Week 5 Homework: Protein Design Part II
Part 1: Generate Binders with PepMLM
The target for this study is the human SOD1 protein (UniProt P00441), specifically focusing on the A4V mutation, which is a common cause of Amyotrophic Lateral Sclerosis (ALS).
Mutant SOD1 Sequence (A4V):
ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using PepMLM-650M, four peptides of 12 amino acids were generated and compared against the known SOD1-binding peptide FLYRWLPSRRGG.
PepMLM Confidence Scores
| Sequence | Description | Perplexity |
|---|---|---|
| FLYRWLPSRRGG | Real Binder | — |
| WHSPVVAVAHWE | Sim 1 | 10.949699 |
| WSVGWAAIAWWX | Sim 2 | 16.027645 |
| WRSYATAIALWK | Sim 3 | 11.729657 |
| WRYYATGAEWKE | Sim 4 | 13.769973 |
Part 2: Evaluate Binders with AlphaFold3
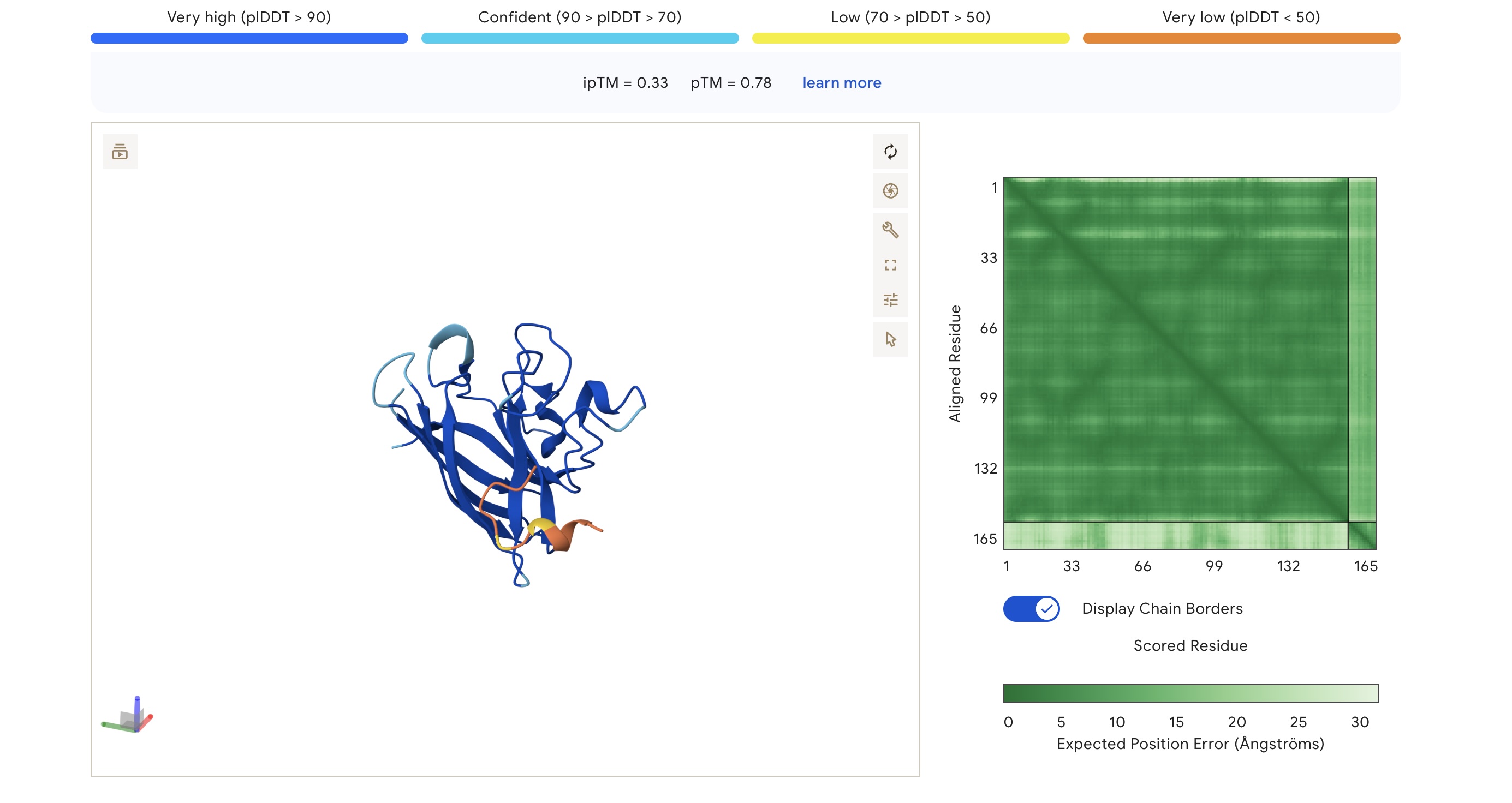
Each peptide was modeled against the mutant SOD1 sequence using AlphaFold3 to assess structural docking and interface confidence (ipTM).
Structural Observations
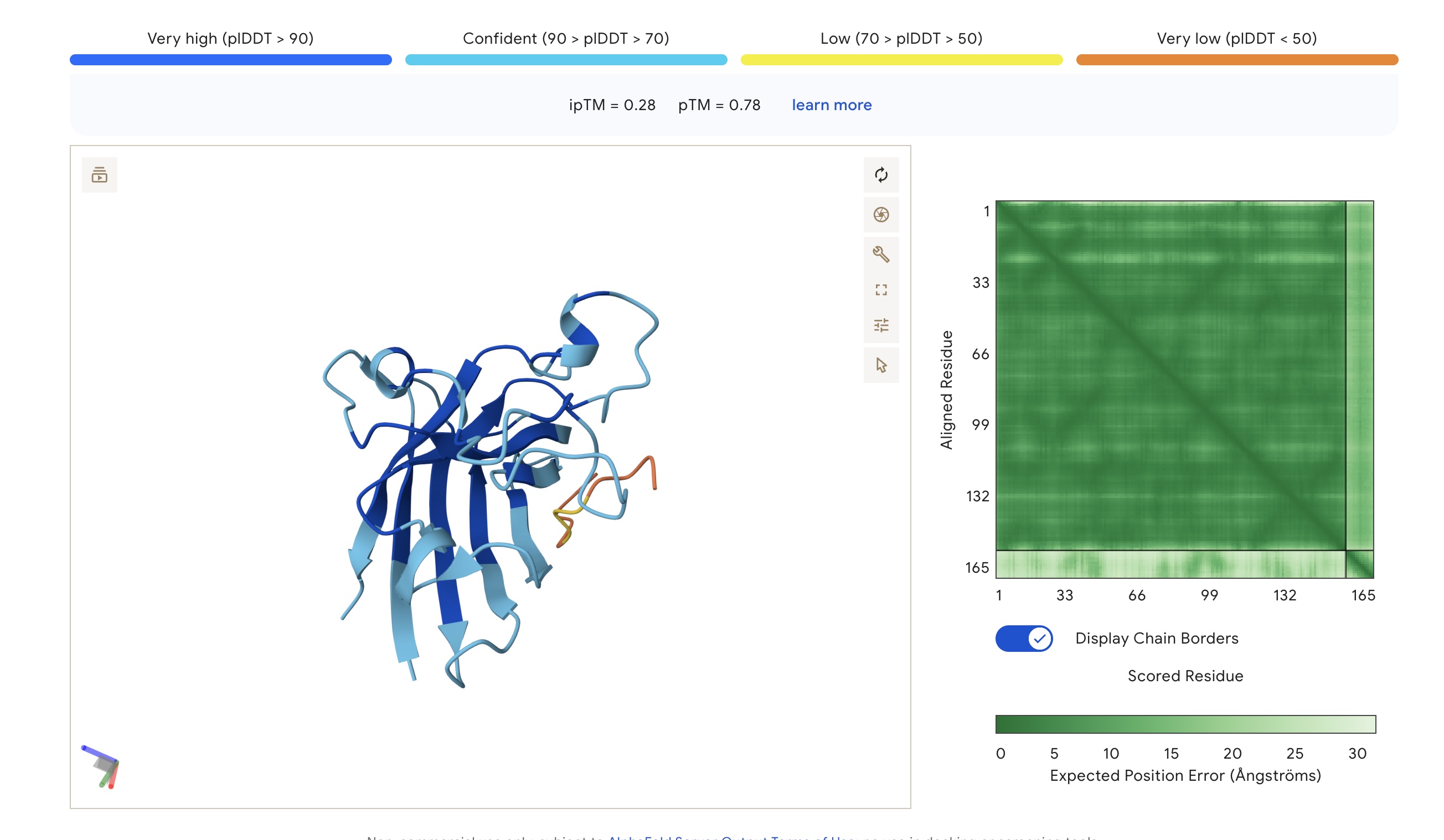
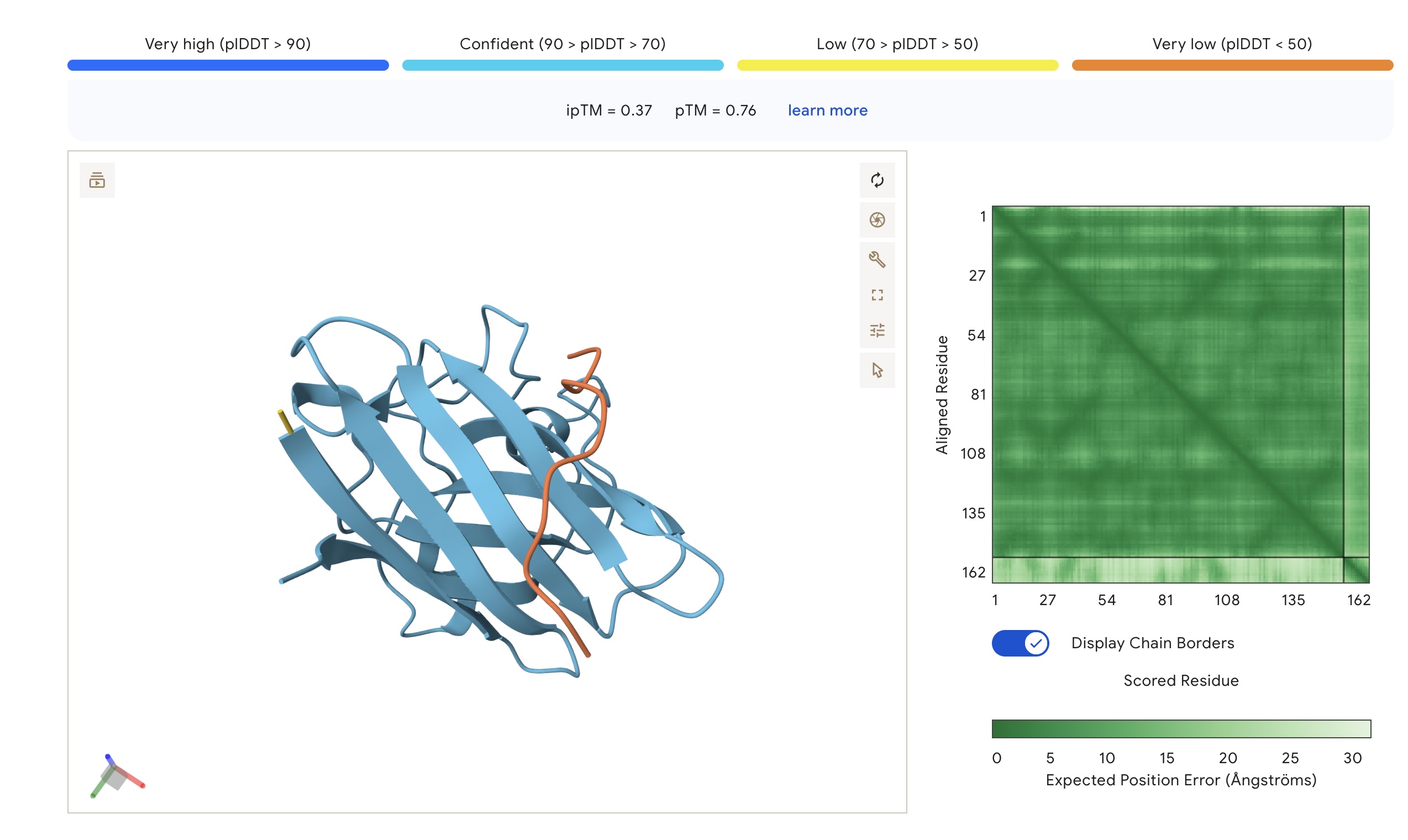
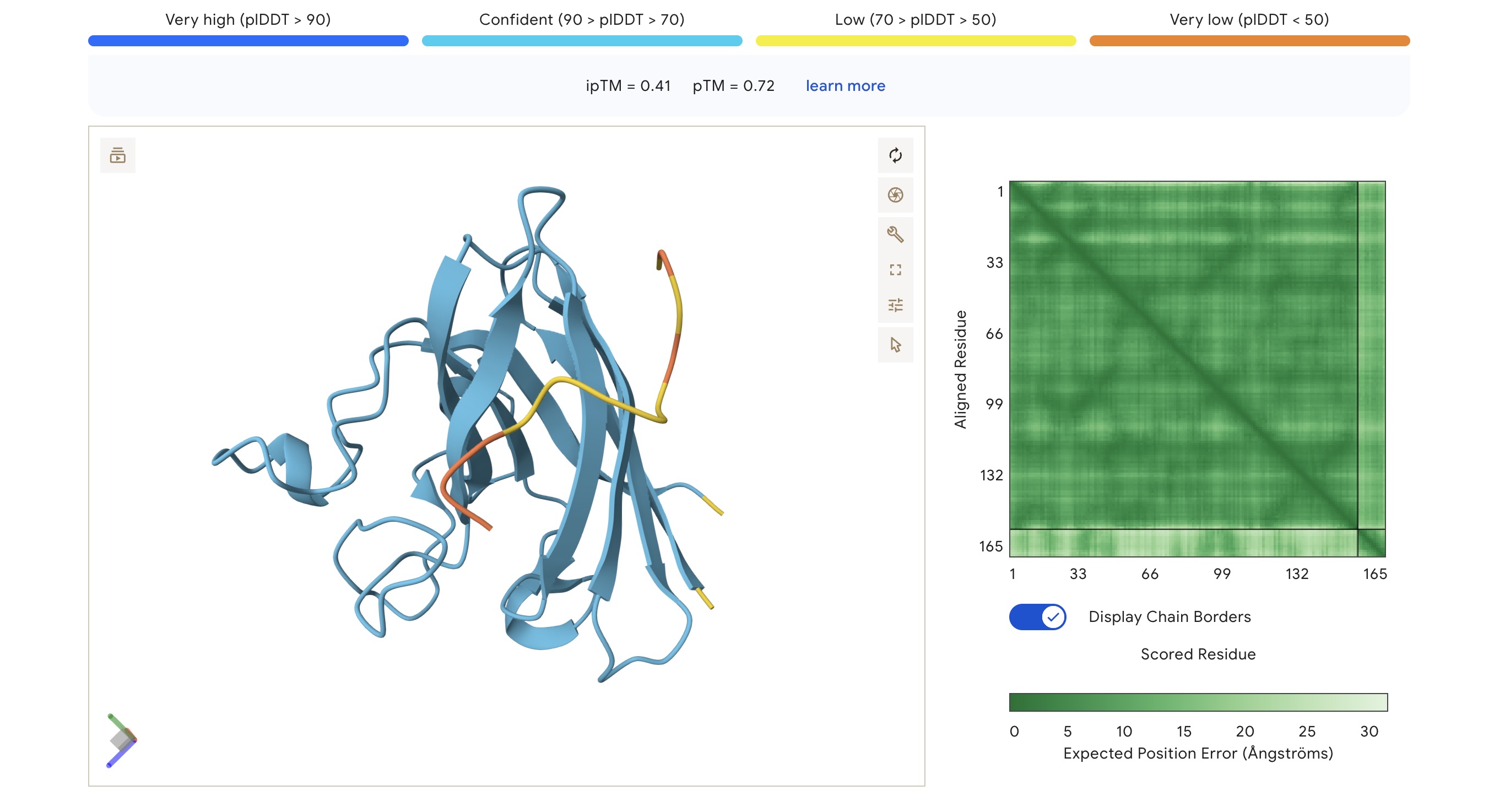
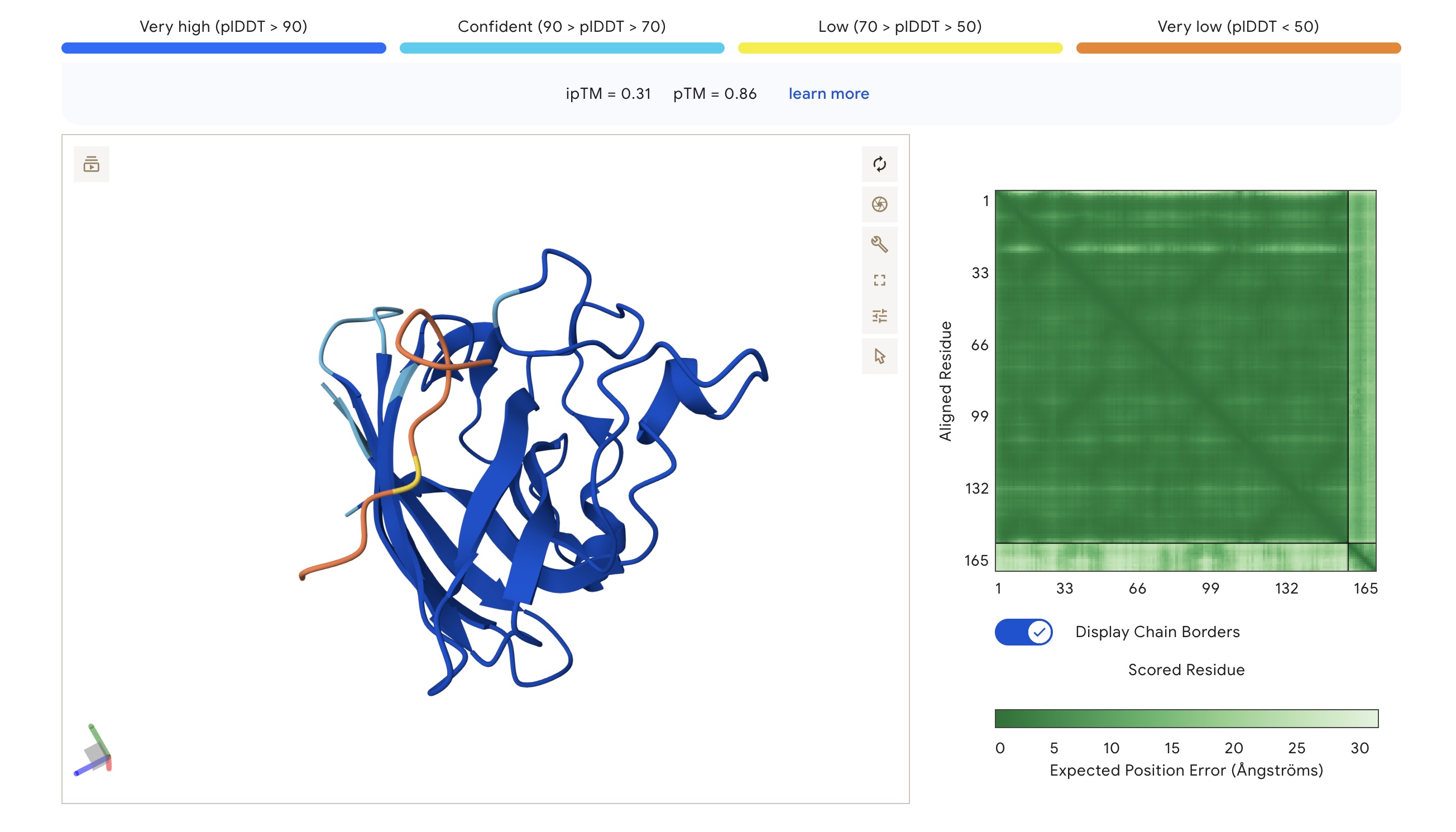
- Localization: Surprisingly, none of the peptides localized specifically to the A4V mutation site at the N-terminus. All peptides were primarily surface-bound.
- Binding Sites: Sim 3 (WRSYATAIALWK) and Sim 4 (WRYYATGAEWKE) both bound to a region that appears to be a potential polymerization site. Sim 2 (WSVGWAAIAWWX) engaged the β-barrel region. The wild-type binder also localized to a suspected polymerization site.
AlphaFold3 Visualizations
Part 3: Evaluate Properties in the PeptiVerse
Beyond structural docking, we evaluated the therapeutic potential (solubility, hemolysis, and affinity) of the generated sequences.
Peptide Property Comparison
| Peptide Sequence | Solubility | Hemolysis | Affinity (pKd) | MW (Da) | Net Charge | pI | GRAVY | ipTM | pTM |
|---|---|---|---|---|---|---|---|---|---|
| FLYRWLPSRRGG | 1.000 | 0.047 | 5.96 (Weak) | 1507.7 | +2.76 | 11.71 | -0.71 | 0.33 | 0.78 |
| WHSPVVAVAHWE | 1.000 | 0.048 | 5.07 (Weak) | 1417.6 | -1.06 | 6.02 | 0.18 | 0.28 | 0.78 |
| WSVGWAAIAWWX | 1.000 | 0.174 | 7.71 (Med) | 1314.6 | -0.24 | 5.53 | 0.78 | 0.37 | 0.76 |
| WRSYATAIALWK | 1.000 | 0.056 | 6.83 (Weak) | 1465.7 | +1.76 | 9.99 | 0.06 | 0.41 | 0.72 |
| WRYYATGAEWKE | 1.000 | 0.063 | 5.71 (Weak) | 1559.7 | -0.23 | 6.28 | -1.44 | 0.31 | 0.86 |
Analysis of Results
The observed ipTM values across the PepMLM-generated candidates range from 0.28 to 0.41, reflecting a low-to-moderate confidence in specific interface orientation. However, two PepMLM peptides outperformed the known binder (ipTM 0.33): WRSYATAIALWK (0.41) and WSVGWAAIAWWX (0.37).
There is a general correlation between structural confidence and affinity; the sequence with the highest affinity (WSVGWAAIAWWX, pKd: 7.71) also showed a strong ipTM. Interestingly, the highest ipTM belonged to WRSYATAIALWK, despite it having a lower predicted affinity, suggesting that AF3 finds this specific binding pose more physically convincing even if the thermodynamic prediction is weaker. Critically, all candidates maintain a 1.000 solubility probability and low hemolysis scores.
Selection Candidate: WRSYATAIALWK While WSVGWAAIAWWX has the highest raw affinity, WRSYATAIALWK offers a superior balance of properties. It provides the highest structural confidence (ipTM 0.41) while maintaining a much safer hemolysis profile (0.056) compared to the “riskiest” sequence in the group (0.174).
Part 4: Generate Optimized Peptides with moPPIt
Unlike the sampling method of PepMLM, moPPIt was used for controlled, multi-objective design to steer generation toward specific residues (dimer interface/A4V site) and optimize therapeutic properties.
moPPIt Generated Peptides
| Sequence | Solubility | Hemolysis | Affinity | MW (Da) | Net Charge | pI | GRAVY |
|---|---|---|---|---|---|---|---|
| GGTTTDDTKAES | 1.000 | 0.054 | 4.20 | 1182.1 | -2.24 | 4.05 | -1.42 |
| ATTGYCGCTMQN | 1.000 | 0.018 | 5.39 | 1249.4 | -0.22 | 5.55 | -0.21 |
| DEGYKKQKGQIQ | 1.000 | 0.041 | 4.75 | 1421.6 | +0.76 | 8.43 | -2.23 |
Comparison and Evaluation
The PepMLM peptides generally focus on high-confidence structural docking and balanced biophysical properties, leaning toward a safe profile while improving affinity over the baseline. In contrast, the moPPIt peptides prioritize diverse chemical spaces and targeted binding.
The moPPIt set introduces extreme charge variations, such as the highly acidic GGTTTDDTKAES (pI 4.05) and the polar-rich DEGYKKQKGQIQ, which differ significantly from the more hydrophobic PepMLM designs. To evaluate these before clinical studies, we would need to perform experimental circular dichroism (CD) to confirm peptide stability and surface plasmon resonance (SPR) to validate the targeted binding affinity at the specific A4V or dimer interface sites chosen during the moPPIt steering process.
Gemini AI was consulted for formatting and content organization