change the R-group to do something nature can’t. So lets design a “Metal-Sensing Amino Acid” since I am interested in how metals can be bound to proteins. For this we can use Bipyridine as our modified R-group because it loves to grab onto metal ions. And we can use UAG (stop codon) to code for our new AA. I found this paper titled “Rewiring Protein Synthesis: From Natural to Synthetic Amino Acids” and in it they describe that we need to modify two specific biological parts to make our synthetic AA. Firstly we need to evolve the Aminoacyl-tRNA Synthetase that are essential enzymes that catalyse the attachment of a specific amino acid to its corresponding tRNA. We can take a natural one and mutate its active site to fit our Bipyridine so it ignores the other 20 natural AA. The resulting tRNA is delivered to the ribosome by an elongation factor which we will have to modify to deliver our bulky new AA.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before enzymes existed, chemistry had to synthesise amino acids through abiotic chemistry fueled by the geological and cosmic energy. The main theories of our time come from:
Miller-Urey Primordial Soup: This theory suggests that the early atmosphere of earth was a reducing environment rich in methane, ammonia and water vapor. Miller and Urey found that passing electrical sparks through these gases produced several amino acids including glycine and alanine
Deep-Sea Hydrothermal vents: As we remember watching deep sea thermal vents footage from BBC’s planet earth documentary. These cents provide a constant stream of superheated, mineral rich water and reducing gases like H2 and H2S which provides extreme pressure and temperature gradients which combined with the mineral catalysts could drive the synthesis of organic molecules. This is refferred to as the iron-sulfur world theory.
Panspermia: One of my favorite theories. Amino acids may not have started on earth at all but were delivered by comets and meteorites. Like a message of life from beyond. Who wrote it? How long ago? Haha. I am gonna write a sci-fi story one day with this premise. The evidence for this theory came from Murchison meteorite which fell in Australia in 1969 and was found to contain over 70 different amino acids.
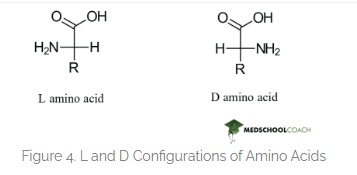
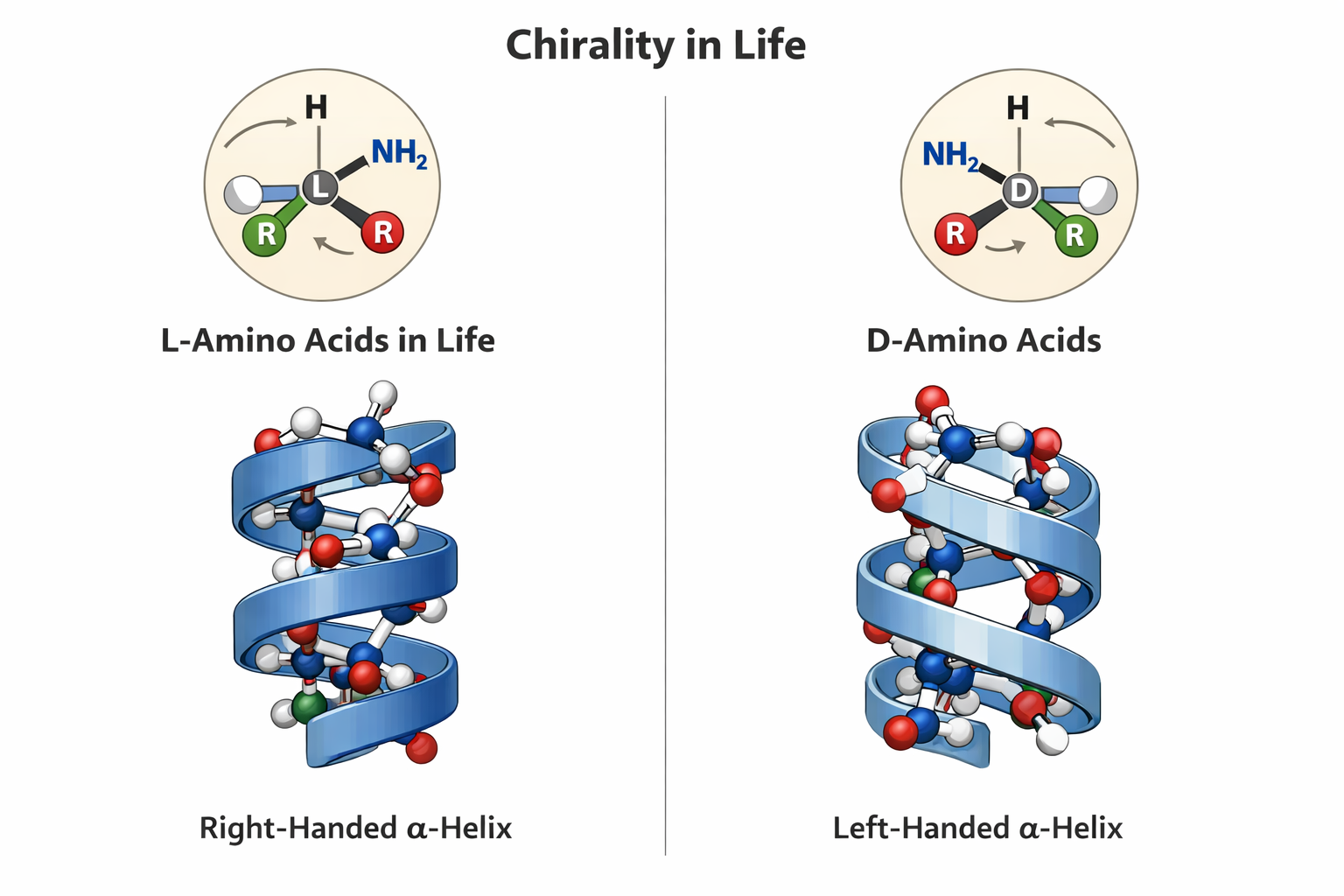
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
This question somehow scares me. Since this relates to Chirality in life. And how fascinatingly all life on this planet is formed by L-amino acids. Contemporary notion is that if we mess around with this and make life from the mirror image which is D-amino acids then we will form helices with a mirrored spin and life that will be incompatible with the enzymes of L-AA oriented life and how catastrophic something like this will be in the wild. But I hold an optimists approach. I feel like this is going to be a huge area of research in synbio and we should approach this problem with good ethics and positivity.
Here is the correct difference between L-D Amino acids.
Here is the hallucinating ChatGPT image that cant draw a correct Amino Acid for the life of me. I have tried many times. But they say its gonna take over the world. It got some parts right.
prompt: ChatGPT please draw me an image showing L and D aminoacids showing chirality of life and how they form mirror alpha helices
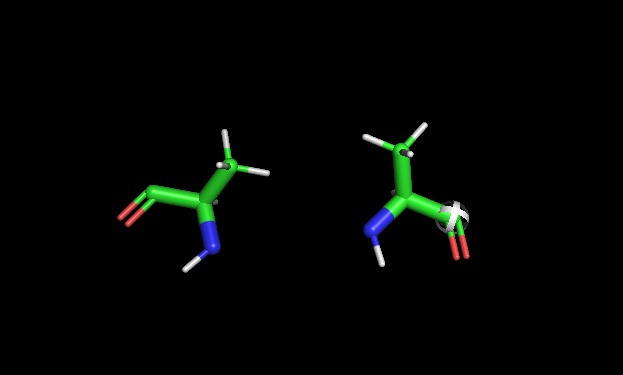
PyMOL chirality: Later used pymol to show L and D Alanine side by side
7. Can you discover additional helices in proteins?
Yes, additional helical structures besides the standard alpha delix can be found in proteins. Studies show that other types of helices occus in many proteins, but they are often overlooked or mistaken for small distortions in alpha helices. These helices are especially common in memberane proteins and are found in a significant number of known protein structures. (Vieira-Pires & Morais-Cabral, 2010)
8 Why are most molecular helices right-handed?
As shown above they are mostly righ-handed because there is more abundance of L-Amino acids. So when coil righ handed helices avoid steric clashes and are more stable and thus more common
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?(also answers Q11 )
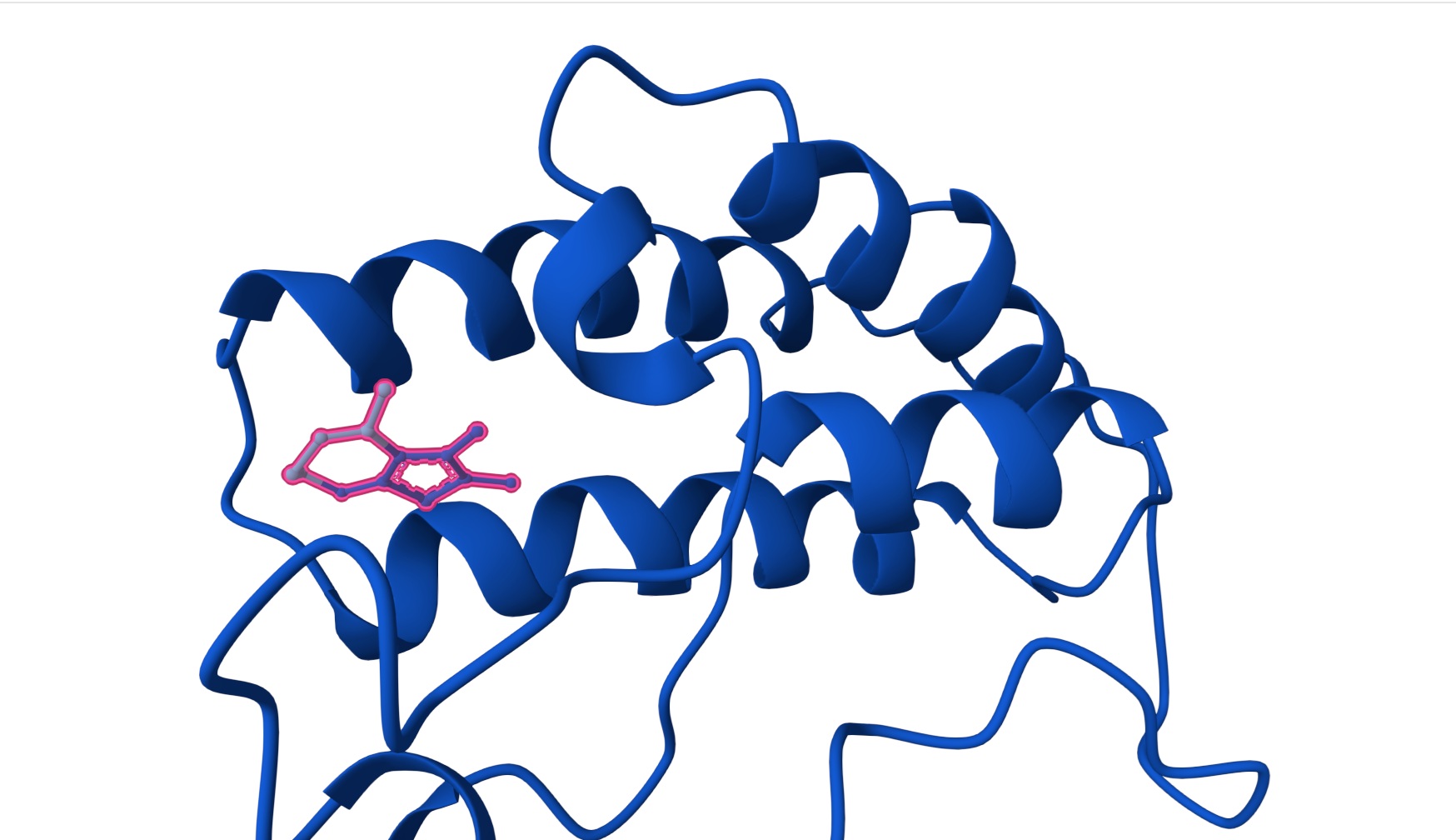
The primary sequences of beta strand and sheet prefer alternating hydrophobic and hydrophilic amino acids. This forms exposed hydrogen bond donors and acceptors on both sides that forms the sheet structure and the hydrophobic side chains interlock between the sheets. This is shown in the image below from pyMol
You can shee in the above image that Hydrogen bonds in grey small dots create horizontal structure and Hydrophobic packing on the inside denoted by the orange surface creates the vertical stacking.
Part B: Protein Analysis and Visualization:
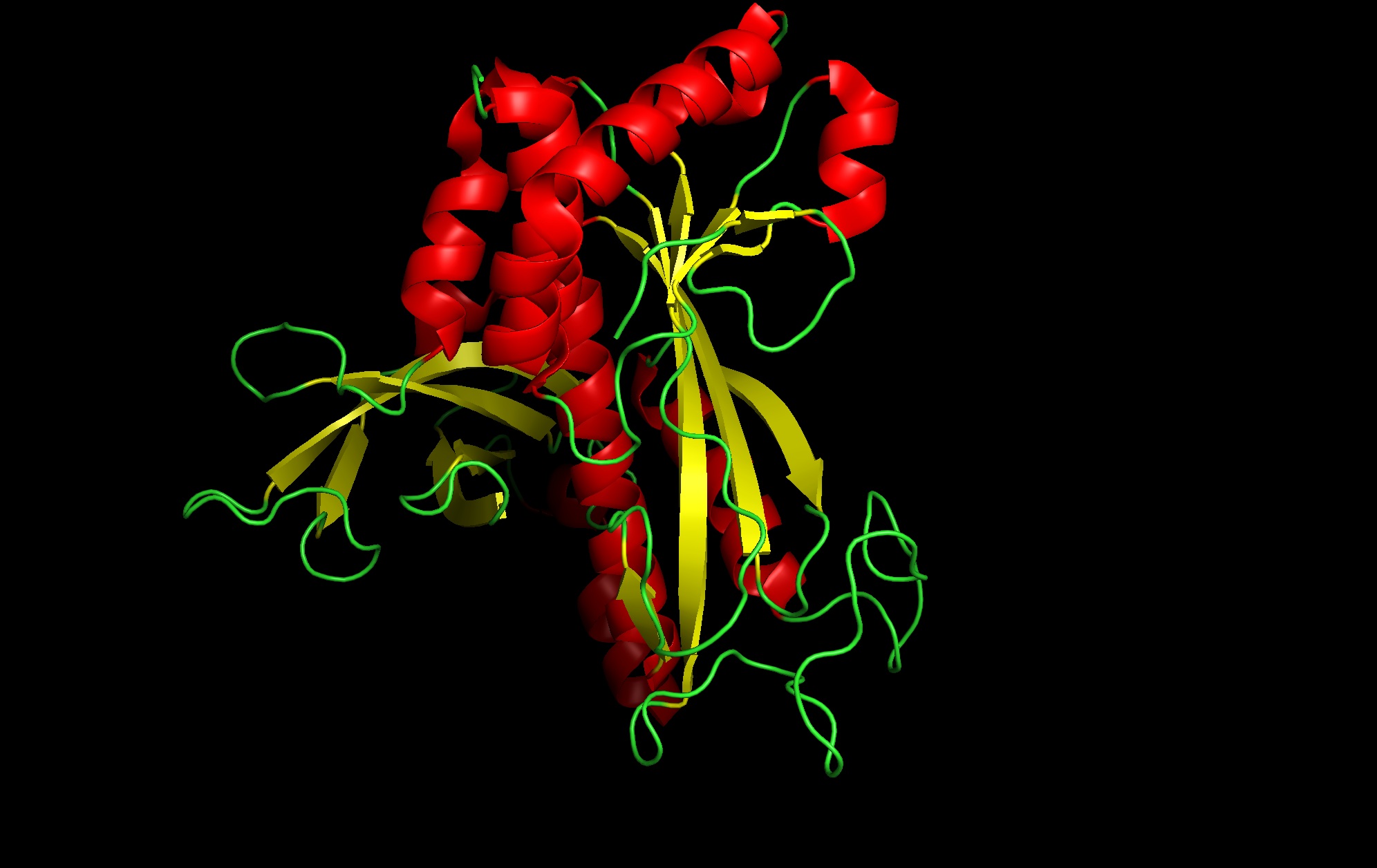
1. Protein Selection and Rationale
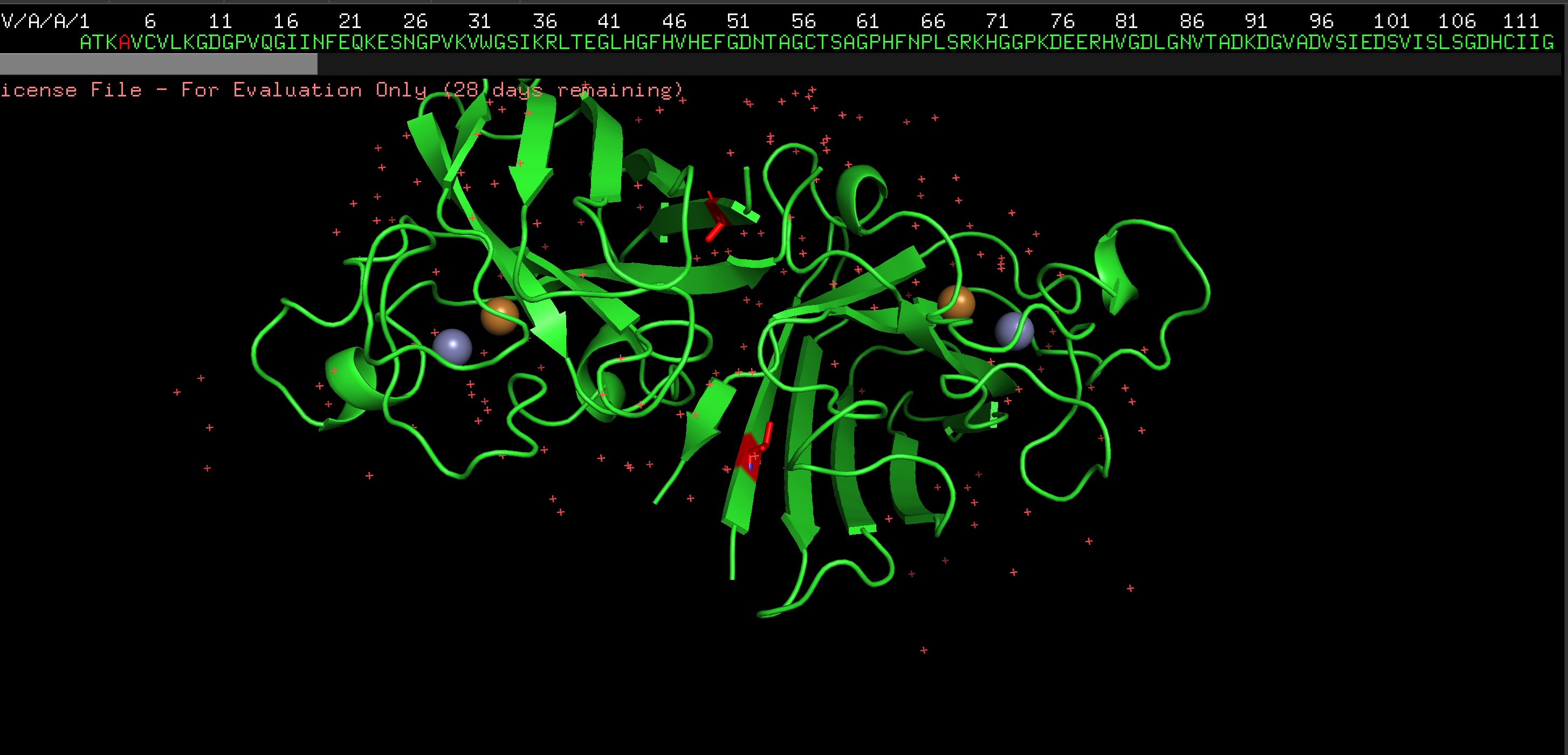
Protein: I selected 4IS4, the Glutamine Synthetase from the model plant Medicago truncatula. I chose this because GS is the primary engine for nitrogen assimilation in plants. By studying a plant-specific decameric structure, I can better understand how to engineer nitrogen uptake in duckweed—a key goal for my PACT proposal to reduce agricultural fertilizer runoff in Canada.
Rationale: GS is the “gatekeeper” for nitrogen assimilation. It converts ammonium (found in high concentration in Canadian hog/dairy manure) into amino acids.
The Goal: To see of we can engineer a version of this protein that works faster or remains stable in the fluctuating temperatures of a canadian lagoon.
2. Amino Acid Sequence:
The length of the protein is: 378 aminoacids.
The most common amino acid is: G, which appears 42 times.
Sequence Homologs: There are thousands of homologs of the protein as this is a highly conserved protein across species.
4. The structure was released on 2014-04-09. The resolution is 2.35 Å so its a very good resolution.
seems like a pretty equal representation of sheets and helices which is a common feature in enzymes
Color by Residue Type: Hydrophobic: Orange; Hydrophilic: Blue
Since this is an enzyme it has tons of pockets
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
a. Unsupervised Deep Mutational Scans
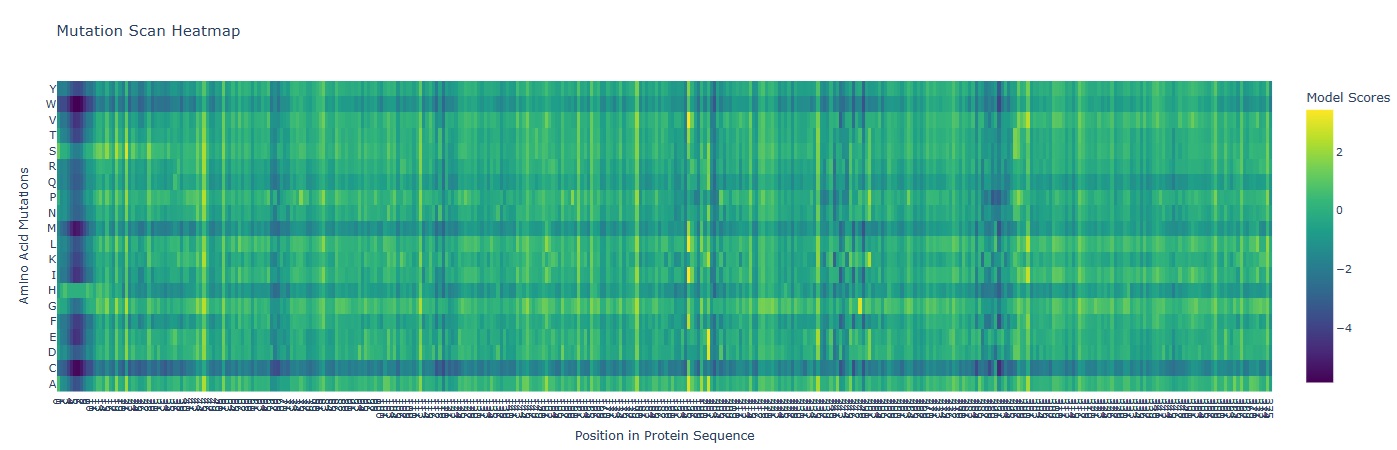
A mutation scan is basically asking “If I mutate every position to every other amino acid, which ones does the model think are tolerated… and which ones are catastrophic?”
In the heatmap the X-axis represents the AA position in the protein (1-378). And the Y-axis represents the 20 possible amino acids.
Deep Blue/ Purple Columns:
These are “Cold” regions. they represent positions where almost any mutation results in a significantly lower model score. This indicates High Conservation. In glutamine synthetase these are usually residues critical for binding ATP, Glutamate or forming the decameric ring interface.
Yellow/Green Spots:
These are “Neutral” or “Favorable” regions. The model suggests the protein could tolerate these changes without losing its structural integrity.
Mutation that Stands out:
I observed a distinct pattern of high sensitivity around Position 2-12 which probably is the catalytic site of the enzyme.
TBD: (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
b Latent Space Analysis
The Latent Space analysis was cool to visualise but the neighbourhood doesnt give anything useful unfortunately.
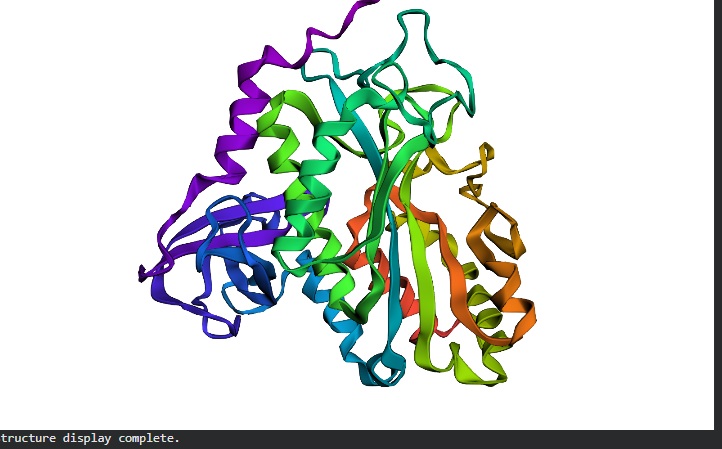
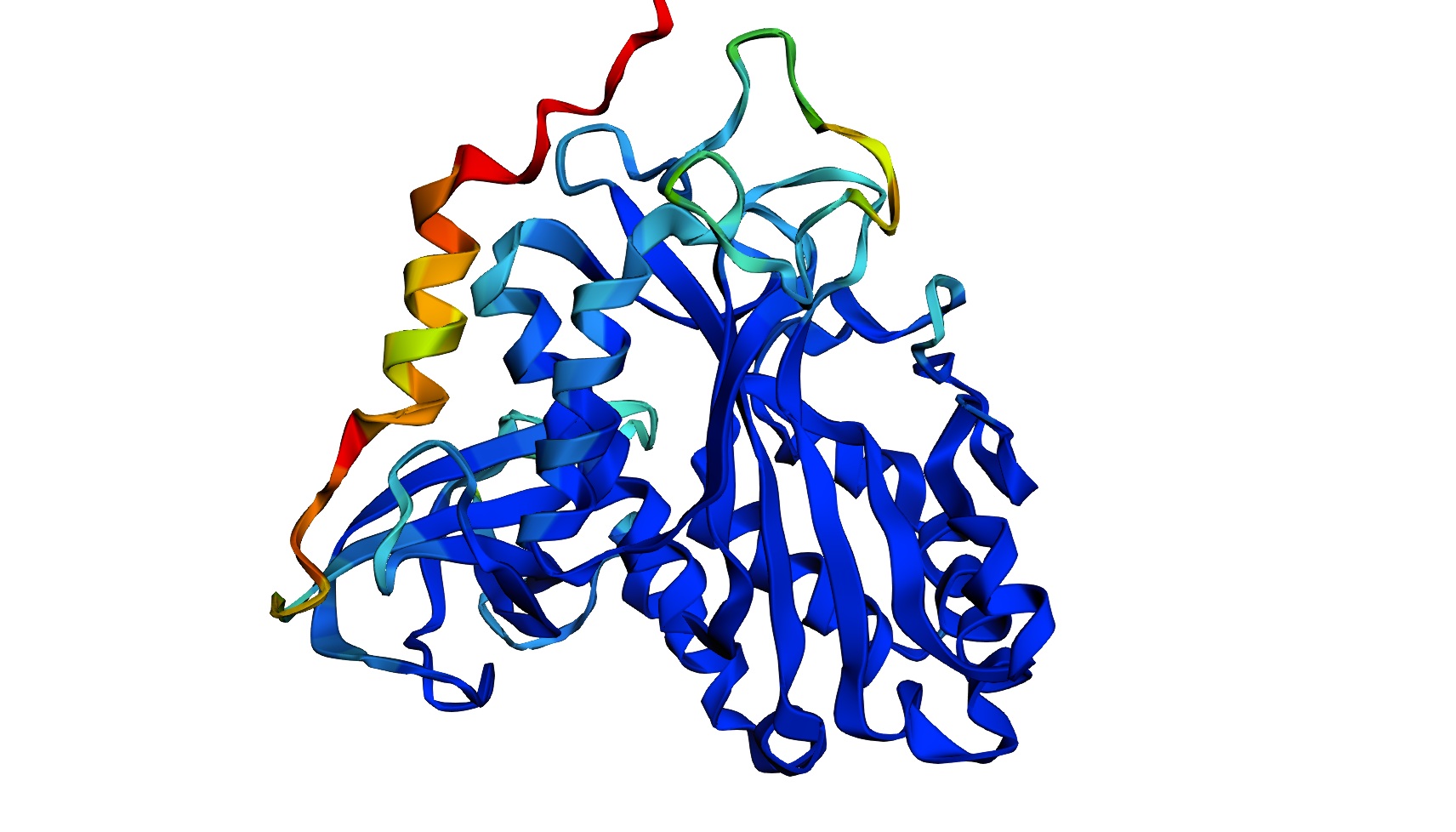
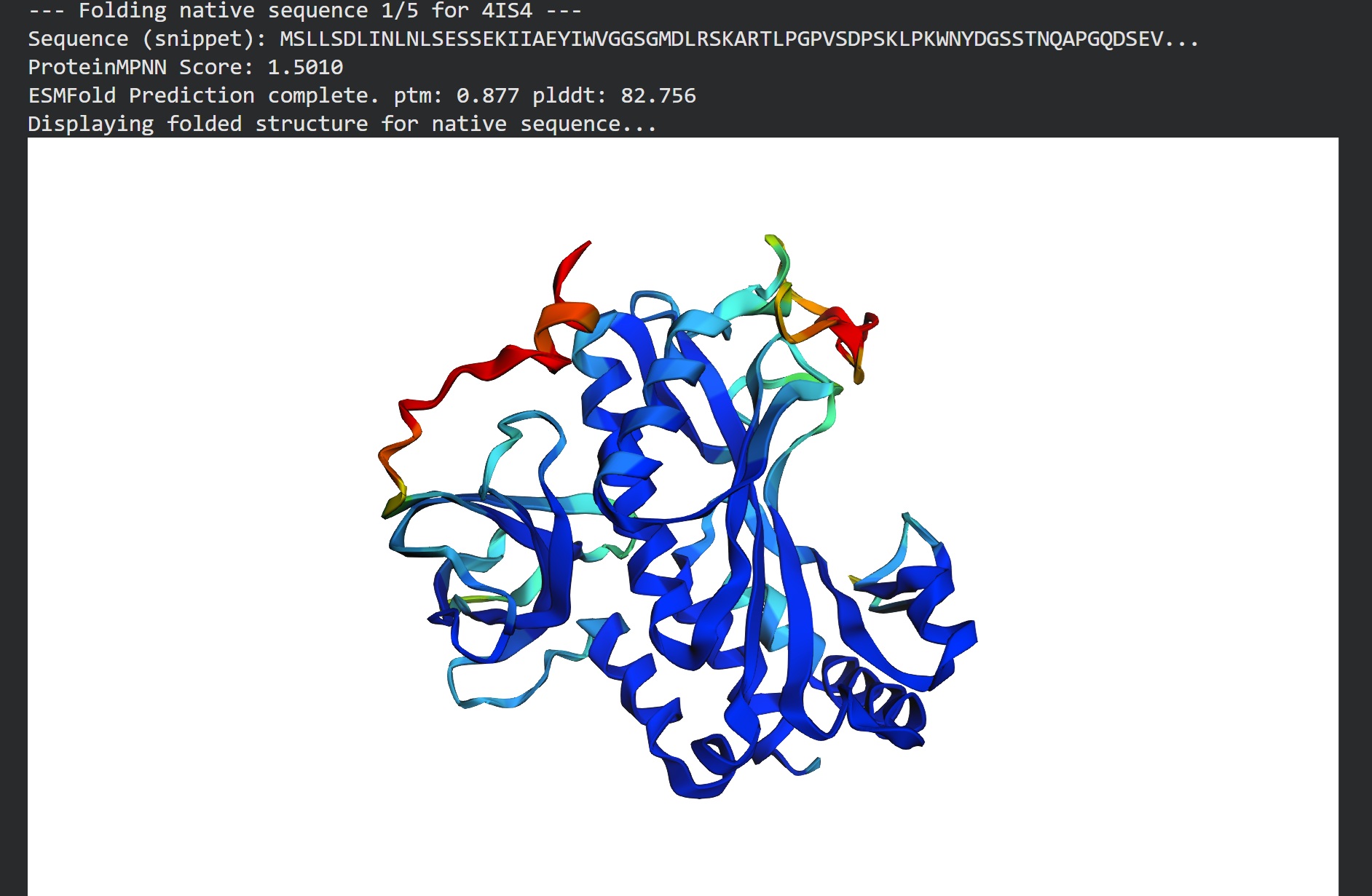
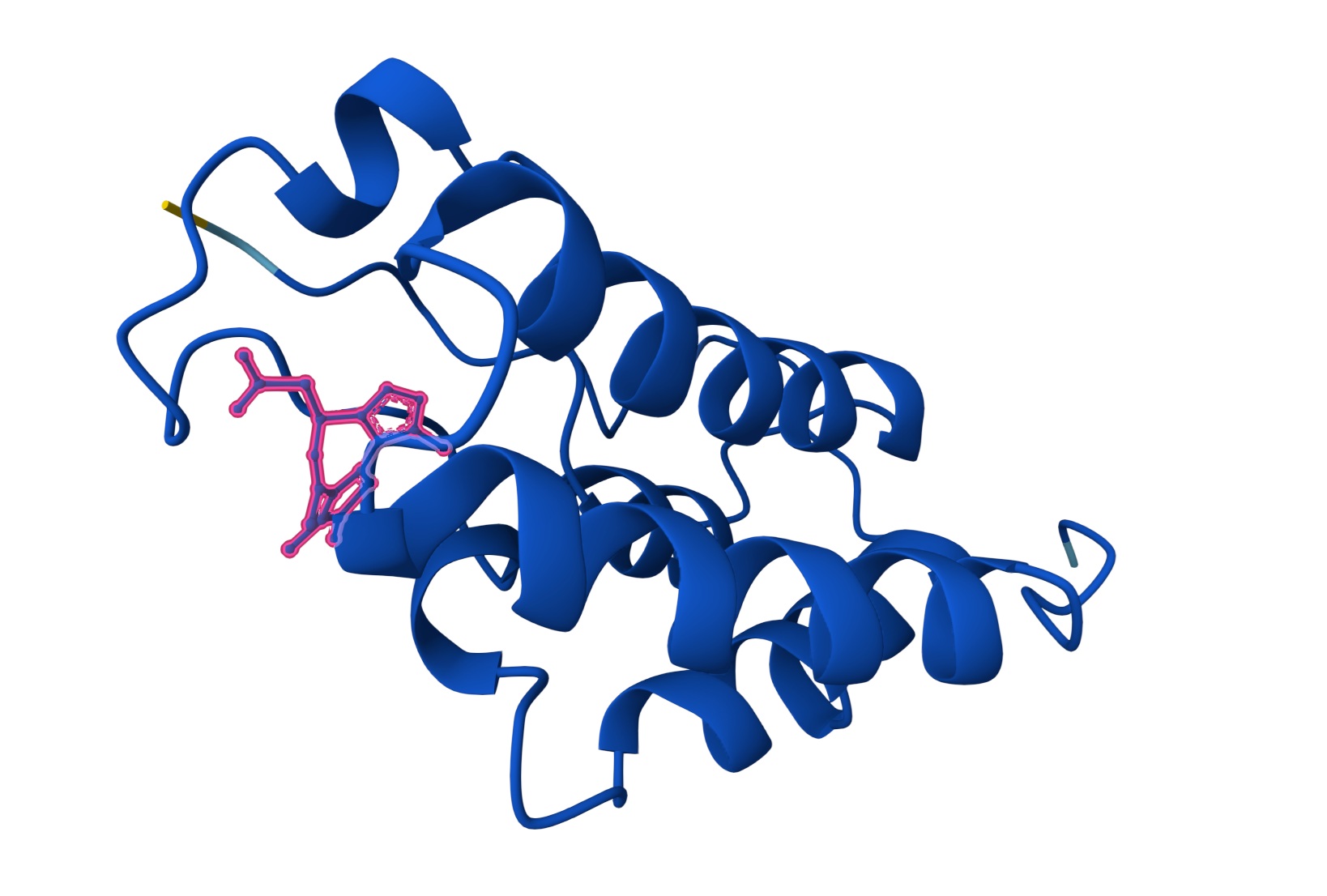
C2. Protein Folding
I got a pyMol structure of a monomer of my protein and compared it to the ESM Fold predicted structure and I have to say these snapshots show a lot of similarities of you focus on the number of parallel B-sheets and their location
Confidence score: Mostly Blue so ESM has a high confidence in this structure which it should
C3. Protein Generation
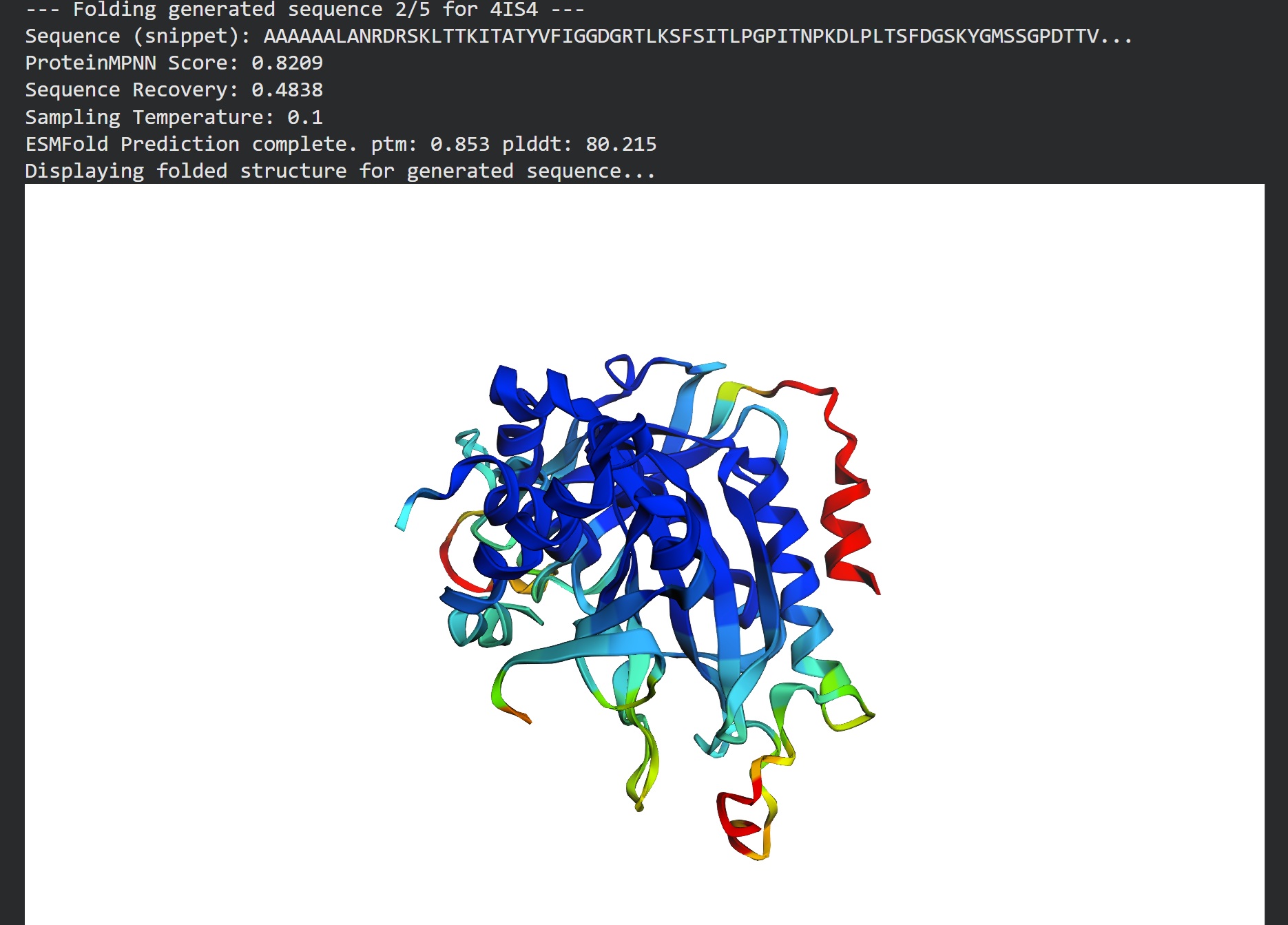
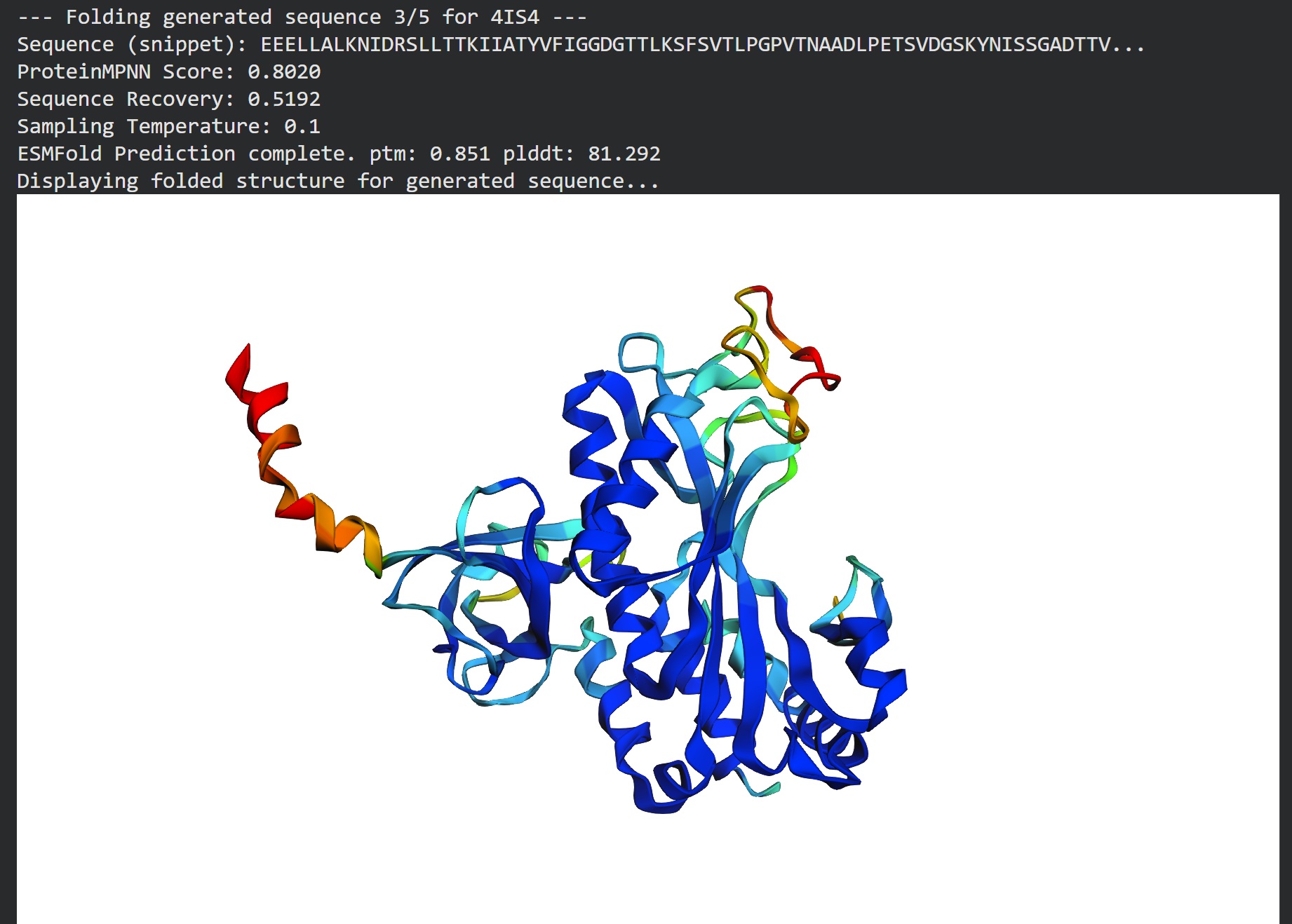
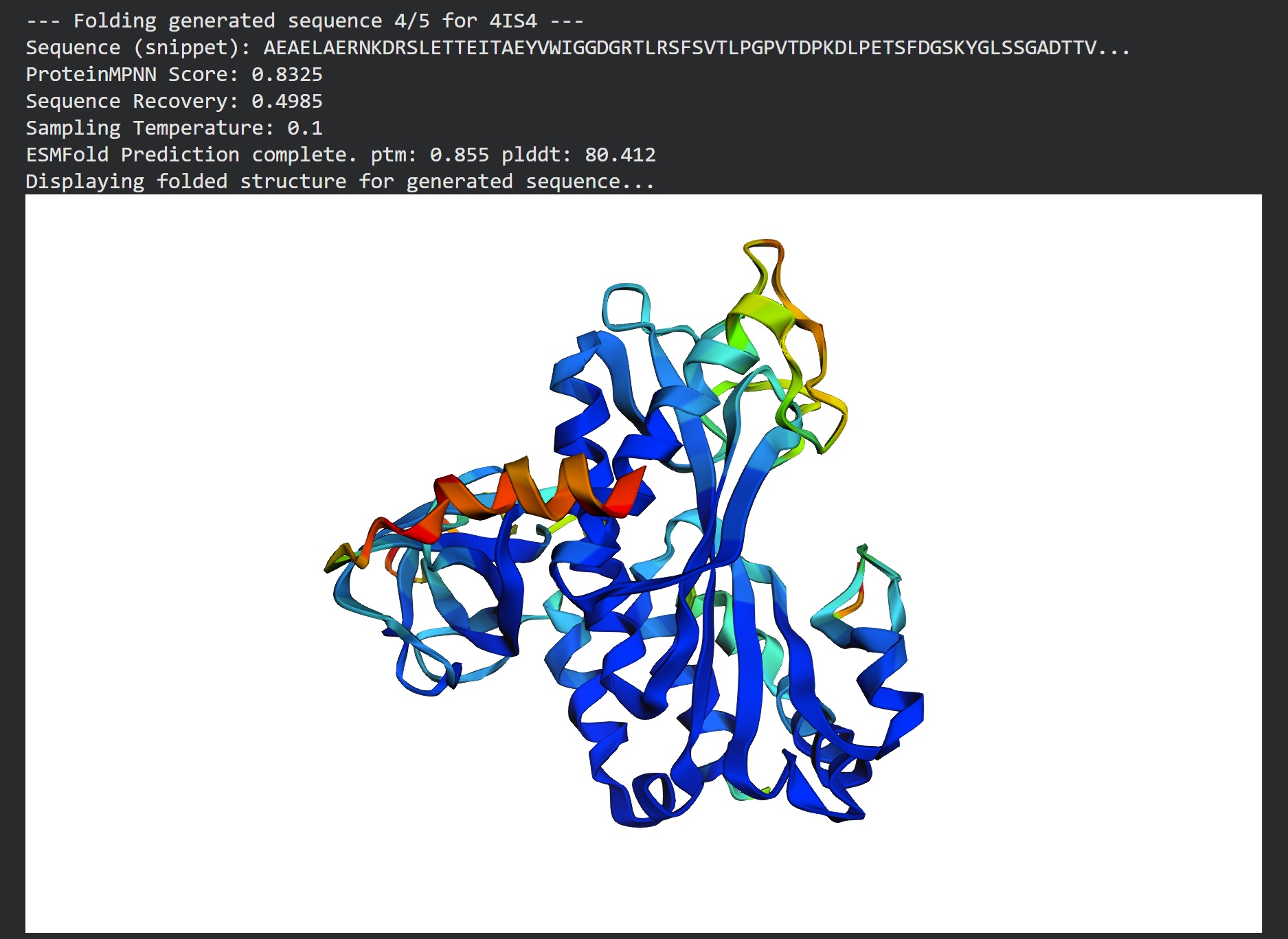
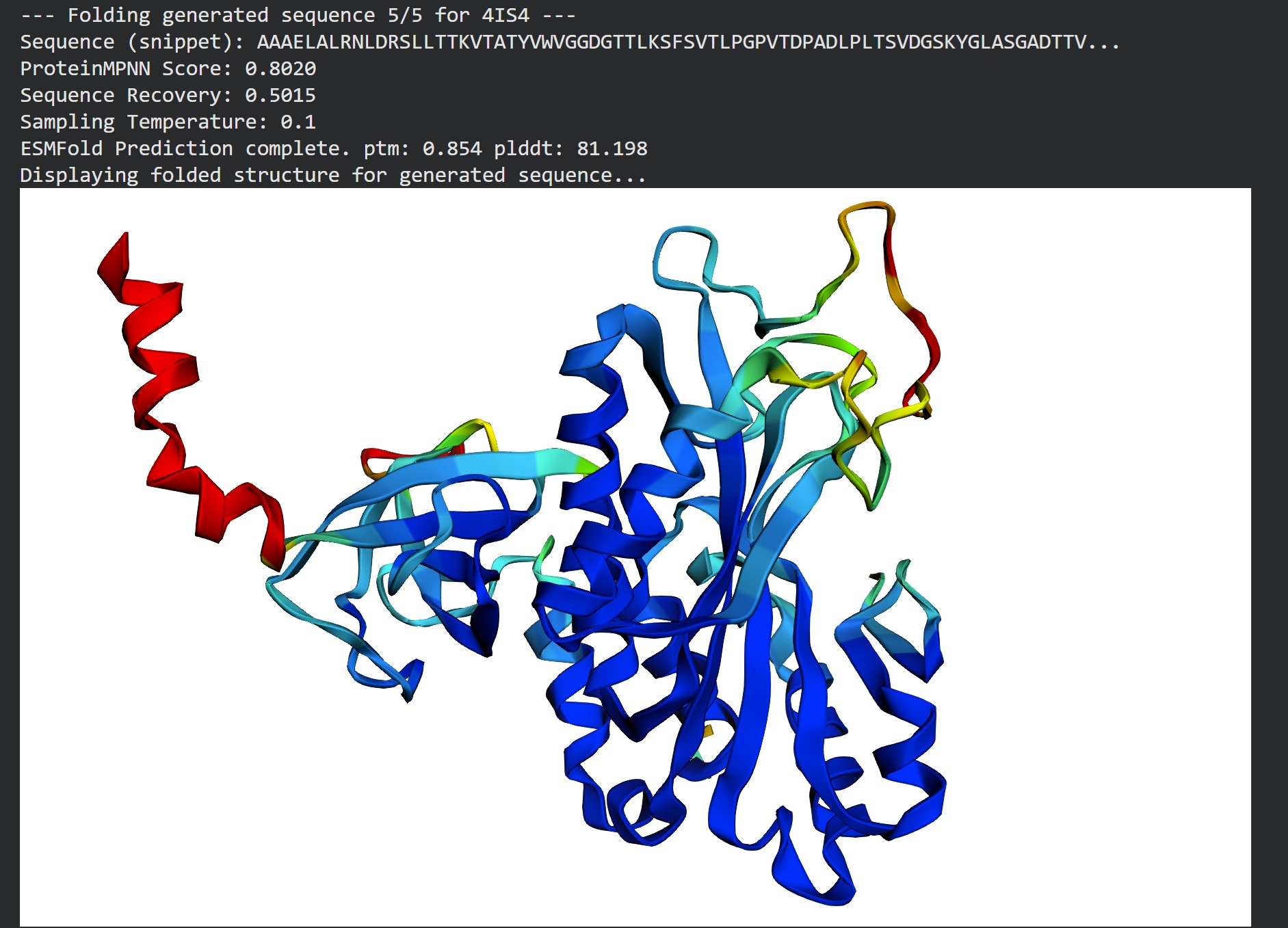
Inverse folding using protein MPNN
Following were 4 generated sequences along with the 1st being the native sequence
These generated sequences closely resemble our original structure so ProteinMPNN did a great job at finding various sets of sequences giving the same PDB structure we entered.
Interesting find: I didnt know that all protein synthesis starts with amino acid Methionine(Met) which is cleaved off by Methionine Aminopeptidases as an essential process in a large majority of cells. It also has a condition where only the sequences with these small uncharged AAs as a second AA can be functionally folded instead of being degraded. (Nguyen et.al.2019)
Introducing SOD1 Seq with A4V Mutation (removing Alanine and adding Valine) associated with the most aggressive form of the ALS disease (and removing the initial methionine) :
ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. PepMLM Generation Results
Candidate
Sequence
Pseudo Perplexity
Design 1
WRSPATGARHKK
13.60
Design 2
WRVPAVAVRHKK
12.17
Design 3
HRYPVVGAEWKK
15.56
Design 4
WRYYAAAIAHKK
13.00
Control
FLYRWLPSRRGG
22.53
Note: Lower perplexity indicates higher model confidence in the sequence’s fit for the SOD1 mutant.
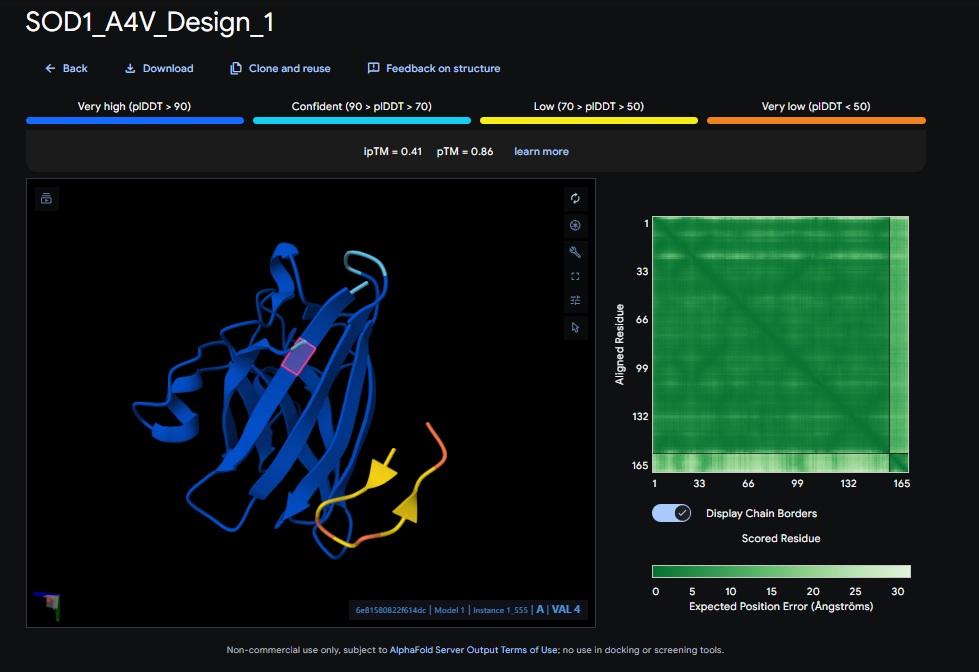
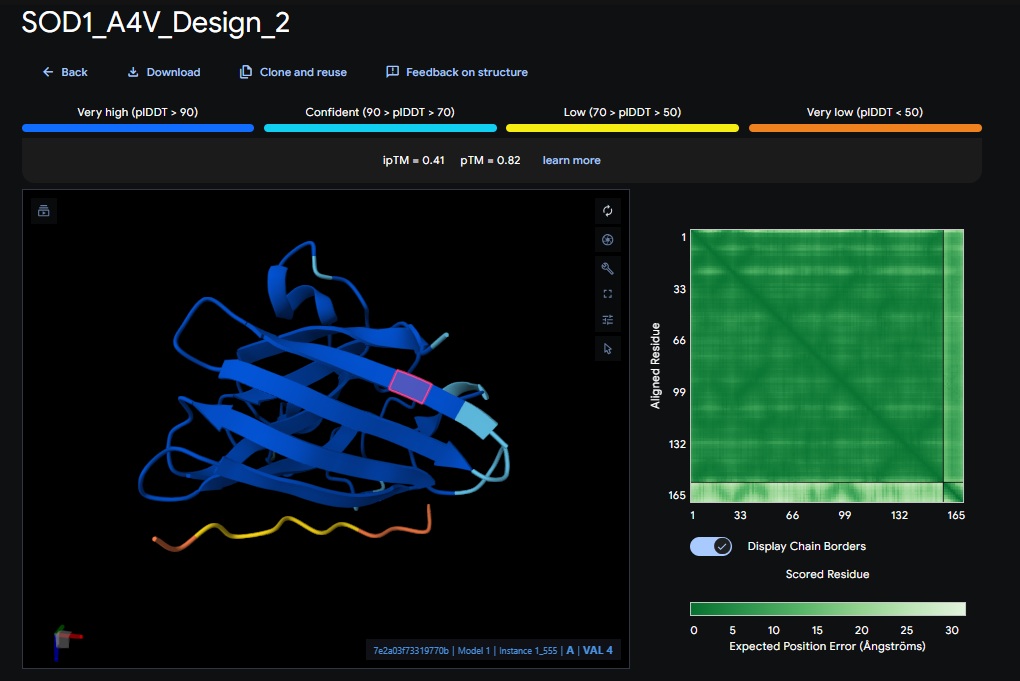
Part 2: Evaluate Binders with AlphaFold3:
AlphaFold3 Evaluation Table
Candidate
Sequence
ipTM Score (Good Score > 0.80)
pTM Score (Good Score > 0.50)
Avg. pLDDT
Binds at A4V?
Design 1
WRSPATGARHKK
[0.41]
[0.86]
[low]
No
Design 2
WRVPAVAVRHKK
[0.41]
[0.82]
[low]
No
Design 3
HRYPVVGAEWKK
[0.25]
[0.87]
[low]
No
Design 4
WRYYAAAIAHKK
[0.34]
[0.72]
[low]
No
Control
FLYRWLPSRRGG
[0.41]
[0.78]
[low]
No
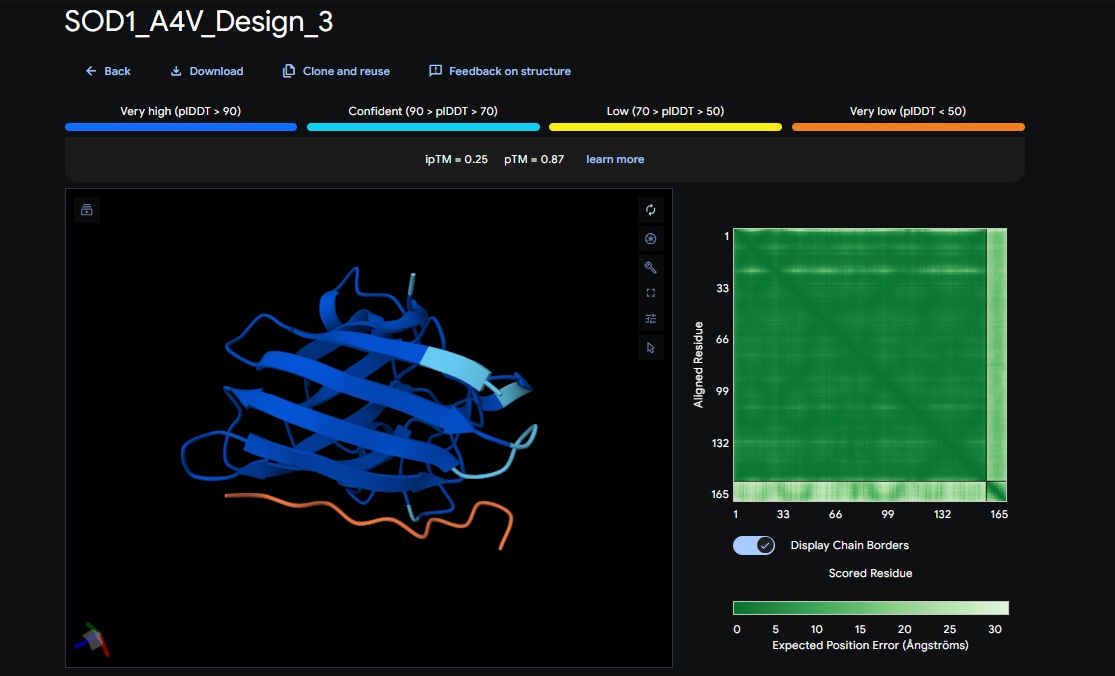
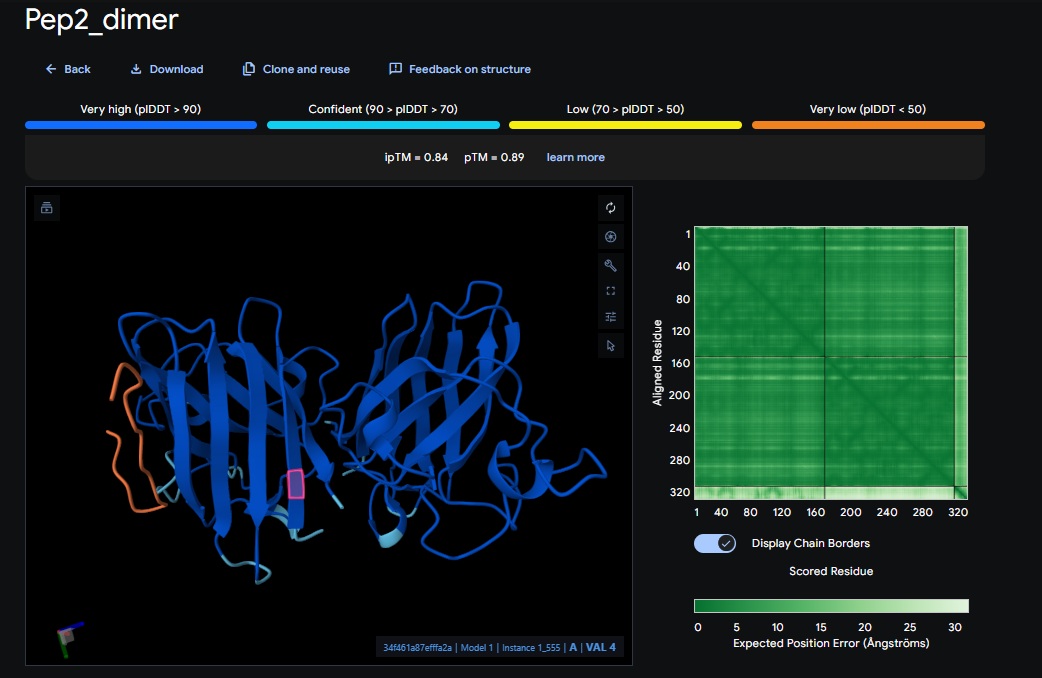
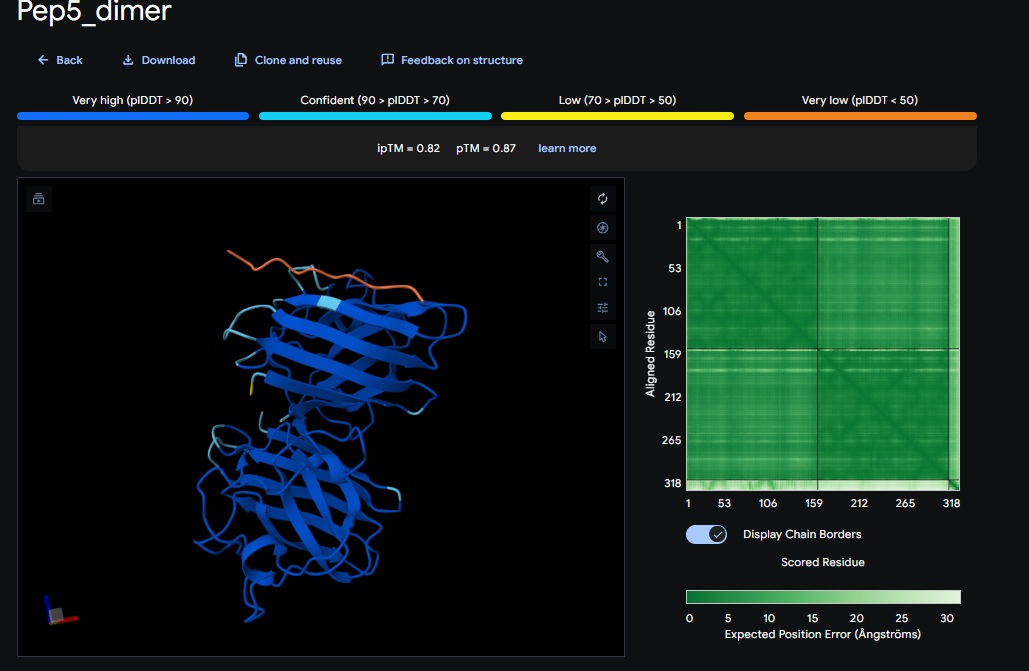
1. Design_1 (WRSPATGARHKK) + SOD1:
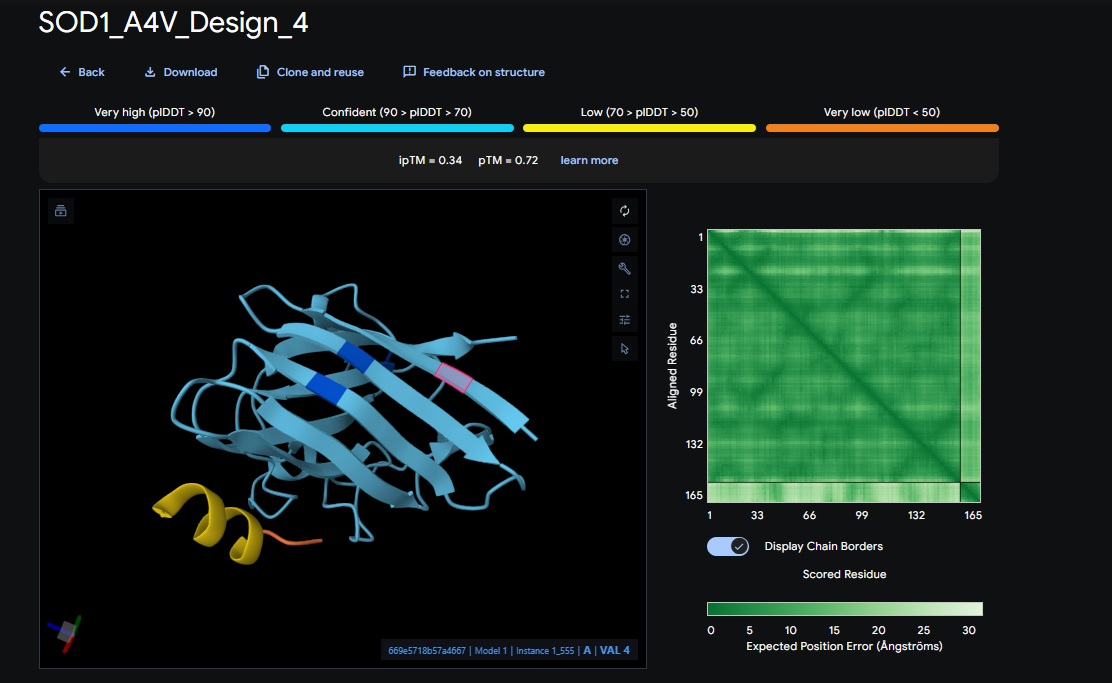
2. Design_2 (WRVPAVAVRHKK) + SOD1:
3. Design_3 (HRYPVVGAEWKK) + SOD1:
4. Design_4 (WRYYAAAIAHKK) + SOD1:
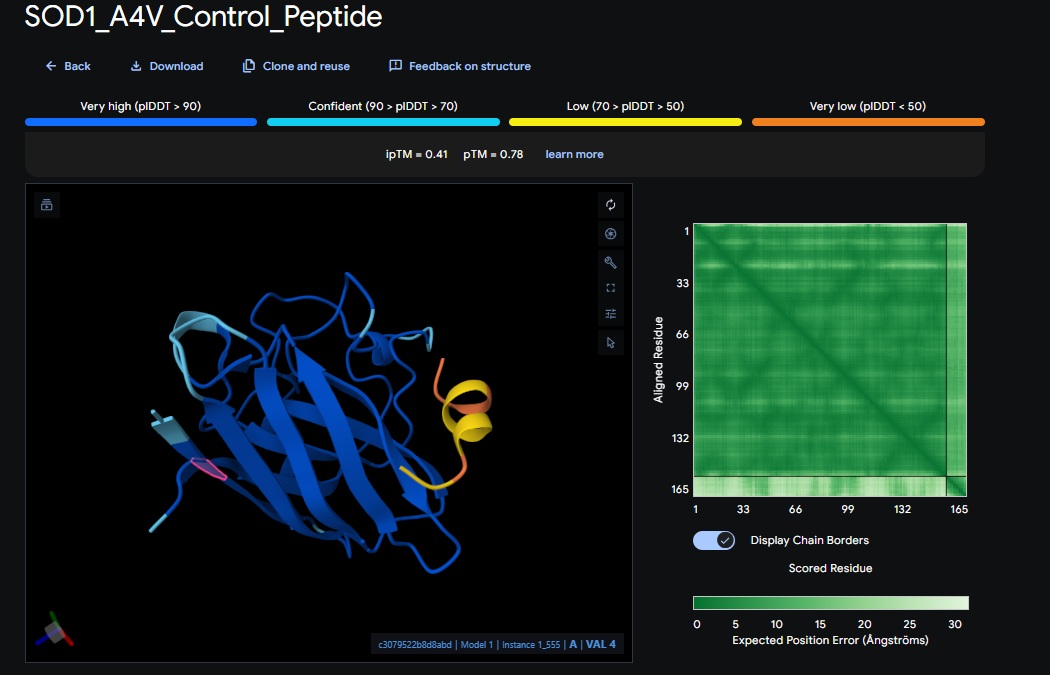
5. Control_Peptide (FLYRWLPSRRGG) + SOD1
At this point I realised that I was only doing the pepMLM and alpha fold with the monomer of the protein so the binder I am trying to design is for the enzymes natural dimer state, So I am going to redo the above sections and see if I get different scores.
2. PepMLM Generation Results for Dimer and 15 AA Peptide length (first 4 designs):
Candidate
Sequence
Pseudo Perplexity
Design 1
WTWVVHVATHKHHLK
15.02
Design 2
DTVVHHHATHEHKKK
14.28
Design 3
WTVEHHLVTKQEKKK
10.65
Design 4
DTWDHHLATKEHKKK
10.52
Design 5
WTRVHAVVEKKK
13.10
Design 6
ATHVHVAIHHKK
7.738
Control
FLYRWLPSRRGG
31.133
Note: Lower perplexity indicates higher model confidence in the sequence’s fit for the SOD1 mutant.
Part 2: Evaluate Binders with AlphaFold3:
AlphaFold3 Evaluation Table
Candidate
Sequence
ipTM Score (Good Score > 0.80)
pTM Score (Good Score > 0.50)
Avg. pLDDT
Binds at A4V?
Design 1
WTWVVHVATHKHHLK
[0.81]
[0.86]
[low]
No
Design 2
DTVVHHHATHEHKKK
[0.84]
[0.89]
[low]
No
Design 3
WTVEHHLVTKQEKKK
[0.75]
[0.83]
[low]
Closest Yet
Design 4
DTWDHHLATKEHKKK
[0.67]
[0.79]
[low]
No
Design 5
WTRVHAVVEKKK
[0.82]
[0.87]
[low]
No
Design 6
ATHVHVAIHHKK
[0.88]
[0.91]
[low]
Almost
Control
FLYRWLPSRRGG
[0.88]
[0.91]
[low]
No
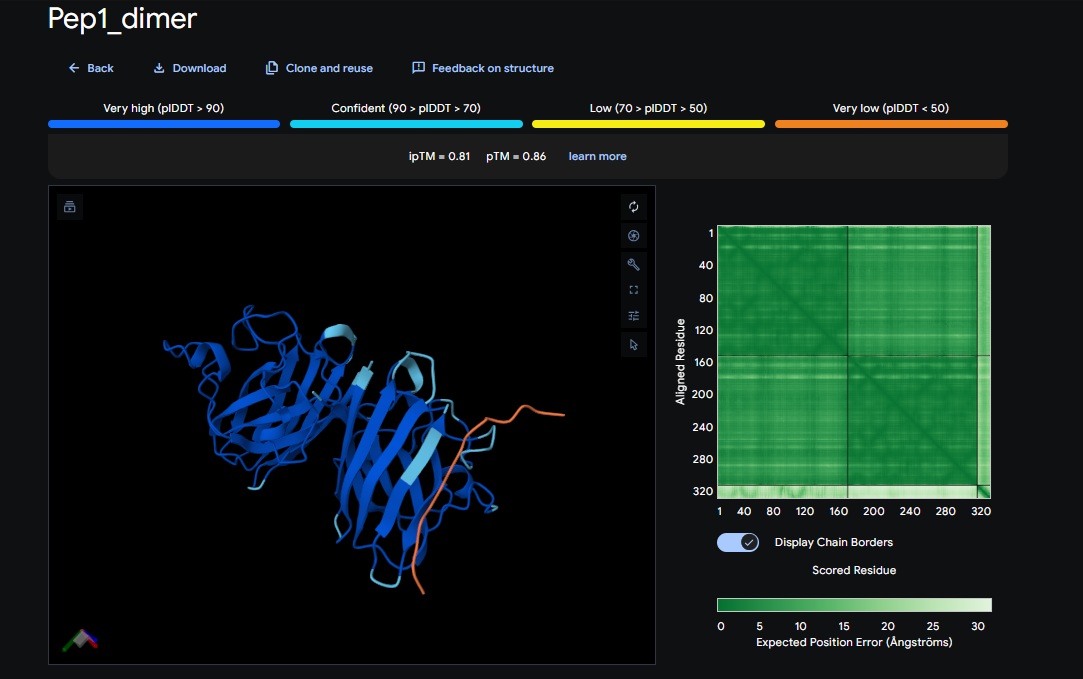
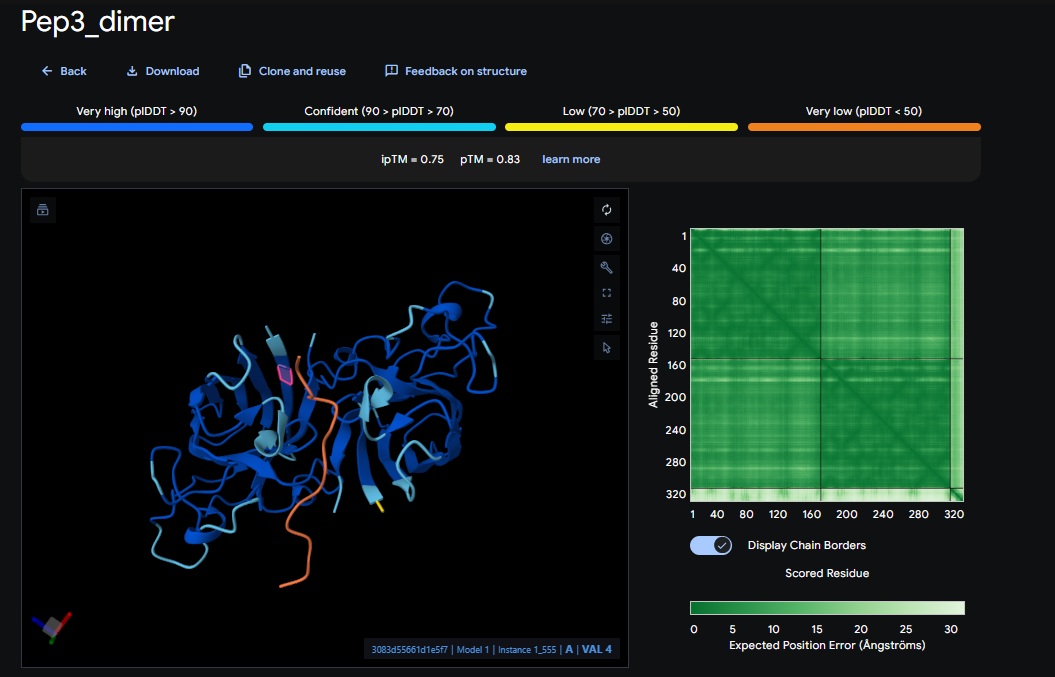
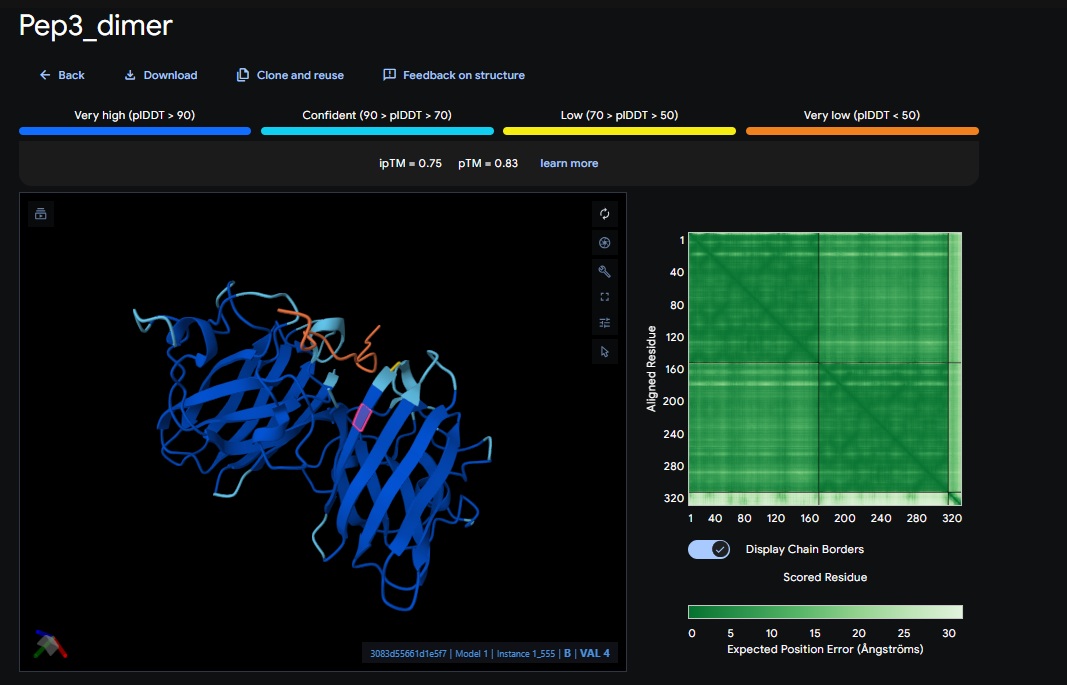
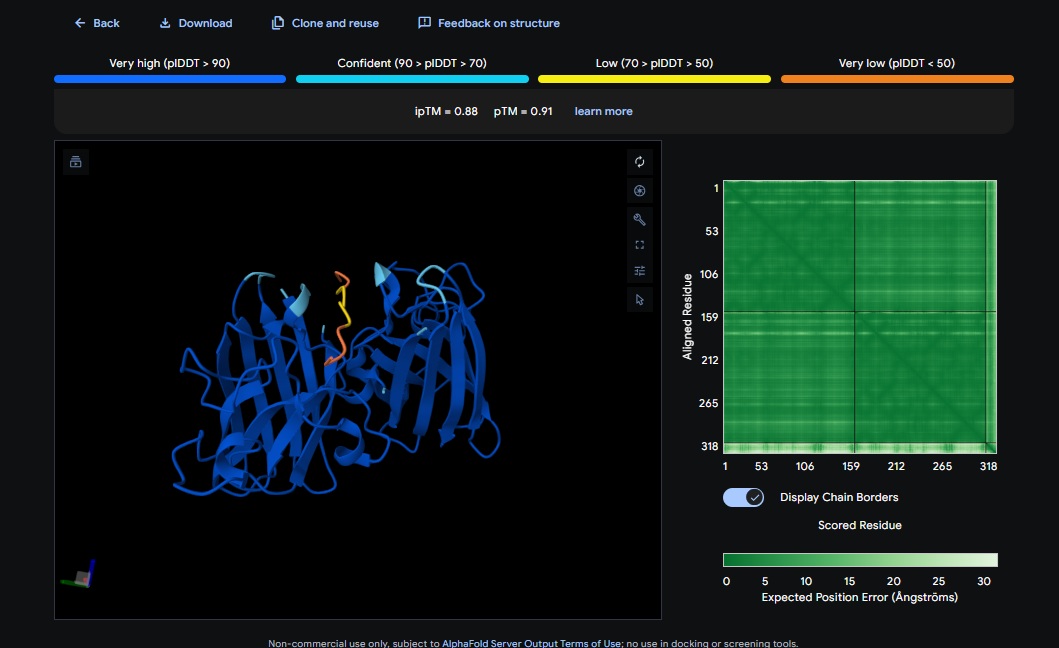
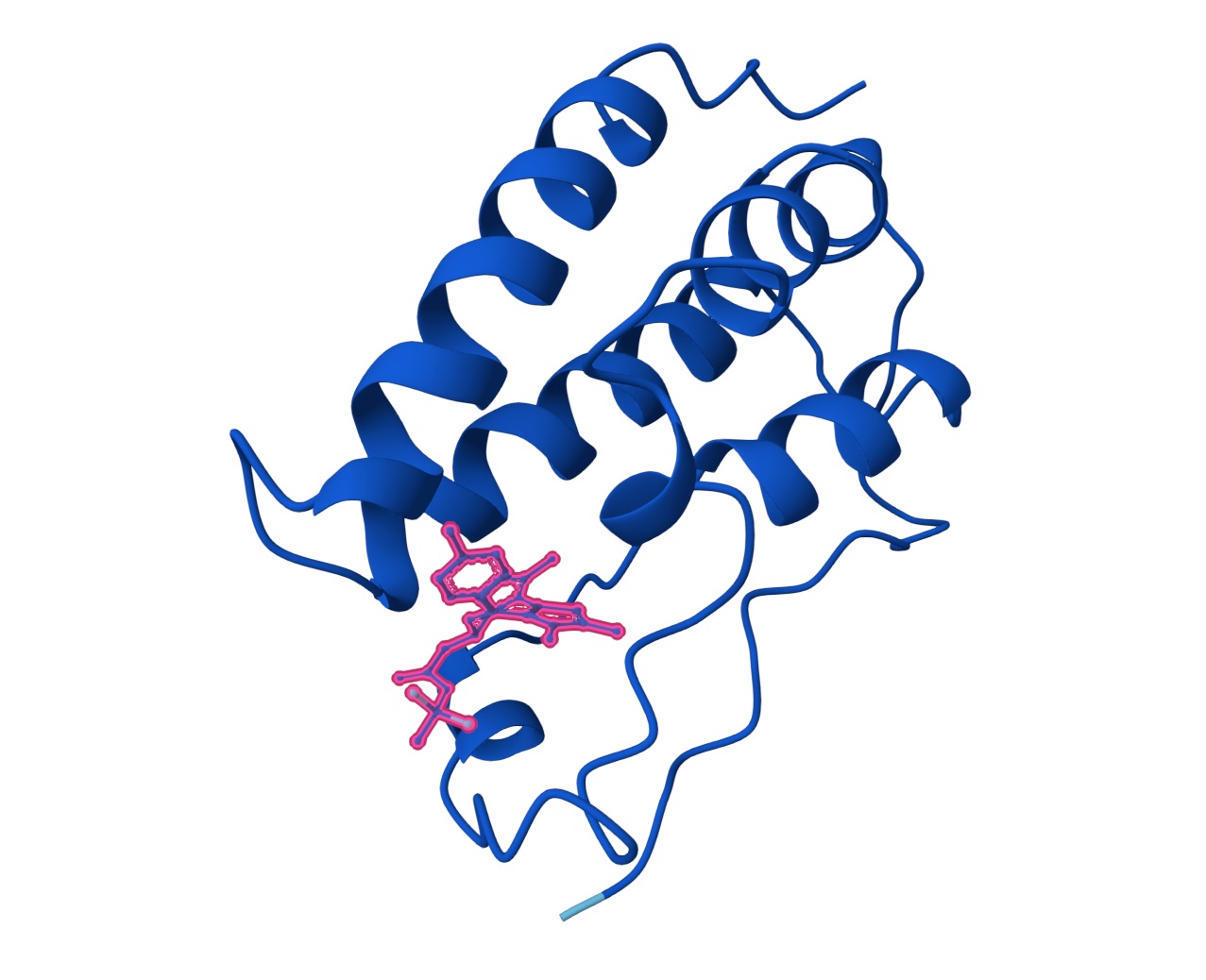
1. Design_1 (WTWVVHVATHKHHLK) + SOD1:
2. Design_2 (DTVVHHHATHEHKKK) + SOD1:
3. Design_3 (WTVEHHLVTKQEKKK) + SOD1:
You can see the peptide burried in the active site for the first time and both ends closest to the A4V Mutation sites of both monomers of the enzyme
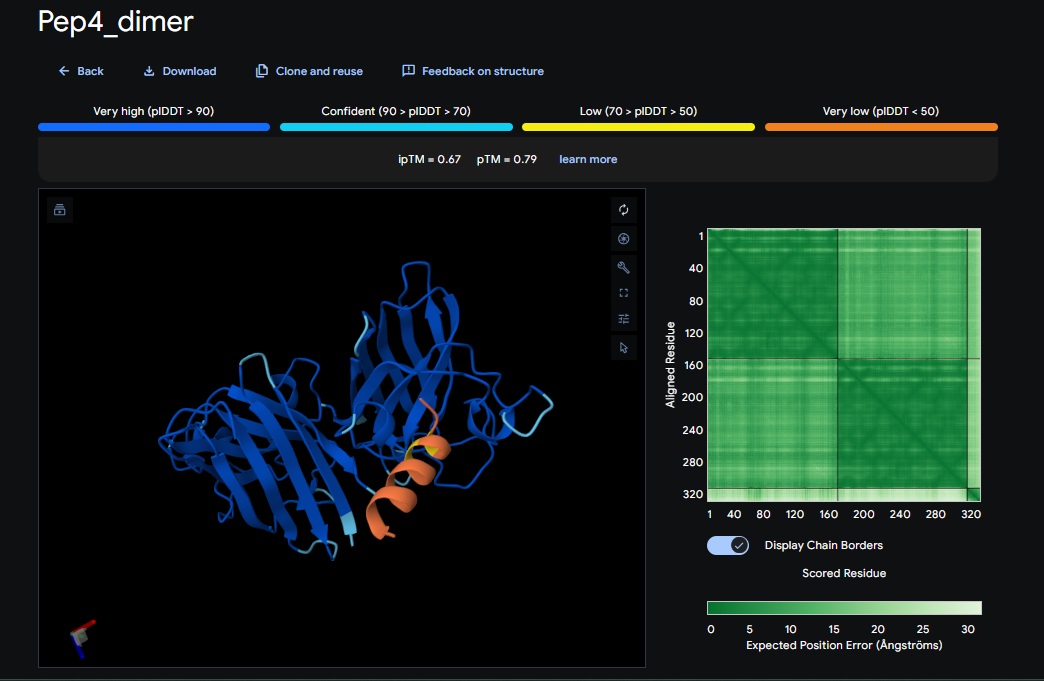
4. Design_4 (DTWDHHLATKEHKKK) + SOD1:
5. Design_5 (WTRVHAVVEKKK) + SOD1:
6. Design_6 (ATHVHVAIHHKK) + SOD1:
burried peptide
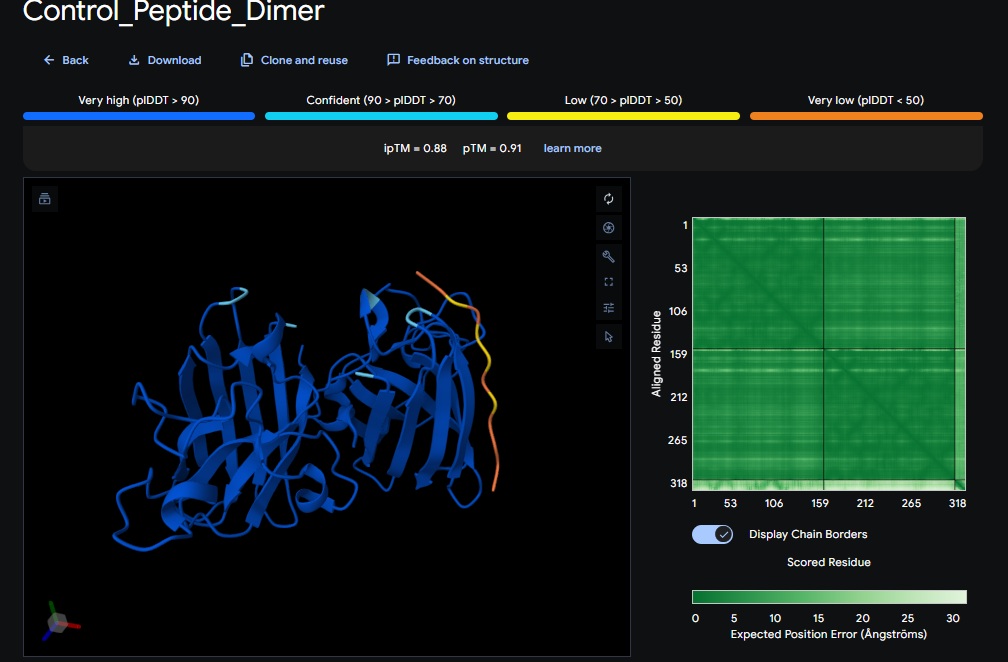
Control_Peptide (FLYRWLPSRRGG) + SOD1
PepMLM and AlphaFold Results Interpretation:
Significant improvement in Confidence (ipTM):
Dimer breakthrough: Switching to a homodimer target in PepMLM was the turning point for better AplphaFold scores.
Perplexity vs structure correlation:
Visually the best peptides for the dimer were Design 3 (WTVEHHLVTKQEKKK), Design 6 (ATHVHVAIHHKK) and Design 4 (DTWDHHLATKEHKKK). Interestingly these also had the best perplexity score (10.65, 7.73 and 10.52 respectively) but not necessarily the best TM scores.
The Design 6 peptide with the dimer (ATHVHVAIHHKK) got the highest ipTM (0.88) and pTM (0.91) scores closest to the control (FLYRWLPSRRGG) which got the same ipTM (0.88) and pTM (0.91) scores. The control didnt get a good perplexity scores and interestingly the control peptide and several designs seem to be on the same spot not near the N-terminus but near one side of the b-sheets. .
Both scores dont always seem to align. It seems that perplexity measures how natural the sequence feels to the language model, but ipTM measures the physical docking.
Targeting the A4V Mutation:
While the Control Peptide and designs showed high general binding scores, Design 6 and 3 were the only ones that were localized near the N-terminus, which is the actual site of the A4V mutation. This suggests that raw scoring (ipTM) doesn’t always guarantee site-specific binding.
Alpha fold likes the control peptide, pepMLM doesnt.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Candidate
Sequence
Solubility (Prob)
Hemolysis (Prob)
Binding Affinity (pKd/pKi)
Net Charge (pH 7)
MW (Da)
Design 1
WTWVVHVATHKHHLK
[1.000 (Soluble)]
[0.035 (non-hemolytic)]
[5.742 (weak)]
[+2.11]
[1879.2]
Design 2
DTVVHHHATHEHKKK
[0.995]
[0.032]
[4.652 ]
[+1.2]
[1804.0]
Design 3
WTVEHHLVTKQEKKK
[1.000]
[0.020]
[4.990 ]
[+1.94]
[1891.2.1]
Design 4
DTWDHHLATKEHKKK
[1.000]
[0.068]
[6.092]
[+1.02]
[1874.1]
Design 5
WTRVHAVVEKKK
[1.000]
[0.023]
[6.168]
[+2.85]
[1480.8]
Design 6
ATHVHVAIHHKK
[1.000]
[0.019]
[4.846]
[+2.14]
[1377.6]
Control
FLYRWLPSRRGG
[1.000]
[0.047]
[6.098]
[2.76]
[1507.7]
After comparing all the results seems to me that the best binders (binder 3 and 6) with respect to the perplexity scores and AlphaFold dont necessarily have the best binding affinity. The best affinity seems to be of Design 5 which actually had a good AlphaFold scores as well (ipTM :0.82) but not far more than the other peptides. They all seem to have good solubility and are non-hemolytic.
Based on all the calculations above I will choose Design 6 (ATHVHVAIHHKK) with the best perplexity score, best AlphaFold score and good therapeutic properties.
Part 4: Generate Optimized Peptides with moPPIt:
Candidate
Binder
Hemolysis
Solubility
Affinity
Motif
Design 1
GEKVCYKLKCMH
0.959324
0.750000
9.463325
0.597243
Design 2
CQDWYKSYRKYR
0.942937
0.916667
7.473063
0.467137
Design 3
RQYDTYYEKCVS
0.948101
0.916667
8.280322
0.467137
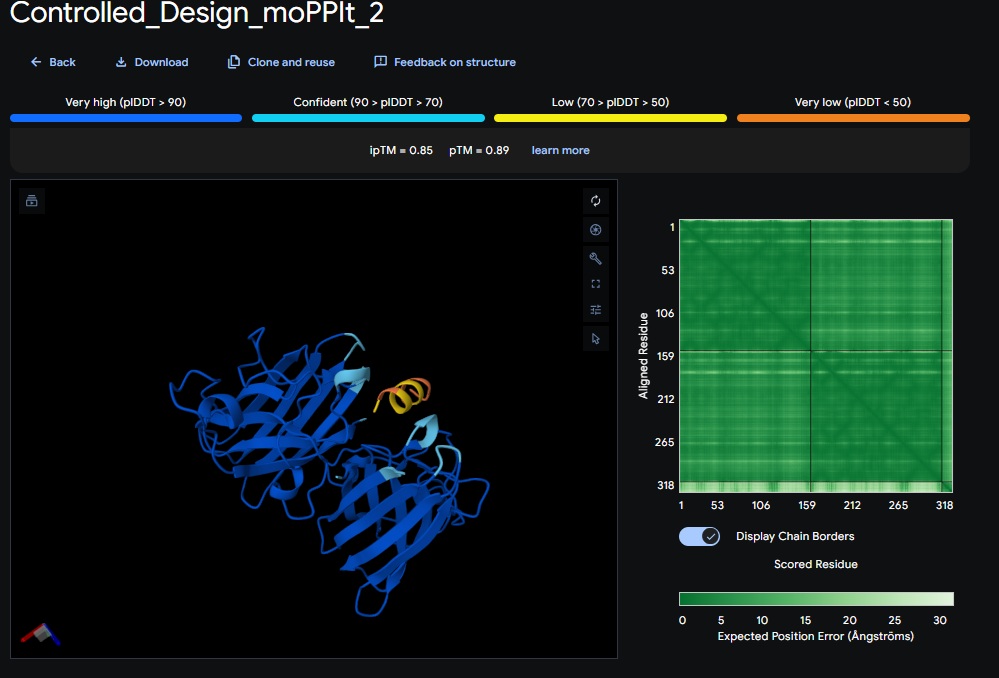
All three peptide binder designs from moPPIt-v3 just knocked the binder design out of the park compared to the PepMLM binders. In no time it created stable binders at the right site (as confirmed with alphafold results below) with great affinity (highest @ 9.4 vs 4.8 from our best design above). All three are very soluble, non-hemolytic and decent binding to our target motif residues in the SOD1 protein (I chose motif residues 2-6 and 143-154).
Design 1:
Design 2:
Design 3:
Part C: Final Project: L-Protein Mutants
Objective: Improve the stability and auto-folding of the lysis protein of a MS2-phage
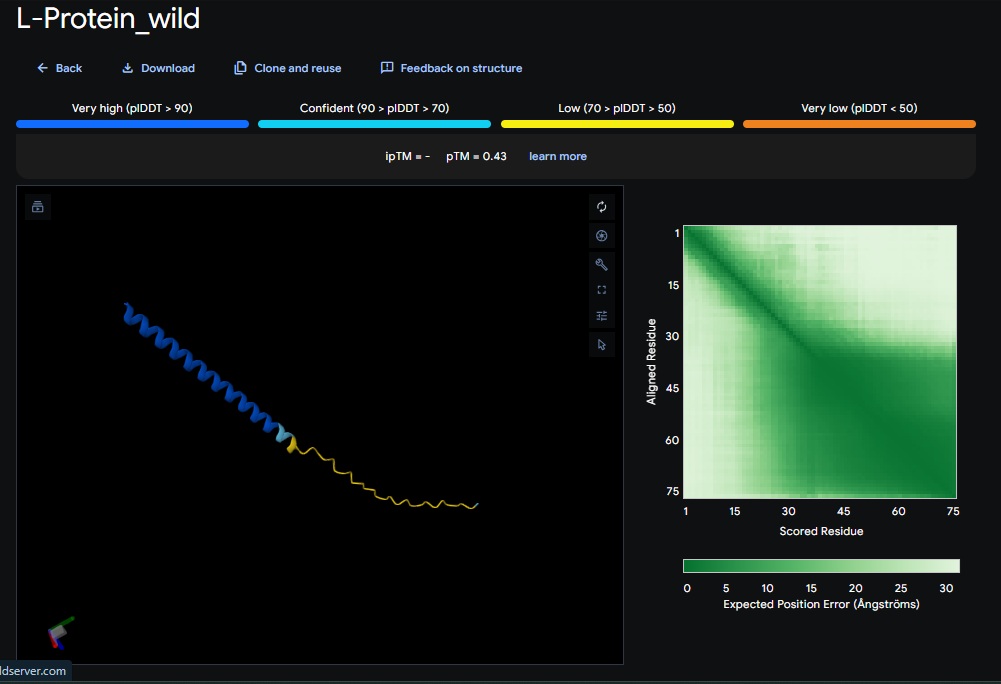
Wild-Type L-protein: Here is what we are working with
Key Summary of findings:
The mutational analysis of MS2 L identifies the highly conserved LS motif (Leu 48 - Serine49) as the functional “trigger” for lysis. Experimental data also shows that even trace amounts of functional protein can cause lysis, but the protein is currently limited by its dependency on the DNA J host chaperone which the bacteria uses by P330Q mutation to get resistant(Chamakura et al., 2017).
De Novo L-Protein Strategy:
To bypass the DNAJ dependency, I am using moPPIt to redesign the L-protein as a self-folding antimicrobial agent.
Targeting the N-terminus:
I am replacing the disordered, basic N-terminal domain (residues 1-35) with a stable, de novo- designed alpha-helix. This removes the regulatory domain that normally requires DNAj for displacement.
Maintaining Lysis activity:
I am preserving the conserved hydrophobic C-terminal domain and the LS motif to ensure that the protein retains its ability to integrate into the host membrane and trigger autolysis.
Stability optimization: I am using AlphaFold2 to validate that the new sequence achieves a high confidence score in isolation. A high score suggests the protein will fold independently and rapidly, reducing the time window for the host to mount a resistance response.
Tool Pipeline Used:
moPPIt (for multi-objective optimization) ——-> AlphaFold2 (for structural validation of autofolding potential) ——–> PeptiVerse (check for high solubility and binding affinity)
Method:
target protein to modify on moPPIt : METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
binder length to be generated by moPPIt: 75 (original size of the L-protein)
Objective and weights in moPPIt:
Hemolysis: We choose nontoxicity as score 1 at first eventhough we are creating a lysis protein. We can change this to 0 if we get low affinity.
Solubility: We choose 1 because we need good solubility. This will hopefully fix the unstable N-terminal.
Affinity: We choose 1 because we are looking for high affinity in the c-terminal portion.
Motif: Keep the generated sequence anchored to the functional lysis residues (i.e. transmembrane region: 41-75, LS-Motif: 48-49).
Interestingly the AlphaFold3 version that we were using for Pranams Assignment (which is using diffusion models instead of geometry transformers in AF2) gives a slighlty better image
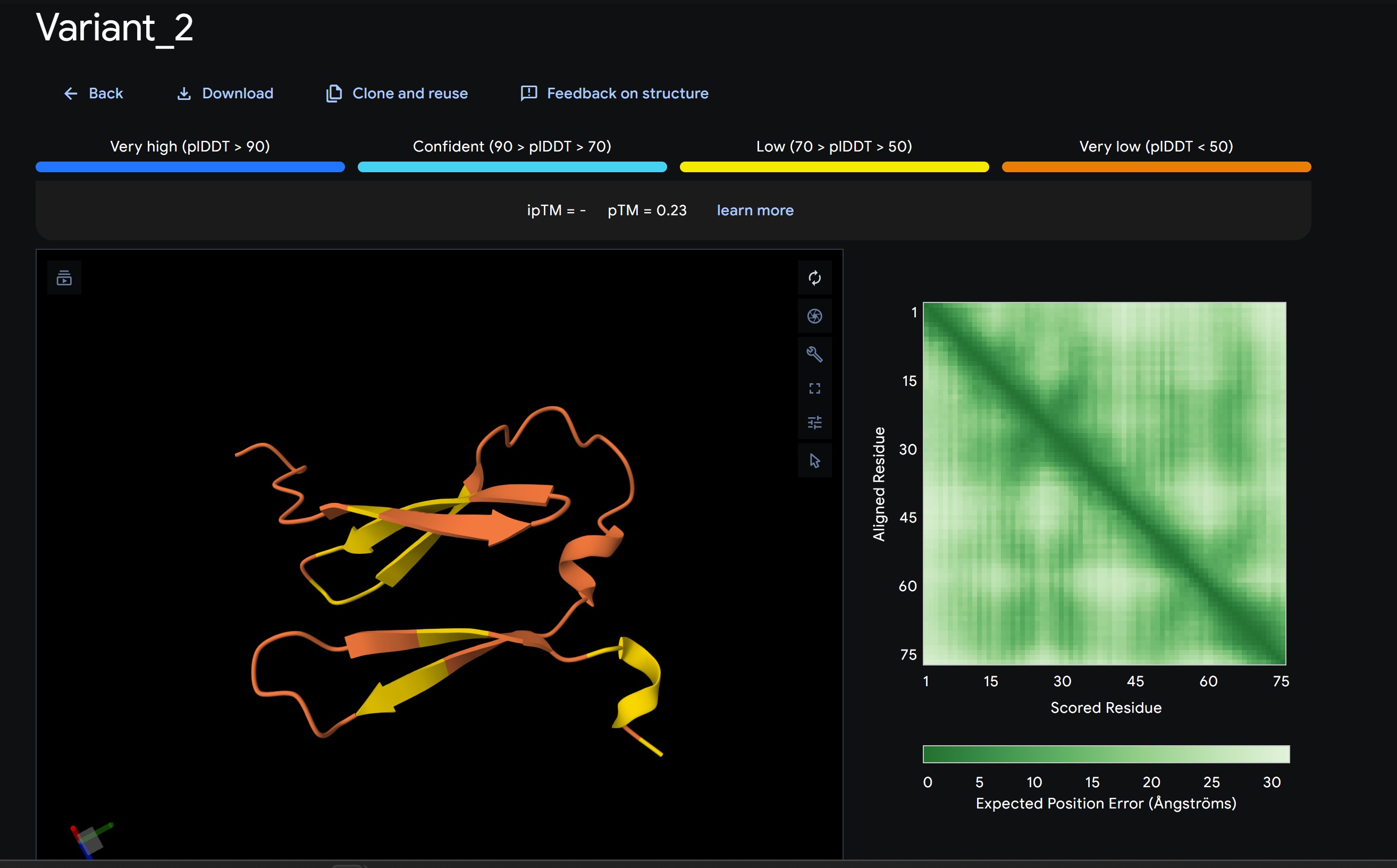
Variant 2:
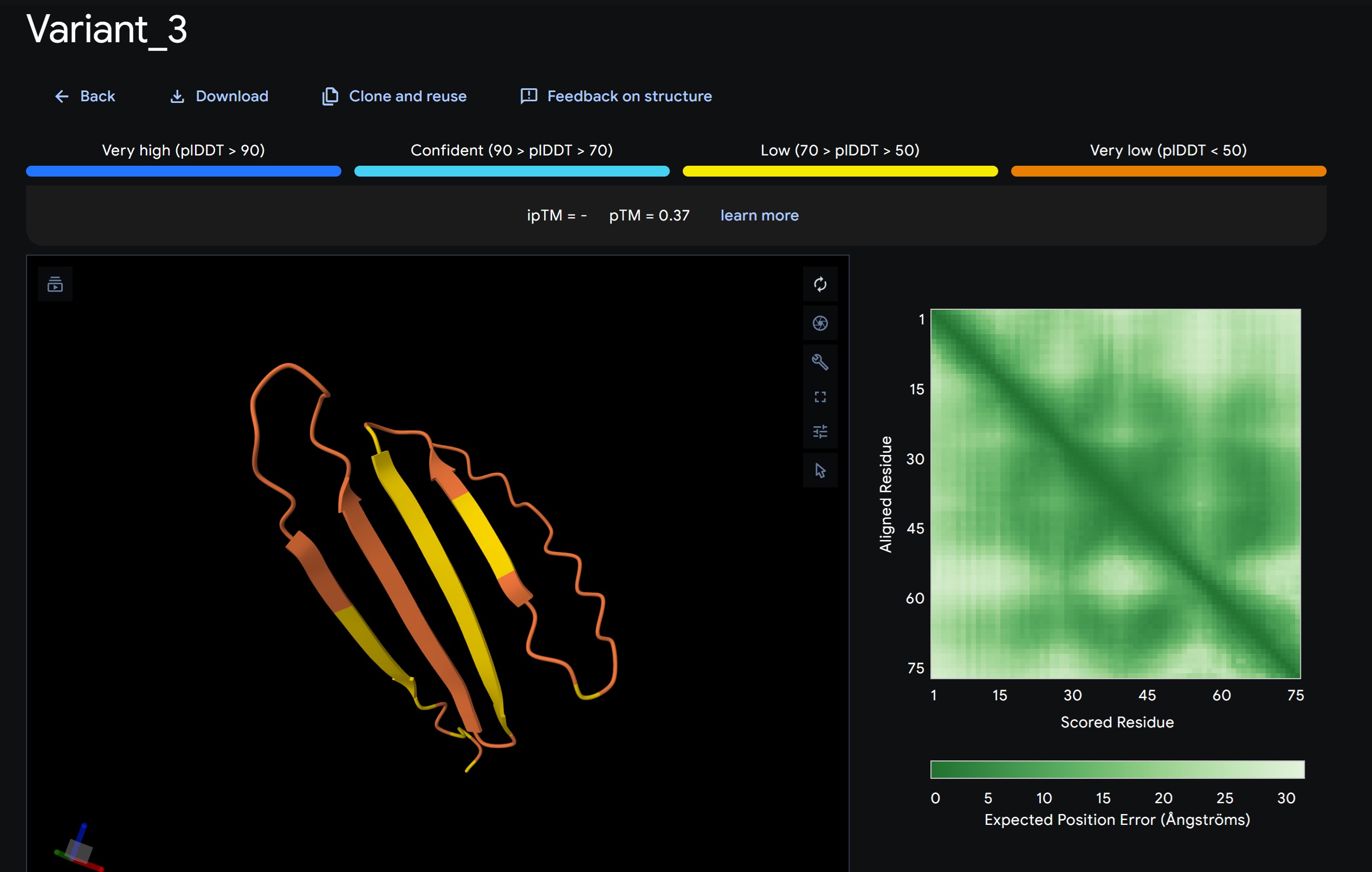
Variant 3:
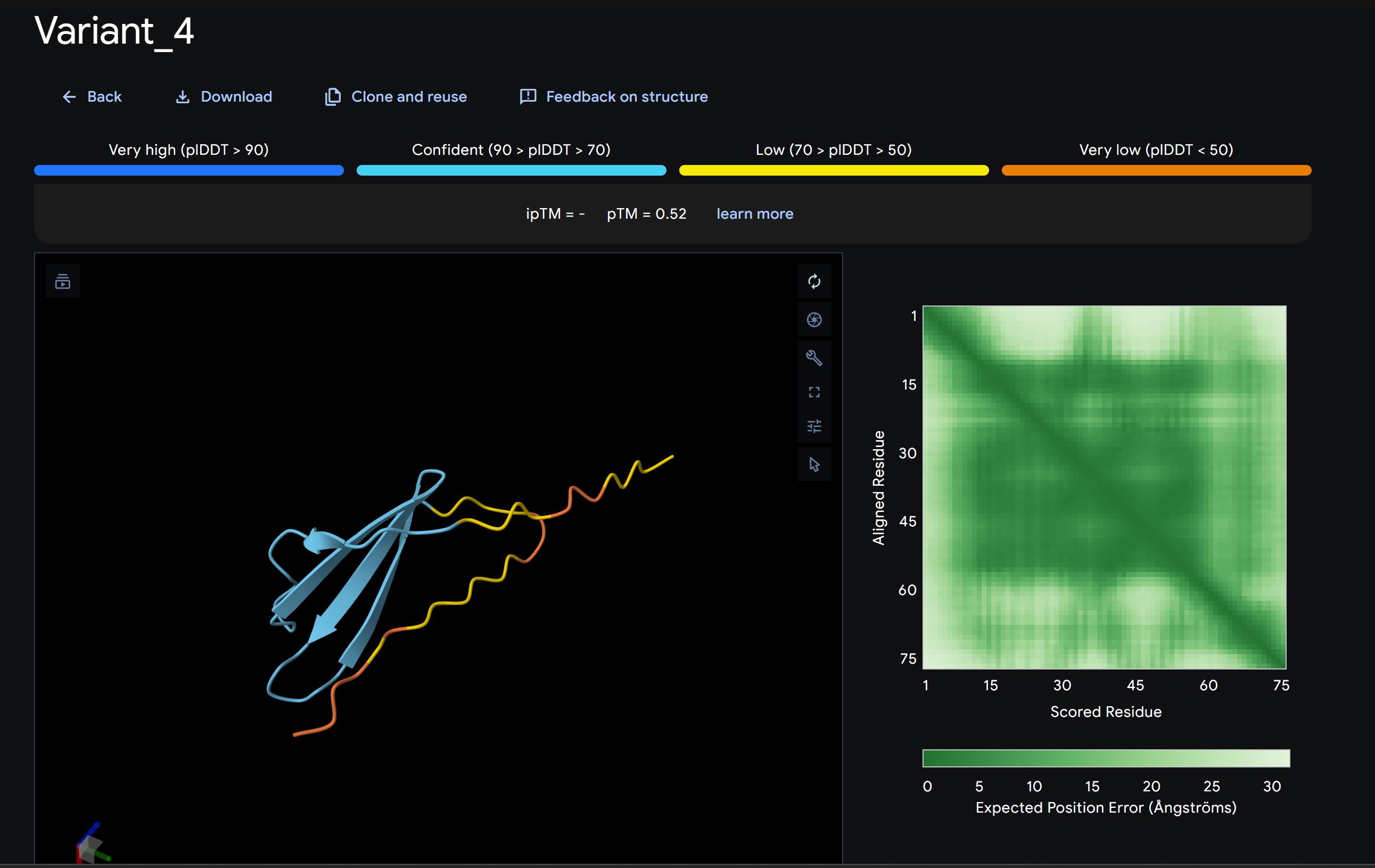
Variant 4:
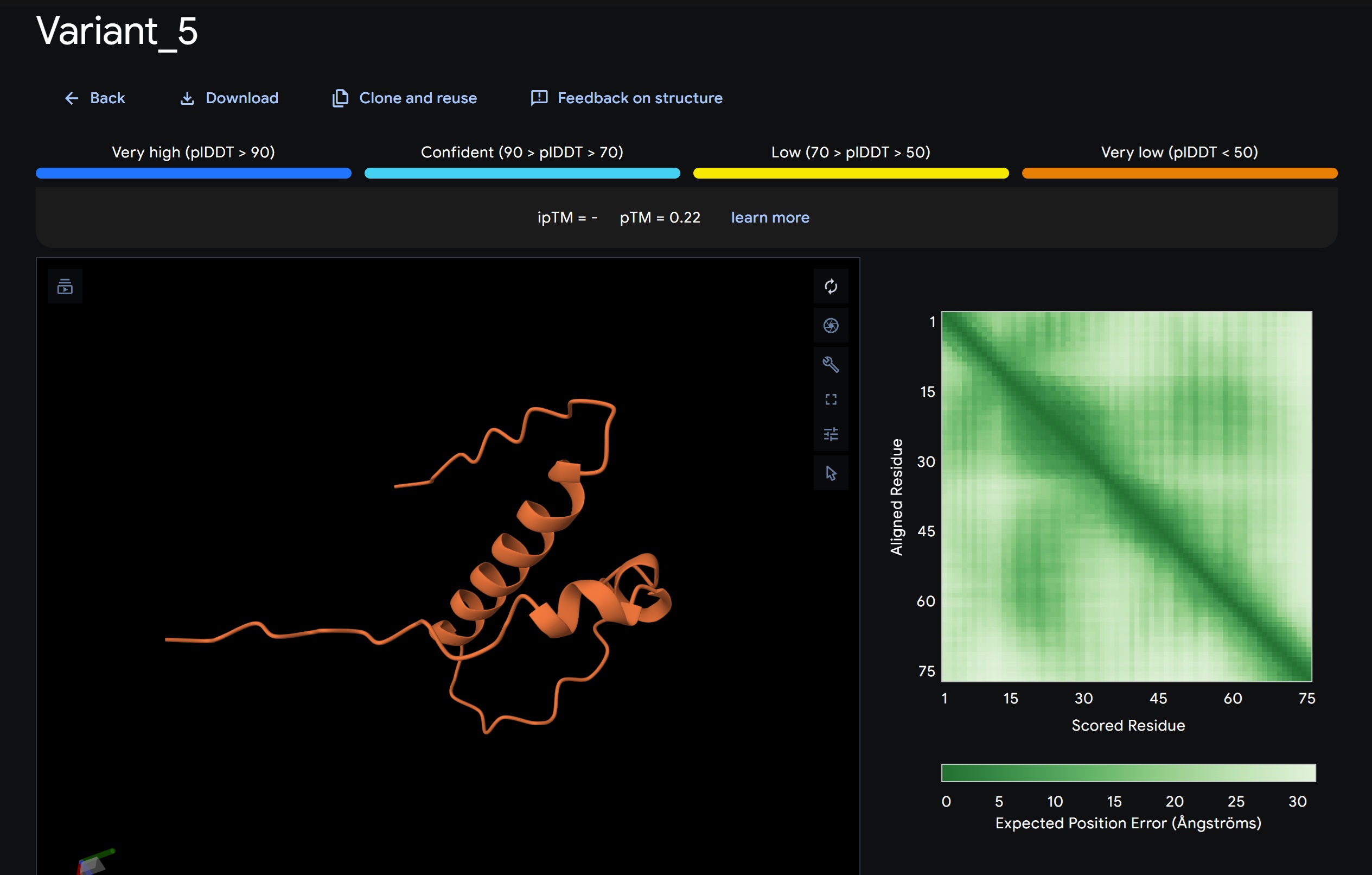
Variant 5:
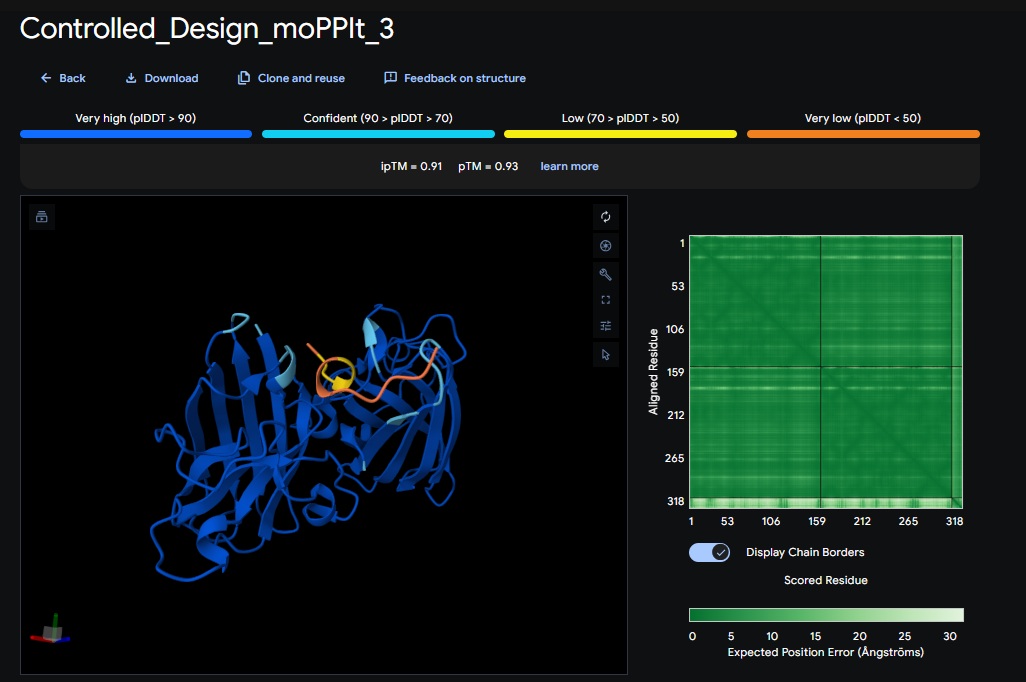
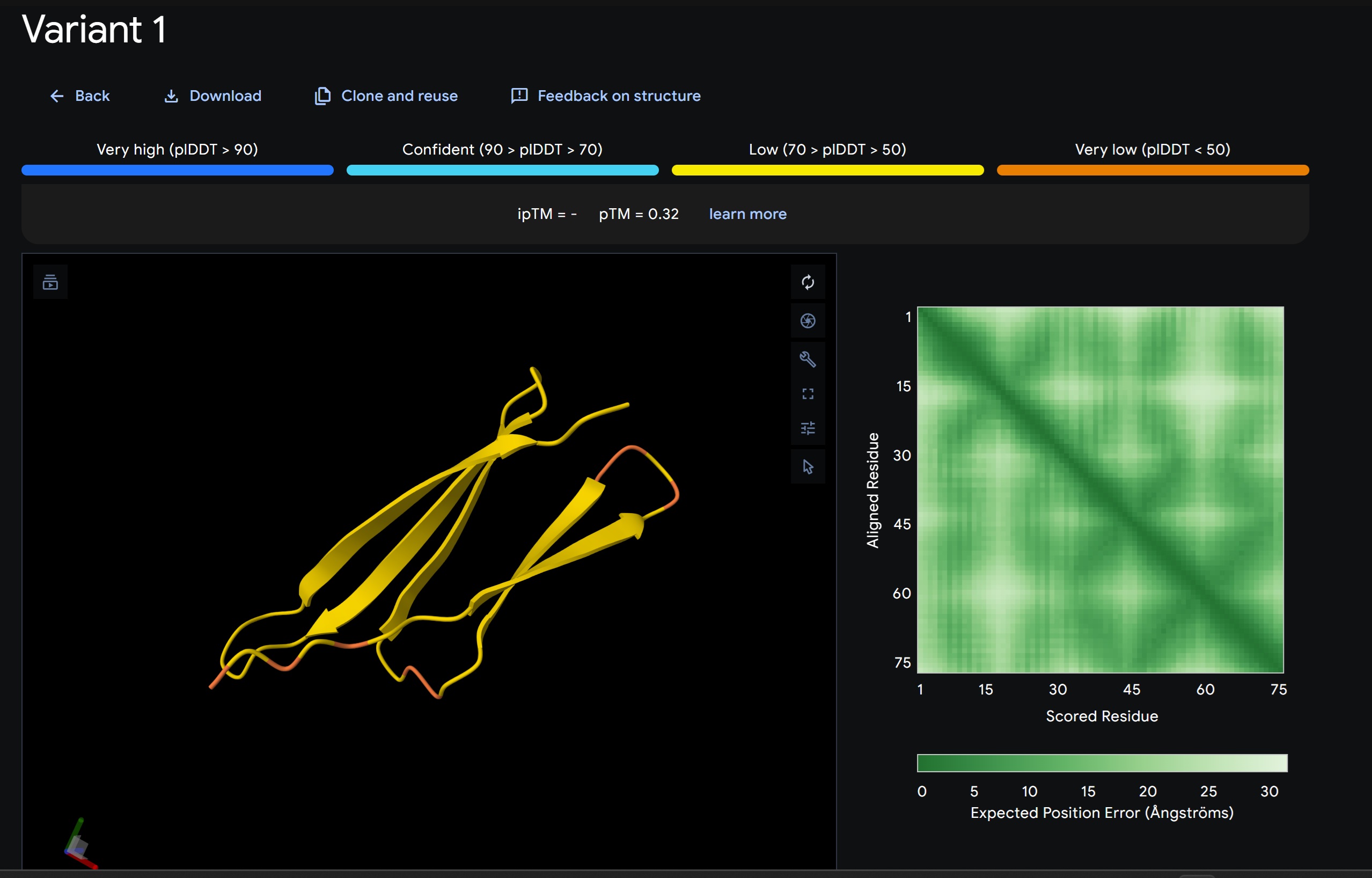
Discussion:
My lead candidate, Variant 1, was generated with a high Affinity score (9.35) and Motif score (0.68). Validation via AlphaFold3 shows the emergence of beta-sheet secondary structures in the redesigned N-terminus. While the overall pTM score is 0.32, the localized folding in the region previously dependent on DnaJ suggests a more rigid, self-stabilizing scaffold. This supports the strategy of using de novo helical/sheet designs to bypass the need for host chaperones like DnaJ (P330Q).
By comparing these variants, it is evident that moPPIt can navigate the trade-offs between solubility (for DnaJ independence) and membrane affinity (for lysis efficiency). Variant 4 emerges as the primary choice for synthesis due to its superior motif preservation and balanced biophysical properties.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele):
Week 6 — Genetic Circuits Part I: Assembly Technologies
Part A: DNA Assembly:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion Master Mix is designed for high accuracy and speed. It consists of:
Phusion DNA Polymeraase: A pyrococcus like enzyme fused to a processivity-enhancing domain. Its purpose is to catalyze DNA synthesis with extremely high fidelity (50x higher than Taq) and speed.
dNTPs (Deoxynucleotide Triphosphates): The building blocks (dATP, dCTP, dGTP, dTTP) used by the polymerase to synthesise the new strand.
Reaction Buffer: This maintains the ideal pH and ionic strength. These have been optimised for a long range of AT and GC content and includes MgCl2 (cofactor for the DNA polymerase)
What are some factors that determine primer annealing temperature during PCR?
The Ta (Annealing temperature) is critical for ensuring primers bind specifically to the target DNA
Melting temperature (Tm): Ta is ususally set 3-5 degree C below the Tm
Primer length and sequence: Longer primers and those with higher GC content have more hydrogen bonds and require a higher Ta
Salt Conc: the concentration of monovalent cations (like K+) and divalent cations like Mg2+ in the bugger stabilizes te DNA duplex affecting the Tm
PCR vs Restriction Enzyme Digest
Both methods create linear DNA, but thet serve differnt roles:
Feature
PCR (Polymerase Chain Reaction)
Restriction Enzyme Digest
Mechanism
De novo synthesis/amplification of a specific region using primers.
Enzymatic “cutting” of existing DNA at specific recognition sites.
Flexibility
Extremely high; you can amplify any sequence and add “tails” (like Gibson overlaps).
Limited to where specific restriction sites already exist in the DNA.
Fidelity
Depends on the polymerase; high-fidelity enzymes (Phusion) are needed to avoid mutations.
Very high; you are simply cutting existing, usually verified, DNA.
When to Use
Best for generating inserts from genomic DNA or adding specific sequences/overlaps to ends.
Best for linearizing a circular plasmid backbone or “snapping” out a known part from a carrier.
Ensuring compatibility for gibson assembly
To make DNA “Gibson-ready”, we must ensure:
Overlapping Ends: Each adjacent fragment must share 15-40 bp of homologous sequence at their ends. This is typically achieved by adding these sequences as “tails” on our PCR primers.
Purification: PCR products and digests must be purified using gel extraction or column cleanup to remove the original template DNA, primers and enzymes (like 5’ -> 3’ exonuclease) that could interfere with the reaction.
Correct Tm of overlaps: The overlapping regions should ideally have a Tm > 48 C to ensure they anneal properly at the isothermal reaction temperature (50 C)
How Plasmid DNA enters E.coli
During Heat shock transformation:
Cells are fiirst treated with calcium chloride (CaCl2). The Ca2+ ions neutralise the negative charges of both the DNA and the cell membrane, allowing the DNA to move close to the surface.
Moving the cells from ice at 0C to 42 C creates a thermal imbalance that increases membrane fluidity and creates temporary pores in the cell membrane.
The plasmid is then pulled into the cell through these pores potentially diven by the change un membrane motential and simple diffusion.
Alternative method: Golden Gate Assembly
Explanation: not as flexible as gibson assembly because it uses restriction enzymes that cut at specific sites.
Pre-lab: Primer Design for gibson assembly:
Acropora millepora chromoprotein (amilCP) variants and Bases:
Color variant
Codon
Amino Acid Change
Original
TGTCAG
(Wild Type Cys - Gln )
Orange
ACTGCT
Thr - Ala
Blue (aeBlue style)
CAGTAC
Gln - Tyr
Pink
TACTGG
Tyr - Trp
Primers
1. Universal Forward Primer:
This primer is designed to facilitate the seamless assembly of the genetic construct into the destination vector. Sequence B (5’ -cacatccccctttcgccag -3’) serves as the homology tail derived from the pUC19 plasmid, providing the 19bp overlap required for the gibson exonuclease to “chew back” and join the fragments. Sequence A (5’-gaattcggtctctatatgcaggtg-3’) is the annealing segment that specifically targets the mUAV backbone. This 3’ segment has been optimized for a melting temperature of 55.1 C ensuring high fidelity during the PCR amplification step.
mUAV region after terminator: gcagggtctcaatatgcaggtg (Binds to the end of the backbone).
pUC10 reverse compliment : gtcgggaaacctgtcgtgccag (Homology for the downstream junction)
Full Primer Sequence:gtcgggaaacctgtcgtgccaggcagggtctcaatatgcaggtg
Week 7 — Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
Practice NW:
High Pass
Low Pass
Dual Region
1. Advantages of IANNs over Boolean Circuits
Traditional Boolean circuits are limited to binary “True/False” logic, which struggles with the “fuzzy” nature of biological environments. IANNs provide:
Noise Filtering: By using weighted thresholds, IANNs can ignore transient spikes (like exercise-induced miR-26) that would normally trigger a false positive in a Boolean switch.
Analog Resolution: They allow for a graded response. In my DermLogic patch, this means the hydrogel can change density proportionally to the severity of the biomarker signal.
Complex Pattern Recognition: IANNs can integrate multiple inputs (miR-21 AND miR-26) to make a single “calculated” decision, similar to how a neuron fires only when a specific summation of signals is reached.
2. Application: The DermLogic Subtractive Patch
Input/Output: The inputs are miR-21 (pathology signal) and miR-26 (physical activity noise). The output is the expression of ELP hydrogel.
Behavior: The IANN performs a subtraction (Output = miR21 - w . miR26). This ensures the patch only assembles/disassembles when the pathological signal outweighs the background noise of the user’s daily movement.
Limitations: IANNs face “Metabolic Load” limits; running complex neural math in a cell-free system requires high concentrations of Csy4, which can deplete the resources needed for the output protein (ELP).
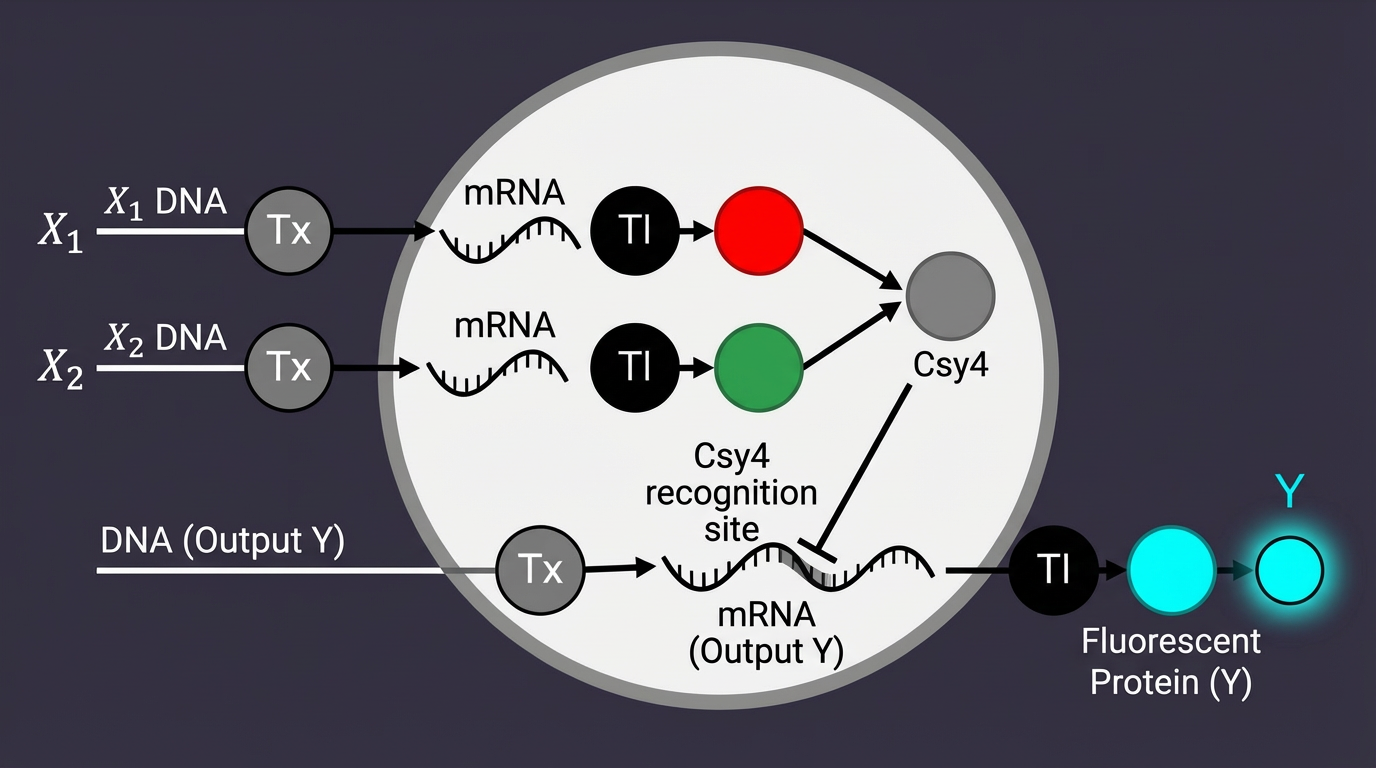
3. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
1. Existing Fungal Materials
Examples include Mycelium-based packaging (e.g., Ecovative) and fungal leather (MycoWorks).
Advantages: They are carbon-negative, biodegradable, and grow on agricultural waste.
Disadvantages: They are currently slower to “manufacture” than plastics and can be sensitive to moisture, leading to premature degradation.
2. Genetic Engineering in Fungi
I would want to engineer fungi to secrete specific therapeutic enzymes upon sensing a skin pathogen.
Why Fungi? Fungi are eukaryotes, meaning they can perform complex “post-translational modifications” (like glycosylation) that bacteria cannot. This makes them better “factories” for human-like proteins.
Advantage over Bacteria: Fungi possess a robust secretory pathway and can form large, physical structures (mycelial mats) that serve as both the “factory” and the “bandage” simultaneously.
Assignment Part 3: First DNA Twist Order
Project Overview: Skin microRNA Receptor Patch
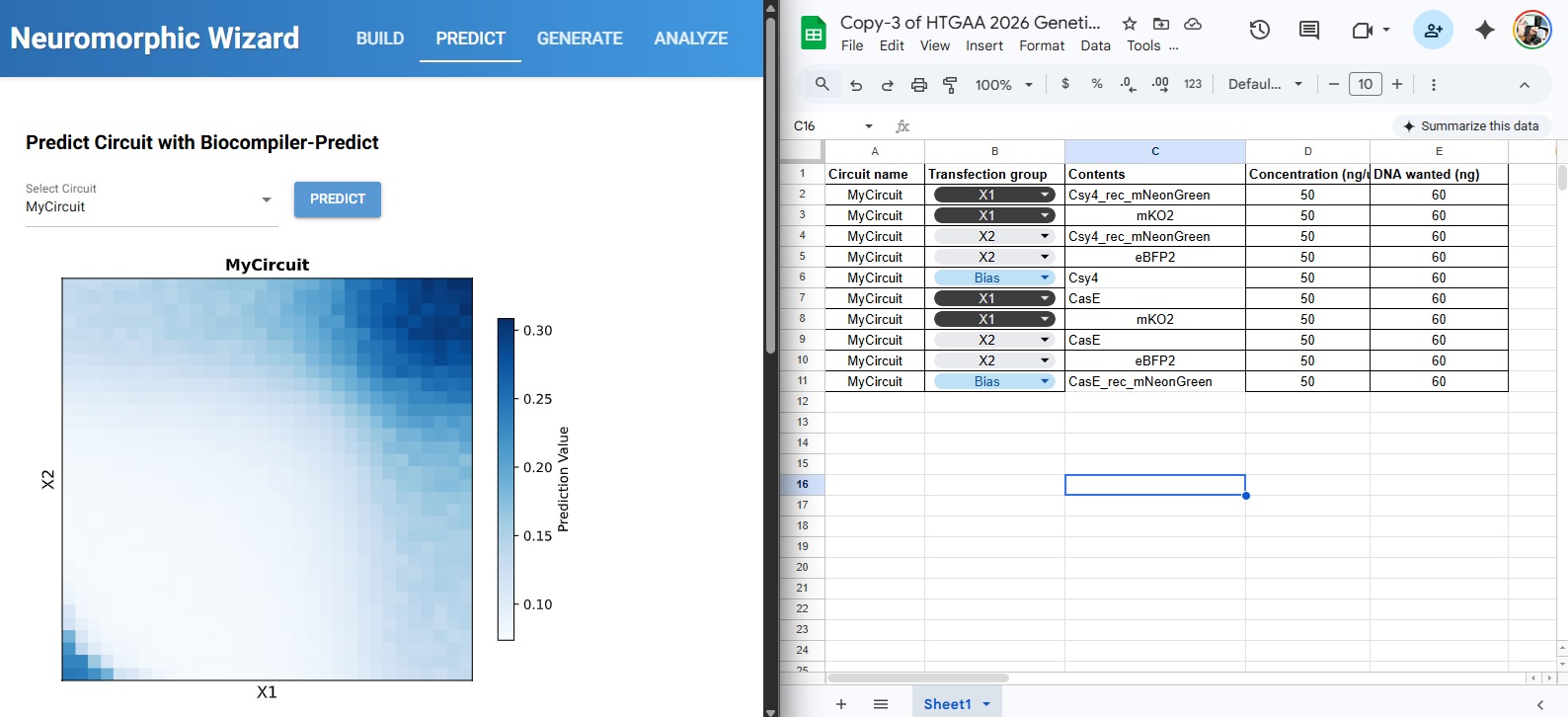
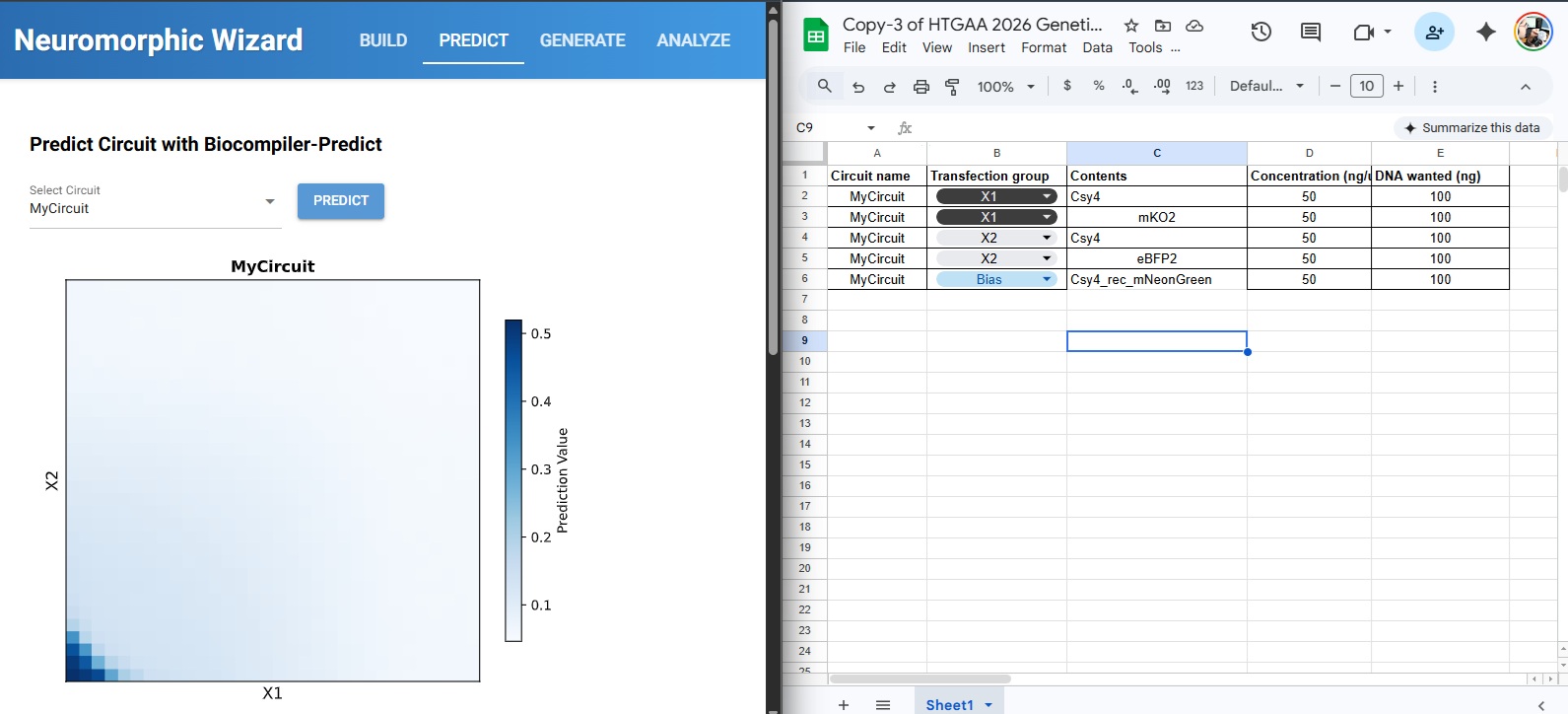
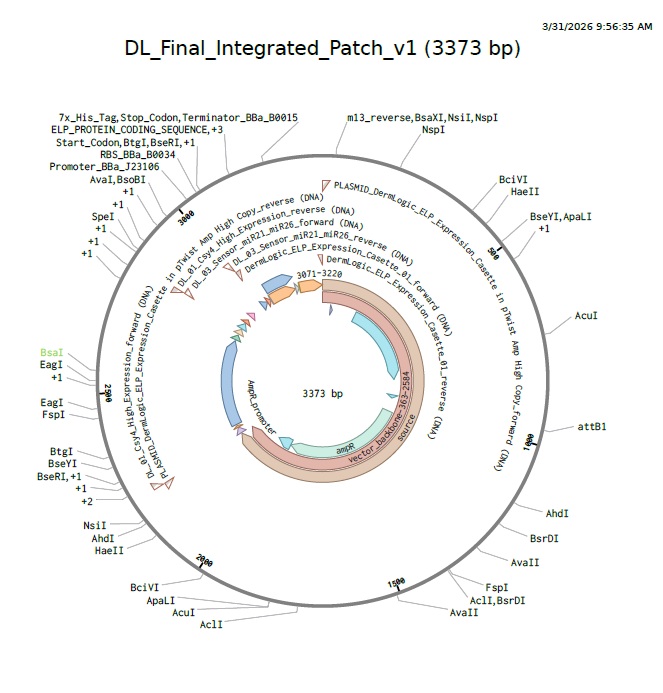
For this week’s assignment, I integrated my Final Project Aim 1 into the neuromorphic circuit framework. I designed a DNA construct (DL_Final_Integrated_Patch_v1) intended to function as a smart, bio-responsive interface.
1. The Design Logic (Neuromorphic Approach)
The circuit is designed to sense specific skin microRNAs (such as miR-21 or miR-26) which serve as “weighted inputs.” Unlike traditional digital logic (0 or 1), this circuit aims to emulate Intracellular Artificial Neural Networks (IANNs) by:
Analog Sensing: Responding to varying concentrations of microRNA rather than a simple on/off state.
Thresholding: Using the genetic architecture to trigger a response only when a specific “signature” or threshold of biomarkers is sensed.
Material Output: Expressing ELP (Elastin-Like Polypeptide) to modulate the physical properties of the hydrogel matrix.
2. Genetic Architecture & Components
The current construct includes several key components visualized in my design:
Promoters & RBS: Utilizing parts like J23106 and B0034 to ensure reliable baseline expression.
Csy4 Processing: Used for RNA transcript maturation or gating to clean up the “noise” in the circuit.
ELP Matrix: The ELP sequence allows the genetic output to be translated into a structural change in the hydrogel, effectively creating an Engineered Living Material (ELM).
Reflective Note: I am exploring how to move beyond simple Boolean gates. While the design is in its first iteration, the goal is to create a “weighted” response system where the hydrogel’s state is a direct “calculation” of the skin’s molecular environment.
3. DNA Synthesis & Backbone Specifications
In accordance with the Week 7 Assignment Part 3 requirements, this construct is designed for synthesis in a high-efficiency vector optimized for cell-free protein expression.
In traditional synthetic biology, sensors are often designed as simple “ON/OFF” Boolean switches. However, for a sweat-sensing patch, the biological environment is naturally “noisy”—for example, physical exercise can cause a 40-fold spike in miR-26, which would normally trigger a false positive in a standard gate.
By adopting a neuromorphic architecture, I am treating miR-21 (the signal) and miR-26 (the exercise noise) as weighted inputs to a single-layer perceptron. Utilizing the Csy4 endoribonuclease as a subtractive processor allows the circuit to perform a real-time analog calculation (Output = Signal - Noise) directly within the ELP hydrogel matrix. This ensures that the diagnostic output is a true reflection of skin pathology rather than a byproduct of the user’s physical activity.
Week 9 — Cell-Free Systems
Part A: General and Lecture- Specific
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Flexibility & Control: Unlike living cells, CFPS is an “open” system. You can directly manipulate the reaction environment—adjusting pH, redox potential, or adding non-natural amino acids—without worrying about maintaining cell viability.
Speed: It bypasses the time-consuming steps of transformation, cell culture, and scale-up, allowing for results in hours rather than days.
Beneficial Cases:
Toxic Proteins: Expressing proteins that would kill a living host cell (e.g., certain antimicrobial peptides).
Rapid Prototyping: Testing many different genetic designs quickly in a high-throughput format.
2. Describe the main components of a cell-free expression system and explain the role of each component.
Cell Extract: Provides the molecular “machinery,” including ribosomes, tRNAs, and initiation/elongation factors.
Energy Source: Molecules like phosphoenolpyruvate (PEP) or creatine phosphate used to regenerate ATP and GTP.
Amino Acids: The essential building blocks for synthesizing the protein chain.
DNA Template: The genetic instructions (plasmid or linear PCR product) for the target protein.
Buffer/Salts: Maintains optimal pH and ionic strength (especially Mg2+ and K+) required for ribosomal function.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Criticality: Energy is consumed rapidly during transcription and translation. Without a regeneration system, the reaction stops once the initial ATP pool is depleted, leading to very low yields.
Method: Use an enzymatic substrate system, such as the Creatine Phosphate/Creatine Kinase system, which transfers a phosphate group back to ADP to regenerate ATP in situ.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic (e.g., E. coli): High yield, fast, and inexpensive. However, it lacks complex post-translational modifications (PTMs).
Protein:GFP (Green Fluorescent Protein) for simple reporting or biosensing where complex folding isn’t required.
Eukaryotic (e.g., CHO or Wheat Germ): Lower yield but capable of complex folding and PTMs like glycosylation.
Protein:Human Insulin, which requires specific disulfide bond formation and folding pathways not present in bacteria.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Challenges: Membrane proteins are hydrophobic and often aggregate or misfold when synthesized in an aqueous cell-free mix without a lipid environment.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Template Degradation: - Reason: Presence of RNases or DNases in the extract.
Strategy: Use high-purity DNA and supplement the reaction with RNase inhibitors.
Energy Depletion: - Reason: Fast consumption of ATP/GTP.
Strategy: Increase the concentration of the energy buffer or use a dialysis-based continuous-exchange system.
Codon Bias: - Reason: The DNA sequence uses codons that are rare in the organism the extract was made from.
Strategy: Use a codon-optimized gene sequence or supplement the reaction with a mixture of rare tRNAs.
Homework question from Kate Adamala
1. Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
Function:DermLogic is a dual-channel AND-gate biosensor designed for point-of-care HPV detection and therapeutic antisense RNA delivery.
Input/Output: The input is extracellular HPV L1 and E6/E7 RNA sequences; the output is a dual-fluorescence signal (GFP/mCherry) and the synthesis of therapeutic antisense RNA.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, the core logic functions in a bulk cell-free mix. However, encapsulation is required for the “therapeutic patch” vision to protect the RNA payload from degradation and to concentrate the reagents for faster kinetics.
Could this function be realized by genetically modified natural cell?
It is difficult. Natural cells have complex innate immune responses (like interferon pathways) that might interfere with or degrade synthetic RNA logic circuits and antisense outputs.
Describe the desired outcome of your synthetic cell operation.
A low-cost, decentralized tool that stratifies HPV risk and simultaneously produces a customized therapeutic response without a cold chain.
2. Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
A robust, shelf-stable lipid bilayer composed of POPC and Cholesterol, or potentially a polymeric/hybrid vesicle for better stability during lyophilization.
What would you encapsulate inside?
BL21 DE3 Lysate, the pDermLogic-v1 plasmid, T7 RNA Polymerase, amino acids, and an energy regeneration system (PEP/ATP).
Which organism your Tx/Tl system will come from?
Bacterial (E. coli) is preferred. It is highly efficient for T7-driven transcription and the toehold switches are optimized for bacterial ribosomes.
How will your synthetic cell communicate with the environment?
Through expressed membrane pores like alpha-hemolysin (aHL). These allow the viral RNA “triggers” to enter the cell and the fluorescent/therapeutic outputs to be detected or released.
3. Experimental details
List all lipids and genes.
Lipids: POPC, Cholesterol.
Genes:sfGFP (Channel A reporter), mCherry (Channel B reporter), and a custom antisense RNA sequence targeting the HPV E6/E7 junction.
How will you measure the function of your system?
Using a dual-channel fluorescence readout (Spark Plate Reader) to monitor the AND-gate logic, and denaturing PAGE gel electrophoresis to verify the production of the ~21 nt antisense RNA.
Homework question from Peter Nguyen
Pitch: “Bio-Sensing Textiles: A garment that changes color upon detecting hazardous environmental pathogens.”
How it works: Freeze-dried cell-free systems containing a specific RNA-based biosensor (riboswitch) are embedded into fabric fibers. Upon exposure to a specific pathogen and moisture (sweat or atmospheric water), the system rehydrates, triggers the sensor, and expresses a chromoprotein that visibly stains the fabric.
Societal Challenge: This addresses the need for passive, wearable safety monitoring for healthcare workers or soldiers in environments with invisible biological threats.
Addressing Limitations: The “one-time use” nature is addressed by making the sensor a disposable patch integrated into the garment; activation is solved by leveraging inherent moisture or the user’s perspiration as the rehydration trigger.
Homework question from Ally Huang
Background Information: Microgravity and cosmic radiation cause significant muscle atrophy and DNA damage in astronauts. Monitoring real-time protein expression in space is difficult due to bulky equipment. BioBits® allows for rapid diagnostic tests on the ISS with a minimal footprint.
Target:Myostatin (MSTN) protein levels, which are key regulators of muscle growth and indicators of muscle wasting.
Relation to Space Biology: Tracking Myostatin levels allows researchers to quantify the rate of muscle degradation in microgravity. By using a cell-free biosensor, we can monitor these levels in real-time without needing to return samples to Earth.
Hypothesis/Research Goal:Hypothesis: BioBits® can be engineered to produce a fluorescent signal proportional to the concentration of Myostatin mRNA. The goal is to create a “just-add-sample” diagnostic kit for astronauts to monitor their physical health during long-duration missions.
Experimental Plan: We will test astronaut saliva samples. Control: Rehydrated BioBits® with a known concentration of MSTN DNA. Experimental: BioBits® rehydrated with the astronaut’s sample. Data will be collected using the P51 Molecular Fluorescence Viewer to observe the intensity of the green fluorescence.
Part B: Final Project Integration — DermLogic
Aim 1: Cell-Free Logic Validation
The core of my final project, DermLogic, relies on the E. coli BL21 DE3 cell-free system to execute a complex molecular AND-gate. This week’s focus on cell-free systems directly informs my strategy for:
Signal Processing: Using dual-channel toehold switches (L1-GFP and E6/E7-mCherry) to stratify HPV risk.
On-Demand Therapeutics: Leveraging the “open” nature of cell-free systems to synthesize ~21 nt antisense RNA molecules immediately upon pathogen detection.
Lyophilization Strategy
Following the principles of BioBits®, DermLogic is designed to be a shelf-stable, “just-add-water” (or sample) diagnostic. My validation plan for Aim 1 includes:
Cryoprotectant Optimization: Testing a mix of 100mM Trehalose and 0.1% BSA to ensure the T7 RNA Polymerase and ribosomes remain functional after freeze-drying.
Stability Testing: Comparing the fluorescence kinetics of fresh vs. rehydrated pellets using the Spark Plate Reader to calculate signal retention.
Key Component: By encapsulating this reaction in a POPC/Cholesterol lipid bilayer with alpha-hemolysin pores, the system transforms from a bulk reaction into a “synthetic cell” capable of localized therapeutic delivery.
1. Identify at least one aspect of your project that you will measure.
For the DermLogic v2 platform, the primary measurement is the fluorescence intensity generated by the reconstitution of split-sfGFP. This serves as a quantitative proxy for the presence of the target HPV16 L1 and E6 mRNA biomarkers. Additionally, I will measure the molecular weight and structural integrity of the transcribed RNA triggers and the synthesized DNA inserts to ensure the “AND-gate” logic components are correct before assembly.
2. Describe the elements you would like to measure and how you will perform these measurements.
Transcriptional Accuracy: I will measure the length and purity of the RNA triggers (L1 and E6) produced via T7 in vitro transcription.
Logic Gate Kinetics (AND-gate): I will measure the rate of sfGFP complementation (fluorescence over time). This requires monitoring four conditions: a negative control, each trigger individually, and the dual-trigger state to confirm that signal only occurs in the presence of both.
Therapeutic Module Validation: I will measure the expression of the 116 nt antisense E6 RNA segment. Since this is co-transcribed with the reporter, I must verify it is not degraded and maintains its predicted size.
Protein Fragment Identity: I need to confirm that the split-sfGFP fragments (N-terminal 1-157 and C-terminal 158-238) are being synthesized correctly within the PURExpress cell-free system.
3. What technologies will you use? Describe in detail.
Fluorescence Spectroscopy (Plate Reader): I will use a microplate reader set to Ex 485nm / Em 507nm to perform real-time kinetic assays at 37°C. This allows for the quantification of the sfGFP reconstitution speed and the establishment of the Limit of Detection (LoD).
Urea-PAGE (Polyacrylamide Gel Electrophoresis): As specified in Aim 3 of my proposal, I will use Urea-PAGE under denaturing conditions to resolve the RNA transcripts. This is critical for confirming the presence of the antisense/scaffold band and ensuring no premature transcriptional termination.
Liquid Chromatography-Mass Spectrometry (LC-MS): Inspired by the Week 10 Lab, I would use Intact Mass Analysis on a system like the Waters Xevo G3 QTof to confirm the molecular weight of the synthesized split-sfGFP fragments.
Peptide Mapping (MS/MS): I would use tryptic digestion followed by tandem mass spectrometry to confirm the amino acid sequence of the split-junction (residues 157/158), ensuring the fragments are prepared exactly as designed for optimal reconstitution.
Week 11 — Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas
My Contribution: I was trying to make a buddha/baby at the bottom center.
What I liked about the project: It was amazing to see it develop as soon as the email went out and during the class. It was a very surreal experience and the spirit of collaboration is really visible when you roll back the history and how it became its final version. So grateful to be a part of this amazing cohort of humans in the loop. Suggestions for improvement: Lets keep this spirit going and make this dream happen; of cloud labs around the world that conduct synthetic biology in the spirit of collaboration and ethics to advance synthetic biology in the least harmful most useful direction. We did it during covid, lets not wait for another pandemic to come together. Thanks Ronan and the team for giving us the taste of this global spirit.
Part B: Cell-Free Protein Synthesis
1. Component Descriptions
Component
Role in Cell-Free Reaction
E. coli Lysate
The guts of the living cell that still work without the shell of a cell. Reminds me of a bug cut into two but still functional since the component parts don’t necessarily know they are “dead”—fascinating for microscopic life. It provides the machinery: Polymerase, Ribosomes, and Enzymes.
Potassium Glutamate
The primary salt for the “biological soup.” It maintains osmotic balance and ensures the ionic strength is just right for protein-DNA interactions.
HEPES-KOH pH 7.5
The buffer system. It keeps the pH steady so the reaction doesn’t crash or get too acidic as metabolic byproducts start to accumulate.
Magnesium Glutamate
A vital cofactor. It’s basically the glue that stabilizes the ribosomes and RNA polymerase; without it, the translation engine stalls.
Potassium phosphate
Provides a phosphate source for energy regeneration and doubles as extra buffering capacity for the system.
Energy/Nucleotide System
The fuel tank. Provides the ATP, GTP, UTP, and CTP required to power transcription and “charge” the tRNAs with amino acids.
Translation Mix
The raw building blocks; the 20 standard amino acids that the ribosomes string together to build your target protein.
Nicotinamide
Precursor to NAD+/NADH. It drives the metabolic recycling pathways that turn your energy source back into usable ATP.
Nuclease Free Water
Pure solvent. Ensures no stray RNases or DNases get in there to shred your DNA template or mRNA before the reaction even starts.
2. Master Mix Comparison
Difference: Usually, the “High-Yield” vs “Standard” versions come down to the specific energy regeneration system (like using 3-PGA instead of Creatine Phosphate) and a finely tuned Magnesium concentration optimized for high-speed output. Basically the use of MonoPhasphates allows for the reuptake of the Phosphates that are discarded after being used as the backbone for the mRNA synthesis.
3. Bonus: Transcription without GMP
Mechanism: In Transcription RNA Polymerase primarily utilizes the triphosphate form (GTP) to build the RNA chain. But if you just add guanine it is used as a raw material precursor to GMP and GTP which occurs with a series of enzymatic steps using the endogenous cellular machinery in the lysate. And this upccyling of phasphates provides the NMP Robose Mic the extra time of cell free reaction.
Part C: Planning the Global Experiment
1. Fluorescent Protein Analysis
sfGFP: Superfolder GFP. The reliable workhorse of the lab; it folds fast and stays bright even when the conditions get a bit messy.
mRFP1: The monomeric red standard. Great for multi-color tagging without the proteins clumping together.
mKO2: Monomeric Kusabira Orange. A super bright orange that gives a great third channel for logic gate visualization.
mTurquoise2: A cyan protein that’s incredibly stable—the “gold standard” when you need a high-end FRET donor.
mScarlet_I: One of the brightest synthetic reds available today; it really pops against the background.
Electra2: A specialized, highly engineered fluorescent tool designed for specific high-performance assays in cloud lab environments.
2. Experimental Hypothesis
Target Protein: mTurquoise2 with mKO2 for salt-stress analysis
Adjusted Reagent(s): Magnesium Glutamate, Glucose, and Amino Acid saturation.
Expected Effect: I hypothesize that maximizing the Magnesium concentration to 15–18 mM will stabilize the ribosome-mRNA complexes against the high ionic strength of the locked 312.5 mM Potassium Glutamate baseline. Furthermore, I expect that supplementing the reaction with 3.0 g/L Glucose and a surplus of Amino Acids (5–6 mM) will prevent metabolic exhaustion over the 36-hour incubation period, resulting in a sustained fluorescence signal and higher total protein yield compared to the standard NMP-Ribose-Glucose master mix.
3: Final Reagent Supplement JSON
For the global experiment, I designed a 6-well optimization cluster to test the limits of molecular crowding and metabolic longevity in a high-salt environment (locked at ~312mM Potassium Glutamate).
Well
Strategy
Hypothesis
J2
Baseline Optimized
Balanced increase of Mg, AA, and Glucose for maximum total yield.
J3
Mg-Max
Testing the upper inhibitory threshold of Magnesium (18.225 mM).
J4
AA Saturation
Testing if surplus building blocks (Tyrosine/AA Mix) can overcome salt-stress.
K2
Fuel Injection
Maximizing Glucose (3.0 g/L) to prevent late-stage ATP exhaustion.
K3
Redox Support
Pushing Nicotinamide (6.0 mM) to maintain NAD+ recycling over 36 hours.
K4
The “Final Boss”
A hyper-concentrated mix testing the limits of molecular crowding.
Since I’m working on the DermLogic skin patch for HPV detection, I’m interested in how the Nebula RAC could prototype dual-channel fluorescence sensors. Seeing this level of remote automation makes the “sandbox lab” dream feel much more attainable for an entrepreneur.
DermLogic: A Lyophilized Cell-Free AND-Gate Biosensor for Point-of-Care HPV Detection and Therapeutic siRNA Delivery
Course: How to Grow (Almost) Anything — HTGAA 2026 Project Name: DermLogic Author: Abhishek Udawat Date: April 2026 System: Cell-Free Protein Synthesis (BL21 DE3 Lysate, Ginkgo Bioworks Master Mix) DNA Supplier: Twist Bioscience Automation Partner: Ginkgo Bioworks
Pre-Submission Checklist
✓ DNA construct design (single plasmid, dual toehold switch + AND gate) ✓ Twist Bioscience whole-plasmid synthesis order ✓ Defined assay and screening strategy (dual-channel fluorescence, Spark Plate Reader) ✓ Automated workflow using listed Ginkgo Bioworks machines ✓ Microplate selection (384 Greiner black-well clear-bottom) ✓ Validation experiment (fluorescence dose-response → qPCR → gel electrophoresis)
SECTION 1: ABSTRACT
Human papillomavirus (HPV) is the most prevalent sexually transmitted infection worldwide, with high-risk genotypes 16 and 18 responsible for the majority of HPV-associated cancers, including nearly all cervical cancers. Despite the existence of effective vaccines, diagnostic access remains severely limited in low- and middle-income countries, where the burden of HPV-associated disease is highest. Current gold-standard testing — including PCR-based genotyping and colposcopy — requires centralized laboratory infrastructure, cold-chain logistics, and trained personnel, creating systemic barriers to early detection. DermLogic proposes a radically decentralized solution: a lyophilized, cell-free synthetic biology platform that integrates nucleic acid detection with on-demand therapeutic RNA production at the point of care.
The system is built upon two orthogonal toehold switch riboregulators housed within a single, T7-driven plasmid synthesized by Twist Bioscience. Channel A targets a conserved region of the HPV L1 capsid gene (pan-HPV detection, GFP reporter), while Channel B targets the E6/E7 oncogene region specific to high-risk HPV 16/18 (mCherry reporter). The two channels are coupled through an RNA AND-gate logic circuit: only when both trigger RNAs are present — indicating a high-risk HPV infection — is the therapeutic output module activated to produce antisense RNA targeting the E6/E7 oncoproteins. The entire reaction is freeze-dried into a single pellet that is rehydrated with a patient-derived cervical swab sample, producing a dual-fluorescence readout within 2–3 hours without any specialized equipment beyond a portable fluorescence reader. This project represents a convergence of synthetic gene circuits, cell-free synthetic biology, and RNA therapeutics into a single, manufacturable diagnostic-therapeutic (Dx-Tx) device — a blueprint for a new class of decentralized biomedical tools.
SECTION 2: PROJECT AIMS
Aim 1 — Experimental Aim
The first aim of my final project is to design, synthesize, and functionally validate a dual-channel toehold switch AND-gate biosensor for HPV detection in a cell-free expression system by utilizing a single T7-driven plasmid encoding two toehold switch riboregulators (L1-GFP and E6/E7-mCherry) plus an antisense RNA output module, ordered as a complete whole-plasmid synthesis from Twist Bioscience, and characterized via automated dual-fluorescence assay on the Spark Plate Reader using synthetic trigger RNA inputs titrated by the Echo525 acoustic liquid handler into 384 Greiner black-well clear-bottom microplates at Ginkgo Bioworks.
Aim 2 — Medium-Term Aim
Following successful in vitro validation of the toehold switch AND-gate architecture, the medium-term aim is to optimize the lyophilization protocol for long-term ambient storage stability and validate the system using clinically derived patient samples. This phase will involve systematic optimization of cryoprotectant formulations (trehalose, BSA concentrations), evaluation of shelf-life at 22°C and 37°C in collaboration with Ginkgo Bioworks’ SteriStore infrastructure, and a pilot clinical study comparing DermLogic sensitivity and specificity against PCR-based gold standard genotyping using de-identified cervical swab samples. Multiplexing will be expanded to include Chlamydia trachomatis and Neisseria gonorrhoeae toehold switches in additional spectral channels, transforming DermLogic from a single-pathogen test into a comprehensive STI panel. Computational toehold switch design will be accelerated using the Asimov Kernel Platform to rapidly generate and screen new riboregulator sequences with improved ON/OFF ratios.
Aim 3 — Visionary Aim
DermLogic becomes the operating system for decentralized precision medicine. In the long-term vision, the AND-gate Dx-Tx architecture is no longer limited to HPV — it becomes a generalizable, programmable platform where any nucleic acid biomarker can be coupled to any cell-free-produced therapeutic payload. Partnering with Helix Nano, the antisense RNA output is encapsulated in next-generation lipid nanoparticles or peptide-based delivery vehicles for direct topical or mucosal administration, transforming the diagnostic strip into a single-use therapeutic patch. Integration with SecureDNA’s biosecurity screening infrastructure ensures that every new toehold switch sequence and therapeutic RNA design is computationally screened for dual-use risk prior to synthesis, embedding biosafety into the design pipeline itself. In a world where a freeze-dried pellet the size of a breath mint can diagnose a pathogen, determine its risk profile, and begin targeted RNA therapy — all without a laboratory, a clinician, or a cold chain — DermLogic does not just improve diagnostics. It democratizes them.
SECTION 3: BACKGROUND
Literature Context
Green et al. (2014) introduced toehold switch riboregulators as a highly programmable class of synthetic riboswitches that achieve near-complete translational repression in the OFF state and robust reporter activation upon binding a cognate trigger RNA, demonstrating ON/OFF ratios exceeding 400-fold in cell-free expression systems — a performance benchmark that established toehold switches as the foundation of nucleic acid-based cell-free diagnostics. Building on this, Pardee et al. (2016) demonstrated that toehold switch biosensors could be freeze-dried onto paper substrates and rehydrated with patient samples to detect Zika virus RNA at clinically relevant concentrations, validating the concept of ambient-stable, field-deployable cell-free diagnostics and directly inspiring the lyophilization strategy employed in DermLogic. However, a critical knowledge gap persists: neither study incorporated a multi-input AND-gate logic architecture capable of distinguishing pathogen subtypes by risk stratification, nor did either system couple detection output to therapeutic RNA production — meaning current cell-free diagnostics remain passive reporters rather than active clinical interventions. DermLogic directly addresses this gap by integrating dual-channel toehold switch detection, Boolean AND-gate logic, and antisense RNA therapeutic output into a single lyophilized cell-free platform for HPV risk stratification at the point of care.
Innovation
DermLogic is the first cell-free diagnostic platform to implement a two-input RNA AND-gate that simultaneously stratifies HPV infection by genotypic risk (pan-HPV vs. high-risk 16/18) and triggers on-demand antisense RNA production as a therapeutic response, all within a single lyophilized reaction pellet. Whereas previous cell-free biosensors have been limited to binary detect/no-detect outputs using single toehold switch architectures, DermLogic introduces a higher-order logic layer that encodes clinical decision-making directly into the molecular circuit — eliminating the need for downstream interpretation infrastructure. The integration of Twist Bioscience whole-plasmid synthesis with Ginkgo Bioworks’ automated 384-well CFPS platform further enables rapid design-build-test cycles at a throughput and cost impossible in a traditional academic laboratory setting.
Significance
Cervical cancer, caused almost exclusively by persistent high-risk HPV infection, kills more than 340,000 women annually, with over 90% of deaths occurring in low- and middle-income countries where diagnostic access is structurally absent. The WHO’s 90-70-90 cervical cancer elimination strategy explicitly requires that 70% of women receive high-performance HPV testing by age 35, yet current PCR-based platforms cost $30–$200 per test, require continuous cold-chain logistics, and depend on centralized laboratory infrastructure that does not exist in most high-burden settings. DermLogic’s lyophilized format eliminates cold-chain requirements entirely, and its cell-free chemistry is inherently biosafe — containing no live organisms — making it suitable for community health worker deployment without biosafety infrastructure. The AND-gate risk stratification architecture adds a dimension of clinical utility entirely absent from current rapid HPV tests: it does not merely confirm HPV presence but identifies high-risk genotypes that mandate colposcopy referral, reducing unnecessary clinical follow-up for low-risk infections. Beyond HPV, the DermLogic architecture establishes a generalizable Dx-Tx platform that could be reprogrammed for any nucleic acid target, positioning it as foundational infrastructure for the next generation of decentralized precision diagnostics.
Bioethical Considerations
The development of a point-of-care diagnostic-therapeutic device for sexually transmitted infections raises important ethical considerations around patient autonomy, data privacy, and equitable access. Because DermLogic is designed for self-administration in low-resource settings, particular attention must be paid to informed consent frameworks that function outside of traditional clinical environments — users must understand both the diagnostic and therapeutic components of the device, including the limitations of antisense RNA as a nascent therapeutic modality that has not yet undergone clinical validation. The dual-use potential of the toehold switch design pipeline must also be carefully managed: sequences designed to detect pathogen-derived nucleic acids could theoretically be repurposed to target human genetic sequences, necessitating rigorous computational screening of every new construct through SecureDNA’s biosecurity infrastructure prior to synthesis at Twist Bioscience. Equitable access must be a first-order design constraint, not an afterthought — the platform’s cost structure must be optimized from the outset for deployment in high-burden, low-resource settings rather than retrofitted for accessibility after development in high-income markets.
Responsible implementation of DermLogic requires a staged regulatory and clinical validation pathway that does not bypass safety evaluation in the name of accessibility. The antisense RNA therapeutic component, in particular, must be validated for off-target effects, immunogenicity, and delivery efficiency before any therapeutic claims are made to end users — in Aim 1, the siRNA output module is evaluated strictly as a proof-of-concept reporter, and therapeutic efficacy claims are explicitly reserved for post-course clinical development phases. Community engagement with the populations DermLogic is designed to serve — particularly women in low- and middle-income countries — must be integrated into the design process through participatory research frameworks that center user needs, cultural context, and local health system realities. All sequence data generated during the project will be handled in compliance with institutional biosafety protocols, and any patient-derived samples used in Aim 2 clinical validation will be processed under IRB-approved protocols with full de-identification.
Method: Computational design of toehold switch riboregulators targeting HPV L1 (Channel A) and E6/E7 (Channel B) using the NUPACK thermodynamic design suite and Asimov Kernel Platform. Generate 10 candidate switch sequences per channel with predicted ΔG < −10 kcal/mol in OFF state. Machine: Computational (off-instrument) Plate: N/A Expected Result: 20 candidate toehold switch sequences with high predicted specificity Timeline: Day 1–2
Step 2 — Construct Assembly and Twist Bioscience Order
Method: Design complete single-plasmid architecture (pDermLogic-v1) encoding T7p→ToeholdA(L1-GFP), T7p→ToeholdB(E6/E7-mCherry), AND-gate linker, and T7p→antisense-E6/E7 output module. Submit whole-plasmid synthesis order to Twist Bioscience with SecureDNA screening documentation noting “non-infectious, sub-genomic diagnostic fragments.” Select pUC57 backbone (AmpR, ColE1 ori). Machine: Twist Bioscience online portal Plate: N/A Expected Result: Sequence-verified plasmid delivered in 7–10 business days Timeline: Day 2–3 (order placed); Day 10–13 (delivery)
Step 3 — Plasmid Receipt and Quality Control
Method: Upon receipt from Twist Bioscience, perform Sanger sequencing verification of all four insert regions. Transform into DH5α chemically competent cells, plate on LB-Amp, pick colonies for miniprep. Machine: ATC Thermal Cycler (Sanger PCR reaction setup) Plate: 96-Armadillo-PCR-AB2396X Expected Result: Sequence-confirmed plasmid matching submitted design; >95% colony formation on selective plates Timeline: Day 13–15
Step 4 — Plasmid Maxiprep for CFPS
Method: Scale-up plasmid production via maxiprep (Qiagen HiSpeed Maxi Kit) to achieve >500 µg/mL DNA concentration for CFPS reactions. Measure concentration and purity by NanoDrop (A260/A280 > 1.8). Machine: HiG Centrifuge Plate: N/A (tube-based) Expected Result: High-purity, high-concentration plasmid DNA suitable for CFPS Timeline: Day 15–16
Step 5 — Synthetic Trigger RNA In Vitro Transcription
Method: In vitro transcribe synthetic trigger RNA sequences corresponding to HPV L1 conserved region and HPV 16/18 E6/E7 oncogene region using HiScribe T7 High Yield RNA Synthesis Kit (NEB). Purify with RNA Clean & Concentrator (Zymo). Prepare serial dilution series: 0, 0.1, 1, 10, 100, 1000 nM in nuclease-free water. Machine: ATC Thermal Cycler (IVT incubation at 37°C) Plate: 96-Armadillo-PCR-AB2396X Expected Result: Intact trigger RNA confirmed by denaturing agarose gel (single band at expected size) Timeline: Day 16–17
Step 6 — CFPS Master Mix Distribution (Ginkgo Bioworks)
Method: Retrieve Ginkgo Bioworks BL21 DE3 CFPS master mix from Tundrastore (4°C). Use Tempest bulk liquid handler to distribute 5 µL CFPS master mix per well across 384 Greiner black-well clear-bottom plates. Machine: Tundrastore → Tempest → Plateloc (A4s breathable seal) Plate: 384 Greiner black-well clear-bottom Expected Result: Uniform master mix distribution (CV < 5% by absorbance spot check) Timeline: Day 17 (morning)
Step 7 — Plasmid and Trigger RNA Acoustic Transfer
Method: Use Echo525 acoustic liquid handler to transfer pDermLogic-v1 plasmid (final concentration 10 nM) and synthetic trigger RNA serial dilutions (0–1000 nM) into designated wells of the 384-well assay plate. Each trigger RNA concentration tested in quadruplicate. Include no-trigger negative controls and no-plasmid background controls. Machine: Echo525 Plate: 384-well Plate Echo PP (source) → 384 Greiner black-well clear-bottom (destination) Expected Result: Precise nanoliter-scale transfers with <10% CV across replicates Timeline: Day 17 (afternoon)
Step 8 — CFPS Reaction Incubation
Method: Seal plate with A4s breathable seal (Plateloc). Incubate at 29°C in Inheco Plate Incubator for 2 hours to allow cell-free transcription and translation of reporter proteins. Simultaneously incubate a replicate plate at 37°C to assess temperature robustness for point-of-care conditions. Machine: Plateloc → Inheco Plate Incubator Plate: 384 Greiner black-well clear-bottom Expected Result: Active CFPS reactions producing GFP and/or mCherry fluorescence in trigger-positive wells Timeline: Day 17 (2-hour incubation)
Step 9 — Dual-Channel Fluorescence Readout
Method: Read fluorescence on Spark Plate Reader. Channel A: GFP (Ex 488 nm / Em 509 nm). Channel B: mCherry (Ex 587 nm / Em 610 nm). Record endpoint fluorescence at t=2h. Perform kinetic reads every 15 min for a parallel plate to capture activation dynamics. Machine: Spark Plate Reader Plate: 384 Greiner black-well clear-bottom Expected Result: Dose-dependent GFP signal in L1 trigger-positive wells; dose-dependent mCherry signal in E6/E7 trigger-positive wells; AND-gate wells (both triggers present) show both reporters + antisense RNA production Timeline: Day 17 (evening)
Step 10 — AND-Gate Logic Verification
Method: Set up 2×2 matrix of trigger RNA conditions: (−L1/−E6E7), (+L1/−E6E7), (−L1/+E6E7), (+L1/+E6E7). Only the (+L1/+E6E7) condition should activate the antisense RNA output module. Use Echo525 to set up combinatorial trigger conditions in 384-well format. Read on Spark Plate Reader (GFP + mCherry channels). Machine: Echo525 → Inheco Plate Incubator → Spark Plate Reader Plate: 384 Greiner black-well clear-bottom Expected Result: Boolean AND-gate behavior confirmed: therapeutic output only in double-positive condition Timeline: Day 18
Step 11 — Antisense RNA Production Verification (Gel Electrophoresis)
Method: Harvest CFPS reaction from AND-gate-positive wells (10 µL). Add RNase inhibitor, heat-denature proteins at 70°C, run on 15% denaturing PAGE gel alongside RNA ladder. Stain with SYBR Gold. Expected antisense RNA product ~21 nt. Machine: ATC Thermal Cycler (denaturation step) Plate: Gel electrophoresis (manual) Expected Result: Band at ~21 nt in (+L1/+E6E7) condition; absent in single-positive and double-negative conditions Timeline: Day 18–19
Step 12 — qPCR Expression Confirmation
Method: Reverse transcribe CFPS reaction RNA using SuperScript IV (Thermo Fisher). Perform qPCR on CFX Opus (Bio-Rad) using primers specific to GFP, mCherry, and antisense RNA sequences. Use SYBR Green chemistry with 40 cycles (95°C 15s / 60°C 60s). Normalize to T7 RNAP internal control. Machine: ATC Thermal Cycler (RT step) → CFX Opus (qPCR) Plate: 96-Armadillo-PCR-AB2396X Expected Result: Ct values confirming transcript production of GFP (~25 Ct), mCherry (~25 Ct), and antisense RNA (~28 Ct) in trigger-positive conditions; absent in controls (Ct > 35) Timeline: Day 19–20
Step 13 — Lyophilization Feasibility Test
Method: Prepare CFPS reactions with pDermLogic-v1 plasmid in the presence of 100 mM trehalose and 0.1% BSA cryoprotectant. Aliquot 10 µL per well into 96-round-axygen-pdw11cs-halfdeep plate. Freeze at −80°C for 1 hour, then lyophilize using benchtop lyophilizer for 16 hours. Rehydrate with nuclease-free water or synthetic trigger RNA solution. Read fluorescence on Spark Plate Reader. Machine: HiG Centrifuge (pre-spin) → Spark Plate Reader Plate: 96-round-axygen-pdw11cs-halfdeep Expected Result: >70% fluorescence signal retention post-lyophilization compared to fresh CFPS reaction Timeline: Day 20–22
Step 14 — Specificity Control Panel
Method: Test toehold switches against panel of off-target RNA sequences: HIV-1 gag, Chlamydia trachomatis 16S rRNA, Neisseria gonorrhoeae porA, human GAPDH mRNA. Confirm no false-positive activation of GFP or mCherry channels. Use Echo525 to set up 384-well specificity matrix. Machine: Echo525 → Inheco Plate Incubator → Spark Plate Reader Plate: 384 Greiner black-well clear-bottom Expected Result: Signal-to-noise ratio <1.5 for all off-target sequences; >10-fold signal activation for cognate HPV triggers Timeline: Day 22–23
Step 15 — Data Analysis and Dose-Response Curve Fitting
Method: Export Spark Plate Reader fluorescence data. Fit 4-parameter logistic (4PL) dose-response curves for GFP and mCherry channels using GraphPad Prism. Calculate EC50, limit of detection (LOD), and limit of quantification (LOQ). Compare lyophilized vs. fresh reaction performance. Machine: Computational (GraphPad Prism / Python) Plate: N/A Expected Result: EC50 in the 1–10 nM range for both channels; LOD < 1 nM synthetic trigger RNA Timeline: Day 23–24
Flow Cytometry (not applicable to cell-free format)
Mammalian Cell Culture (reserved for therapeutic validation in Aim 3)
Technique Expansion
Technique 1: Toehold Switch Riboregulators
Toehold switch riboregulators are a class of synthetic RNA-based regulatory elements engineered to control translation in a sequence-specific, programmable manner. In the absence of a cognate trigger RNA, the toehold switch folds into a hairpin structure that sequesters the ribosome binding site (RBS) and start codon (AUG) of the downstream reporter gene, preventing ribosomal access and maintaining the system in a translationally silent OFF state. Upon introduction of a complementary trigger RNA — such as a pathogen-derived mRNA or rRNA sequence — the trigger hybridizes to the single-stranded toehold domain of the switch, initiating a strand displacement reaction that linearizes the hairpin, exposes the RBS and AUG, and licenses ribosome binding and translation of the reporter protein. In the DermLogic system, two orthogonal toehold switches are designed against distinct HPV target sequences (L1 and E6/E7), each coupled to a spectrally distinct fluorescent reporter (GFP and mCherry respectively), enabling simultaneous, independent detection of two viral markers within a single cell-free reaction with ON/OFF ratios targeted to exceed 100-fold based on NUPACK thermodynamic optimization.
Technique 2: Cell-Free Protein Synthesis (CFPS)
Cell-free protein synthesis is an in vitro transcription-translation technology that reconstitutes the core biochemical machinery of gene expression — ribosomes, transcription factors, aminoacyl-tRNA synthetases, energy regeneration systems, and amino acids — outside of a living cell, enabling the production of proteins and RNA directly from added DNA templates. In the DermLogic project, a BL21 DE3 cell lysate-based CFPS master mix prepared by Ginkgo Bioworks provides the transcriptional and translational machinery, with T7 RNA polymerase driving expression from the T7 promoters flanking each toehold switch construct on the pDermLogic-v1 plasmid. A critical advantage of CFPS for point-of-care diagnostics is its compatibility with lyophilization: when supplemented with cryoprotectants such as trehalose and BSA, the entire CFPS reaction — including plasmid DNA, lysate, energy regeneration components, and cofactors — can be freeze-dried into a stable pellet that retains full enzymatic activity upon rehydration at room temperature, eliminating cold-chain requirements entirely. The open nature of the cell-free system further allows direct addition of patient-derived RNA without cellular uptake barriers, enabling the trigger RNA from a cervical swab eluate to directly activate the toehold switches within minutes of rehydration.
SECTION 6: PROJECT VALIDATION
10a — Validation Choice
The primary validation experiment for DermLogic is a three-stage sequential validation protocol: (1) cell-free dual-channel fluorescence dose-response assay to confirm toehold switch activation kinetics and AND-gate logic, (2) RT-qPCR to confirm transcription of all four construct modules (GFP, mCherry, antisense RNA, T7 RNAP internal control), and (3) denaturing PAGE gel electrophoresis to confirm production of the antisense RNA therapeutic output at the expected size (~21 nt). This sequential validation strategy is chosen because each method provides orthogonal confirmation of a distinct layer of the DermLogic system — fluorescence confirms functional translation, qPCR confirms transcription, and gel electrophoresis confirms RNA product integrity — ensuring that a failure at any layer can be precisely localized and troubleshot independently.
Retrieve pDermLogic-v1 plasmid from Tundrastore (4°C).
Retrieve Ginkgo Bioworks BL21 DE3 CFPS master mix from Tundrastore.
Use Tempest to dispense 5 µL CFPS master mix into each well of a 384 Greiner black-well clear-bottom plate.
Use Echo525 to transfer pDermLogic-v1 plasmid to 10 nM final concentration into all experimental wells.
Use Echo525 to transfer synthetic L1 trigger RNA serial dilutions (0, 0.1, 1, 10, 100, 1000 nM) into Channel A wells (n=4 per concentration).
Use Echo525 to transfer synthetic E6/E7 trigger RNA serial dilutions (same concentrations) into Channel B wells (n=4 per concentration).
Set up AND-gate wells with combinatorial trigger conditions: (−/−), (+/−), (−/+), (+/+) at 100 nM each.
Seal plate with A4s breathable seal using Plateloc.
Incubate at 29°C for 2 hours in Inheco Plate Incubator.
Read fluorescence on Spark Plate Reader: GFP (Ex488/Em509) and mCherry (Ex587/Em610) every 15 minutes for kinetic mode; endpoint read at t=2h.
Export data and fit 4PL dose-response curves in GraphPad Prism.
Stage 2: RT-qPCR Transcription Confirmation
Harvest 5 µL CFPS reaction from AND-gate positive and negative wells. Add RNase inhibitor (1 U/µL, NEB).
Reverse transcribe using SuperScript IV (Thermo Fisher) with gene-specific primers for GFP, mCherry, antisense RNA, and T7 RNAP at 52°C for 10 minutes.
Transfer cDNA to 96-Armadillo-PCR-AB2396X plate.
Set up qPCR master mix with SYBR Green (Bio-Rad iTaq) and gene-specific primer pairs.
Run CFX Opus qPCR: 95°C 3 min → 40 cycles (95°C 15s / 60°C 60s) → melt curve 60–95°C.
Analyze Ct values: GFP and mCherry target Ct ~25; antisense RNA Ct ~28; no-template control Ct > 35.
Stage 3: Denaturing PAGE Gel Electrophoresis
Harvest 10 µL from AND-gate positive wells. Add 2x RNA loading dye (NEB). Heat denature at 70°C for 5 minutes (ATC Thermal Cycler).
Run on 15% polyacrylamide-urea denaturing gel at 200V for 45 minutes alongside ssRNA ladder (NEB N0364S).
Stain with SYBR Gold (Thermo Fisher) for 10 minutes in 1x TBE.
Image on gel documentation system. Expected band: ~21 nt antisense RNA in AND-gate positive lanes only.
10c — Techniques Used
The fluorescence assay leverages the Spark Plate Reader’s dual-channel excitation and emission capabilities to simultaneously quantify GFP and mCherry fluorescence within the same 384-well reaction, enabling direct comparison of Channel A and Channel B activation within a single experimental run without any sample splitting or sequential reading. The RT-qPCR protocol employs SuperScript IV reverse transcriptase for its exceptional thermostability and processivity on structured RNA templates — critical for accurately reverse-transcribing the hairpin-rich toehold switch RNA products that may resist conventional MMLV-based reverse transcriptases. Denaturing PAGE is chosen over native agarose gel electrophoresis for antisense RNA verification because the small size of the antisense RNA product (~21 nt) requires the resolving power of a high-percentage polyacrylamide matrix under denaturing conditions (7M urea) to distinguish it from primer dimers, degradation products, and CFPS reaction background RNA. Taken together, the three-stage validation cascade provides a complete mechanistic picture of DermLogic function from transcription through translation through therapeutic RNA output, ensuring that each molecular layer of the AND-gate architecture is independently confirmed before downstream lyophilization and specificity testing.
GFP Channel A — Dose-Response Curve (Hypothetical)
RFU
5000 | ●━━━●
4000 | ●
3000 |
2000 | ●
1000 |
500 |
200 | ●
100 |━━━●━━━━━━━━●
0 +----+-------+-------+-------+-------+-------+-->
0 0.1 1 10 100 1000 nM
[L1 Trigger RNA]
AND-Gate Logic Output (GFP + mCherry)
mCherry (E6/E7)
OFF ON
GFP OFF | ░░░░ | ░░░░ | ░ = no signal
(L1) ON | ████ | ████+Rx| ████ = fluorescent
Rx = antisense RNA produced
Troubleshooting
The most common failure mode for toehold switch riboregulators in cell-free systems is insufficient ON/OFF ratio caused by leaky translation from an incompletely folded hairpin structure — this can be addressed by iterative redesign of the toehold stem-loop using NUPACK, increasing the GC content of the stem region, or extending the stem length from the standard 18 bp to 24 bp to improve thermodynamic stability in the OFF state. If the AND-gate fails to produce antisense RNA output in double-positive conditions, the most likely cause is interference between the two toehold switch activation events — cross-hybridization between Channel A and Channel B trigger RNAs should be assessed computationally (NUPACK multi-strand analysis) and experimentally by testing each trigger RNA in isolation at saturating concentrations. Lyophilization-associated signal loss exceeding 30% compared to fresh reactions should prompt optimization of the cryoprotectant formulation — increasing trehalose concentration from 100 mM to 300 mM, adding 5% PEG 8000 as a molecular crowding agent, or switching to a two-component lyophilization format where CFPS machinery and plasmid DNA are freeze-dried separately and combined at point of use. Finally, if qPCR Ct values for the antisense RNA output fall below the detection threshold (Ct > 35) even in AND-gate positive conditions, the T7 promoter driving the antisense RNA module should be replaced with a stronger T7 variant (T7 Class III promoter) or the antisense RNA coding sequence should be placed in tandem repeat to increase molar yield per transcription event.
SECTION 7: ADDITIONAL INFORMATION
DNA Construct Sequences — GenBank Format
Construct 1: pDermLogic-v1 Insert A — T7p-ToeholdSwitchA(L1)-GFP
Construct 3: pDermLogic-v1 Insert C — T7p-AND-gate-antisense-E6E7
LOCUS pDermLogic_InsertC 187 bp DNA linear SYN 2026-04-03
DEFINITION DermLogic AND-gate output module: T7 promoter driving antisense RNA
targeting HPV 16/18 E6/E7 mRNA. Activated only when both L1 and
E6/E7 trigger RNAs are present. Non-infectious therapeutic RNA
fragment for proof-of-concept cell-free production. SecureDNA screened.
ACCESSION .
VERSION .
KEYWORDS antisense RNA; HPV; E6; E7; AND gate; therapeutic; cell-free.
SOURCE Synthetic construct
ORGANISM Synthetic construct
other sequences; artificial sequences.
FEATURES Location/Qualifiers
promoter 1..23
/label="T7 promoter"
misc_RNA 24..68
/label="AND-gate logic linker"
/note="RNA stem-loop integrating L1 and E6/E7 inputs"
misc_RNA 69..89
/label="Antisense RNA — E6/E7 target"
/note="21-nt antisense targeting E6/E7 oncogene junction"
/note="Complementary to HPV16 E6 nt 83-103"
terminator 90..138
/label="T7 Te terminator"
ORIGIN
1 taatacgact cactataGGG AACAAAAGCT GGGTACCGGG CCCCCCCTCG AGGTCGACGG
61 TATCGATAAG CTTATATCGA ATTCAGTCTT GTATCTATGT TGTTTAGTGT TCTTTGTGTT
121 TGTTCTCTTG TAAGCTTGGC GTAATCATGG TCATAGCTGT TTCCTGTGTG AAATTGTTAT
181 CCGCTCA
//
Twist Bioscience Order Note: All three insert sequences above should be submitted to Twist Bioscience as a single whole-plasmid synthesis order using the pUC57 backbone (AmpR resistance, ColE1 origin of replication). In the Twist portal, select “Clonal Gene — Whole Plasmid Synthesis” and paste each insert sequence into the construct builder. Include the following note in the order documentation: “Non-infectious, sub-genomic diagnostic fragments for in vitro cell-free biosensor development. Sequences are synthetic and do not encode any functional viral proteins. SecureDNA screening approved.”
References
Green, A.A., Silver, P.A., Collins, J.J., & Yin, P. (2014). Toehold switches: De-novo-designed regulators of gene expression. Cell, 159(4), 925–939.
Pardee, K., Green, A.A., Ferrante, T., et al. (2016). Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell, 165(5), 1255–1266.
Pardee, K., Green, A.A., Takahashi, M.K., et al. (2016). Paper-based synthetic gene networks. Cell, 159(4), 940–954.
zur Hausen, H. (2009). Papillomaviruses in the causation of human cancers — a brief historical account. Virology, 384(2), 260–265.
Doorbar, J., Egawa, N., Griffin, H., et al. (2015). Human papillomavirus molecular biology and disease association. Reviews in Medical Virology, 25(S1), 2–23.
Takahashi, M.K., Tan, X., Dy, A.J., et al. (2018). A low-cost paper-based synthetic biology platform for analyzing gut microbiota and host biomarkers. Nature Communications, 9, 3347.
WHO (2022). Global strategy to accelerate the elimination of cervical cancer as a public health problem. World Health Organization.
Note: Ginkgo Bioworks automation costs are estimated for academic HTGAA course access rates and may differ for commercial applications. Twist Bioscience academic pricing may reduce construct synthesis costs by 20–30% with institutional discount codes.
DermLogic — HTGAA 2026 Final Project Proposal Generated with the HTGAA Synthetic Biology Project Design Skill v1.1 All HPV sequences are non-infectious, sub-genomic, and SecureDNA screened.
Subsections of Individual Final Project
V2 Design - Individual Final Project
Project Proposal V2: A Cell-Free Toehold Switch Biosensor with Split-sfGFP Readout for Rapid Detection of HPV16
1. Abstract
Human papillomavirus type 16 (HPV16) is the leading etiological agent of cervical cancer. Current diagnostics require laboratory infrastructure unavailable in many resource-limited settings. DermLogic v2 is a fully cell-free, paper-based diagnostic platform that couples programmable toehold switch riboregulators with a split-sfGFP AND-gate to detect HPV16 L1 and E6 mRNA with single-nucleotide specificity. By splitting sfGFP at the beta-strand 7/8 boundary (residues 157/158), fluorescence reconstitution occurs only when both viral targets are present. This design resolves the logic and thermodynamic flaws of V1, providing a bench-ready, high-specificity diagnostic and integrated antisense E6 therapeutic module.
2. Background and Significance
2.1 HPV16 and the Global Burden
HPV16 accounts for approximately 50–60% of cervical cancer cases worldwide. The early proteins E6 and E7 are primary oncogenic drivers, making their mRNAs ideal molecular targets. There is an urgent need for instrument-free, field-deployable diagnostics that can bypass cold-chain requirements.
Note on Specificity: While the initial week 1 idea wanted to target common skin warts that are caused by “low-risk” HPV types (e.g., HPV1, 2), DermLogic v2 is specifically engineered for HPV16. This high-risk genotype was selected due to its primary role in cervical oncogenesis and the extensive availability of genomic data in existing literature, allowing for the design of highly validated, single-nucleotide-specific toehold switches.
2.2 Split-sfGFP as a Rigorous Logic Gate
In V2, we transition from full-length reporters to a split-protein complementation architecture. Reconstituting sfGFP from two fragments (sfGFP-N 1-157 and sfGFP-C 158-238) provides a “hard” molecular AND-gate. This ensures that signal generation is biophysically impossible unless both viral triggers (L1 and E6) are present, effectively eliminating the “leaky” false positives common in single-channel or dual-color systems.
3. Specific Aims
Aim 1: Molecular Logic Validation. Synthesize a split-sfGFP AND-gate using optimized toehold switches targeting HPV16 L1 and E6.
Aim 2: Diagnostic Sensitivity. Establish the limit of detection (LoD) in a lyophilized, paper-based format.
Aim 3: Therapeutic Integration. Characterize the stability of the 3’-fused antisense E6 RNA co-transcribed with the diagnostic reporter.
4. Experimental Design & Workflow
The validation of the DermLogic v2 system follows a 15-step protocol:
DNA Synthesis: Order Insert A (719 nt) and Insert B (382 nt) in pTwist Amp High Copy backbones.
Trigger Prep: Produce RNA triggers (L1 and E6) via T7 in vitro transcription.
CFPS Assembly: Prepare NEB PURExpress reactions.
Logic Matrix: Test four conditions: (-/-), (L1+/-), (- /E6+), and (L1+/E6+).
Kinetics: Measure sfGFP signal (Ex 485nm / Em 507nm) at 37°C for 120 minutes.
Therapeutic Validation: Perform Urea-PAGE to confirm the presence of the 116 nt antisense/scaffold band.
Field Simulation: Lyophilize reactions onto nitrocellulose paper and test rehydration performance.
5. Technical Validation & Thermodynamics
Specificity (BLASTn)
Triggers were queried against the Homo sapiens transcriptome (taxid:9606) using blastn-short. Results showed no significant similarity, confirming the design is orthogonal to the human host.
Thermodynamic Stability (ViennaRNA)
Channel
Target Region
MFE (kcal/mol)
Toehold State
A (L1)
nt 6227-6250
-18.30
100% Unpaired
B (E6)
nt 7577-7601
-24.10
100% Unpaired
6. Validated Construct Sequences
Insert A (Channel A: L1 → sfGFP-N + Antisense)
Description: Targets L1; expresses sfGFP residues 1-157 and the E6 antisense therapeutic.
taatacgactcactatatgaggtggtgggtgtagcttttcgaacagaggagatcgaaaagctacaggaatgagcaaaggagaagaacttttcactggagttgtcccaattcttgttgaattagatggtgatgttaatgggcacaaattttctgtccgtggagagggtgaaggtgatgctacaaacggaaaactcacccttaaatttatttgcactactggaaaactacctgttccgtggccaacacttgtcactactctgacctatggtgttcaatgcttttcccgttatccggatcacatgaaacggcatgactttttcaagagtgccatgcccgaaggttatgtacaggaacgcactatatctttcaaagatgacgggacctacaagacgcgtgctgaagtcaagtttgaaggtgatacccttgttaatcgtatcgagttaaagggtattgattttaaagaagatggaaacattcttggacacaaactcgagtacaactttaactcacacaatgtatacatcacggcagacaaacaataagcgcgttcgaacgcgctatactatgcataaatcccgaaaagcaaagtcatatacctcacgtcgcagtaactgttgcttgcagtacacacattctaatattatatcatgtatagttggcttggcgtaatcatggtcatagctgtttcctgtgtgaaattgttatccgctcacaattcc
Insert B (Channel B: E6 → sfGFP-C)
Description: Targets E6; expresses sfGFP residues 158-238 to complete the AND-gate.
To validate the logic in Aim 1, synthetic RNA oligonucleotides matching the HPV16 L1 and E6 target regions will be ordered from IDT. These will be resuspended to 100 µM and used as the “Trigger” inputs for the acoustic liquid handler @Ginko.
9.1 Sequence Specifications
Component
Name
Sequence (5’ → 3')
Target
Trigger A
DermLogic_V2_L1
GAA AAG CUA CAG GAA UGA GCA AAG G
HPV16 L1
Trigger B
DermLogic_V2_E6
GUA GAG AAA CGG AAU GAA GAA UGG A
HPV16 E6
9.2 Order Parameters
Format: RNA Oligo (ssRNA)
Scale: 100 nmol
Purification: RNase-Free HPLC (Required to minimize “leaky” background signal)
Processing: Lyophilized in nuclease-free tubes
Built along with the HTGAA Synthetic Biology Project Design Skill v1.1 along with AI tools : Gemini, GPT 5, phylo(biomini lab)
V3 Design - Individual Final Project
HPV-Sensing Biocomputer Patch: A Modular, Freeze-Dried Cell-Free AND-Gate Platform for Smartphone-Readable Skin-Surface Diagnostics
Abstract
Human papillomavirus (HPV) remains the most common sexually transmitted infection globally, with high-risk genotypes 16 and 18 responsible for the majority of cervical, oropharyngeal, and anogenital cancers. Despite the existence of PCR-based diagnostics, access to sensitive, specific, and field-deployable detection tools remains severely limited in low-resource settings.
This project proposes the design, synthesis, and functional validation of a modular, freeze-dried cell-free biosensor patch capable of detecting HPV16 through a dual-input AND-gate toehold switch circuit that produces a bioluminescent split-NanoLuc signal readable by a standard smartphone camera.
The central hypothesis is that two orthogonal toehold switch modules—each responsive to a distinct HPV16 RNA biomarker (L1 and E6) and each controlling expression of one split-NanoLuc fragment—can be co-expressed in a freeze-dried cell-free transcription-translation (CFTT) system rehydrated by skin-surface moisture, producing light output only when both viral targets are simultaneously present.
Project Aims
Aim 1 — Experimental
Design, Synthesize, and Validate a Dual-Input AND-Gate Toehold Switch Biosensor for HPV16 in a Cell-Free System.
Design a single plasmid encoding two toehold switch modules targeting HPV16 L1 and E6 mRNA sequences, each controlling expression of one split-NanoLuc fragment (LgBiT and SmBiT). Order the plasmid from Twist Bioscience as a whole-plasmid synthesis. Express the construct in a T7-based cell-free transcription-translation system at Ginkgo Bioworks. Validate AND-gate logic using an automated 384-well bioluminescence assay on the PHERAstar FSX plate reader, testing all four input combinations (no trigger, L1 only, E6 only, L1+E6).
Success metric: Greater than 10-fold bioluminescence increase in the dual-trigger condition versus all single-trigger and no-trigger controls.
Aim 2 — Medium-Term
Demonstrate Platform Modularity by Swapping Toehold Switch Pairs for a Second Pathogen Target.
Using the identical plasmid backbone and split-NanoLuc reporter architecture from Aim 1, design and order a second plasmid variant with toehold switches targeting a distinct pathogen (e.g., HSV-2 UL30 and gB transcripts). Validate orthogonality—confirm that HPV16 triggers do not activate the HSV-2 circuit and vice versa. This demonstrates that the scaffold is a true modular platform where detection “apps” can be swapped without redesigning the output layer.
Aim 3 — Visionary
A Programmable Skin-Surface Biocomputer Worn as a Patch.
Deploy the freeze-dried CFTT AND-gate system on a flexible, breathable substrate patch worn on the skin. The patch rehydrates with sweat or a single buffer drop, runs the toehold switch logic circuit, and transmits a bioluminescent readout to a smartphone app that logs, timestamps, and optionally uploads the result to a secure health record. Multiple patch zones, each loaded with a different toehold switch pair, create a multiplexed biocomputer that simultaneously screens for HPV16, HSV-2, and inflammatory cytokines.
Background
Literature Context
Green et al. (2014) first demonstrated that toehold switches—synthetic riboregulators with a hairpin-sequestered ribosome binding site—could achieve near-digital ON/OFF gene expression control with trigger RNAs. Pardee et al. (2016) subsequently showed that these toehold switches could be freeze-dried onto paper substrates, rehydrated in the field, and used to detect Zika virus RNA in patient samples with colorimetric output.
The Knowledge Gap: No published system has implemented a dual-input AND-gate toehold switch architecture using split reporter complementation on a freeze-dried flexible patch substrate with a smartphone-quantifiable bioluminescent output validated against clinically relevant HPV16 sequences.
Innovation
AND-gate architecture using two independent toehold switches controlling split-NanoLuc fragments reduces false positives.
NanoLuc bioluminescence requires no excitation light source, making it compatible with smartphone camera detection.
Modular scaffold design enables rapid design-order-test cycles for new pathogen targets.
Experimental Design
Step
Action
Method/Tools
1
Target Selection
Identify HPV16 L1/E6 sequences; Design switches via NUPACK.
LOCUS SwitchB_SmBiT 198 bp DNA linear
DEFINITION Toehold Switch B (HPV16 E6 trigger-responsive) controlling SmBiT
expression. T7 promoter, toehold hairpin, RBS, SmBiT CDS, T7
terminator.
ORIGIN
1 taatacgact cactataGGG agaccggcag atctgatatc atcgatgaat
51 tcgagctcgg tacccgggga tccTCTAGAG TCGACCTGCA GGCATGCAAG
101 CTTGTGAGCG GCTGGCGGCT GTTCAAGAAG ATCTCGTAAG CTTGGCGTAA
151 TCATGGTCAT AGCTGTTTCC TGTGTGAAAT TGTTATCCGC TCACAATTCC
201 AC
//
Bioethical Considerations
Ethics: Devices must be designed with explicit informed consent and local-only data storage. The AND-gate logic is an ethical choice to reduce false positives and potential stigma.
Risk Mitigation: The cell-free system contains no living organisms and is naturally inactivated, minimizing environmental biosafety risks.