Week 4 Lab: Protein Design Part I
Protein Selection
Briefly describe the protein you selected and why you selected it. Again, continuing with my idea for designing a carbon sequestration system, I am going to be looking at Rubisco, the most abundant protein on earth. Rubisco catalyzes photosynthesis in plants, converting atmospheric CO2 into an organic three-carbon acid that eventually is built up into sugars for plant growth. It is made of 8 large subunits and 8 small subunits.
Sequence and Composition
Identify the amino acid sequence of your protein.
Large Subunit:
MSPQTETKASVGFKAGVKDYKLTYYTPDYETKDTDILAAFRVTPQPGVPPEEAGAAVAAESSTGTWTTVWTDGLTSLDRYKGRCYHIEPVAGEESQFIAYVAYPLDLFEEGSVTNMFTSIVGNVFGFKALRALRLEDLRIPTSYVKTFQGPPHGIQVERDKLNKYGRPLLGCTIKPKLGLSAKNYGRAVYECLRGGLDFTKDDENVNSQPFMRWRDRFLFCAEAIYKAQAETGEIKGHYLNATAGTCEEMIKRAVFARELGAPIVMHDYLTGGFTANTSLAHYCRDNGLLLHIHRAMHAVIDRQKNHGMHFRVLAKALRLSGGDHIHAGTVVGKLEGEREITLGFVDLLRDDFIEKDRSRGIYFTQDWVSLPGVLPVASGGIHVWHMPALTEIFGDDSVLQFGGGTIGHPWGNAPGAVANRVALEACVQARNEGRDLAREGNQIIREASKWSPELAAASEVWKEIKFEFPAMDTL
Small Subunit:
VVLSKECAKPLATPKVTLNKRGFATTIATKNREMMVWQPFNNKMFETFSFLPPLTDEQISKQVDYILTNSWTPCLEFAASDQAYAGNENCIRMGPVASTYQDNRYWTMWKLPMFGCTDGSQVLSEIQACTKAFPDAYIRLVCFDANRQVQISGFLVHRPPSATDYRLPADRQV
How long is it? What is the most frequent amino acid? The Large Subunit is roughly 475 amino acids long and the small subunit is roughly 175 amino acids long. The most frequent amino acid is Alanine (A) in both subunits with ~13 A’s in the small chain and ~46 in the large chain.
Homologs and Structure
How many protein sequence homologs are there for your protein? There are 8 versions of Rubisco Large and Small subunits that are marginally different and assemble together to create a 16-mer (hexadecamer) protein.
Does your protein belong to any protein family? They belong to the Rubisco Large and Small subunit chain families.
When was the structure solved? Is it a good quality structure? Form I, the 8L, 8S hexadecamer was solved in the 1990s with an initial resolution of 2.2 angstroms, with specific structures like that of Chlamydomonas reinhardtii being solved for 1.4 angstroms in 2001. This is a very good quality structure (smaller resolution is better, typically < 2.70 Å).
Are there any other molecules in the solved structure apart from protein? Yes, magnesium ions and carbamylated lysine.
3D Molecule Visualization (PyMol)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
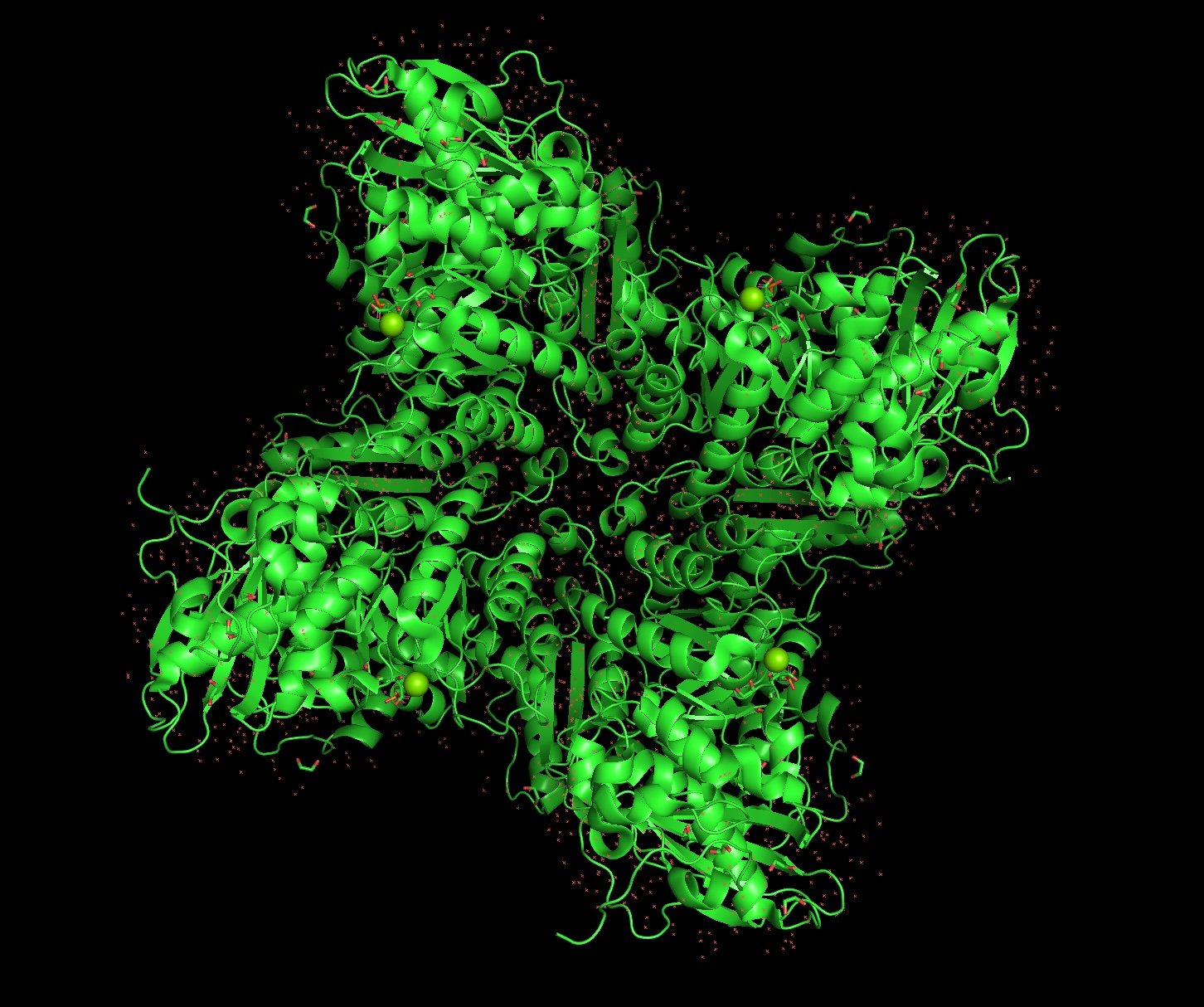
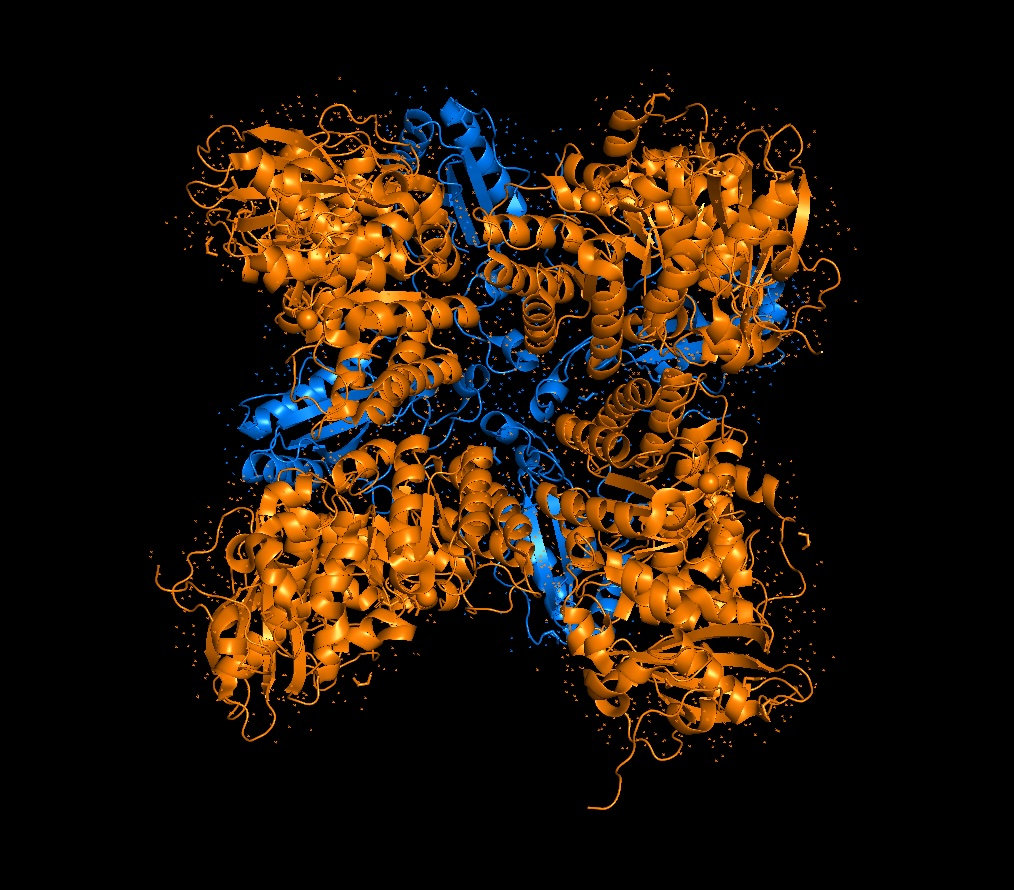
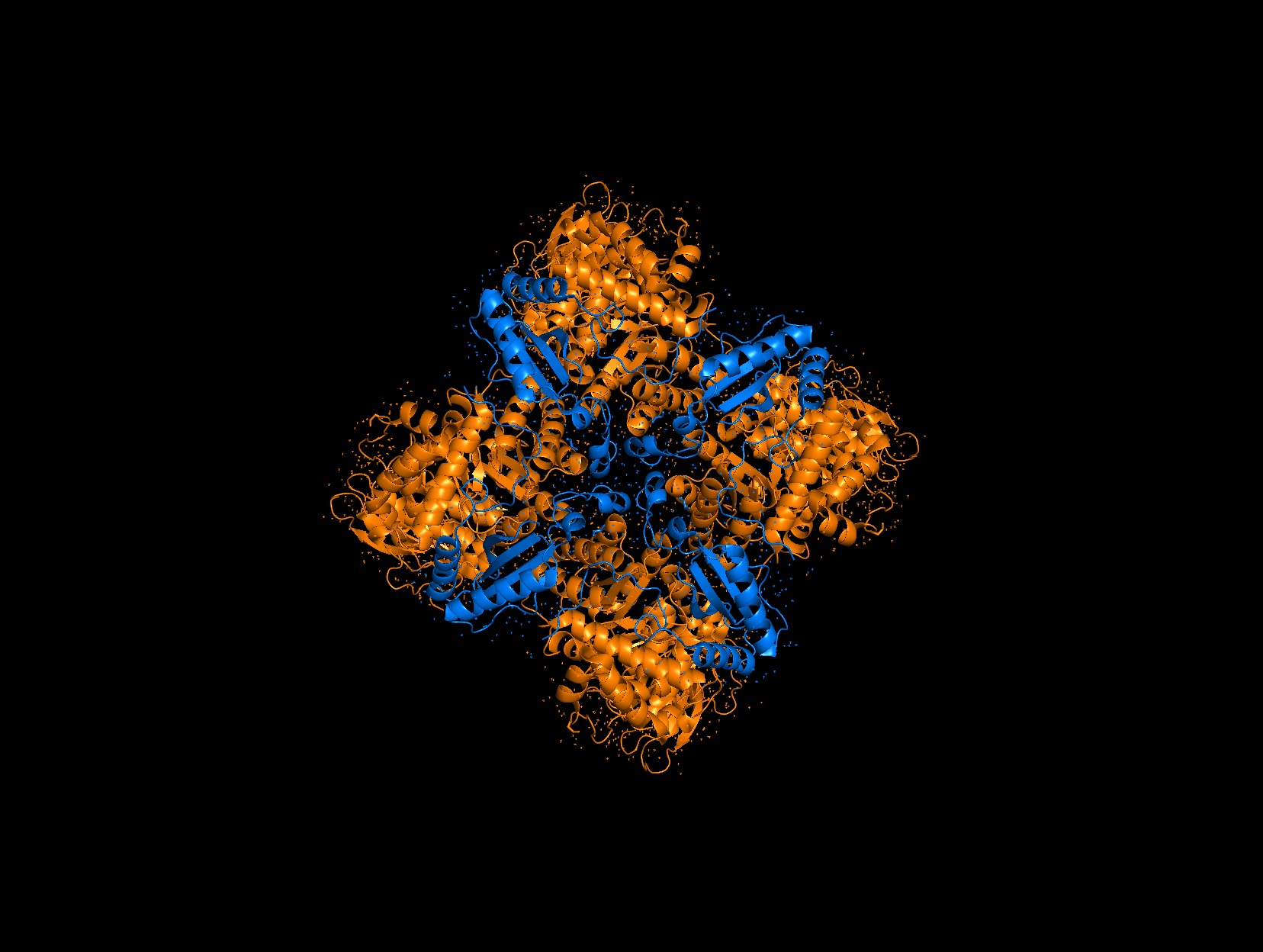
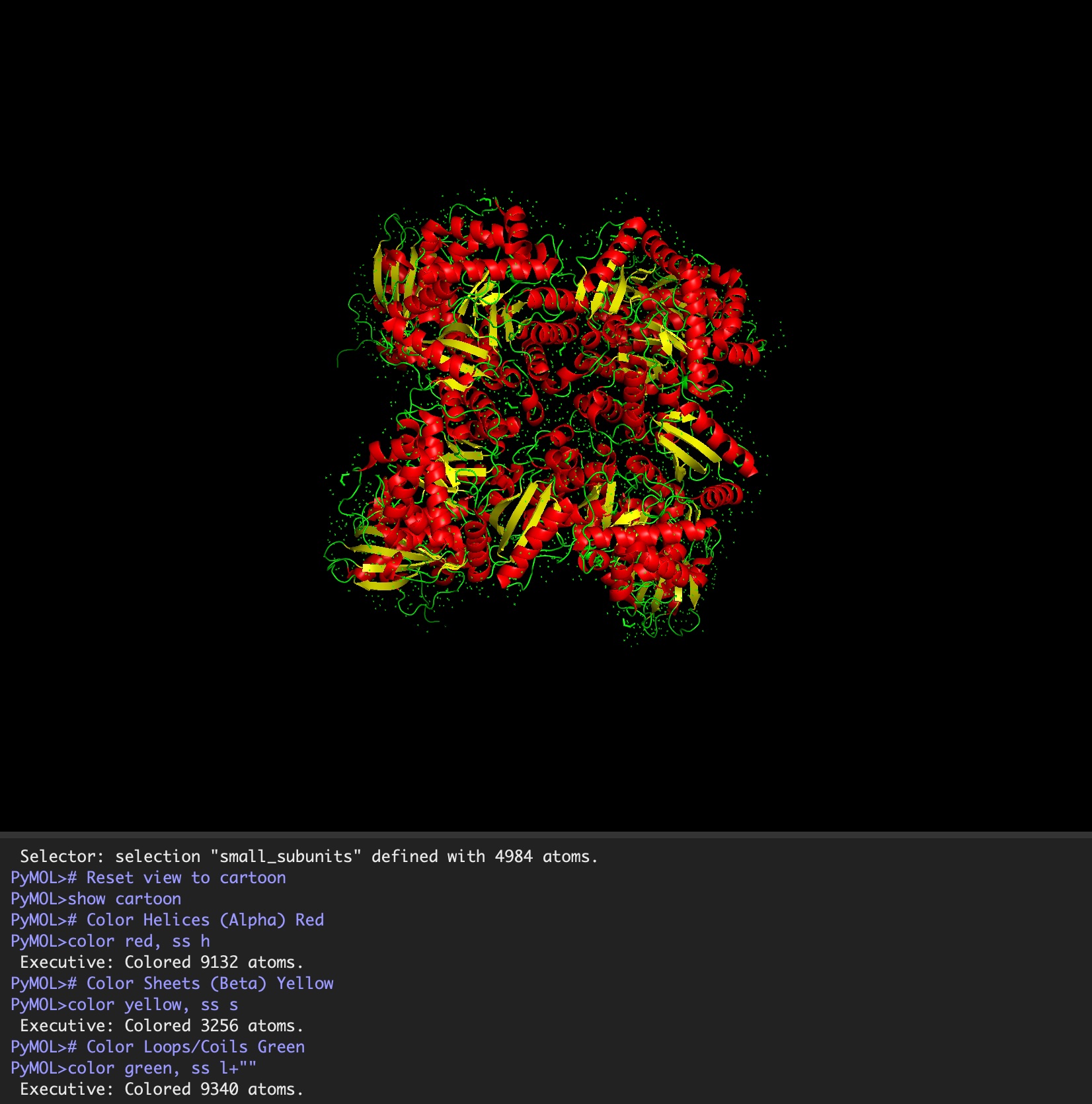
Color the protein by secondary structure. Does it have more helices or sheets?
It has roughly 40% helices and 20% beta sheets.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
It is a mosaic of hydrophobic and hydrophilic residues, making it efficient in aqueous environments but also able to interact with hydrophobic residues.
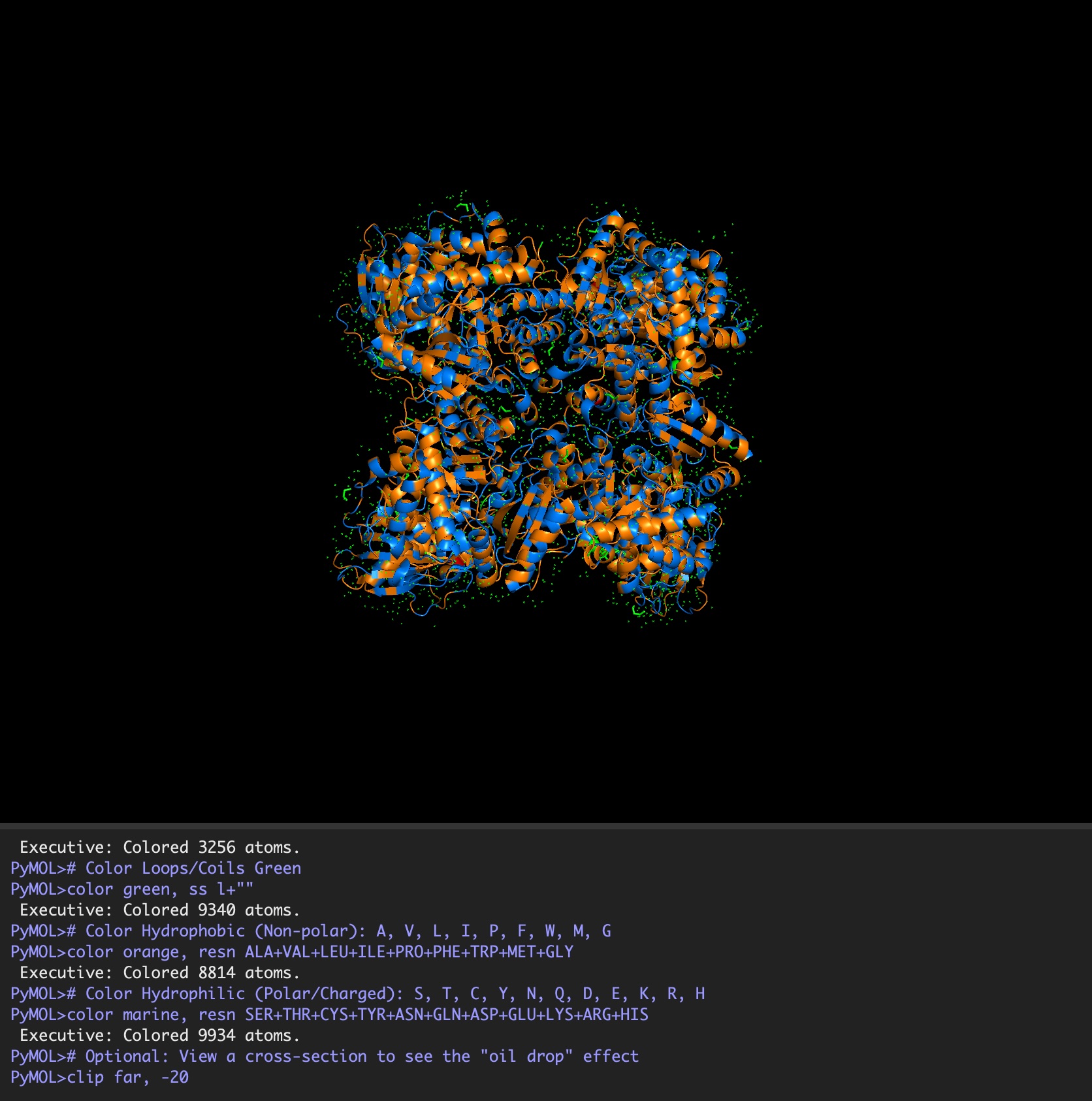
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)? Because the holoenzyme is made of 8 large subunits, it has 8 identical active sites. The active pocket sites contain deep indentations located at the mouth of each TIM barrel. This is where Ribulose-1,5-bisphosphate and CO2 meet to be converted into sugar.
The central pore is visible from the “top” (along its axis of symmetry) and is a prominent hole/channel running through the center of the entire L8S8 complex. There are also smaller crevices between the small subunits and the large subunit core, which some researchers believe might play a role in gas or metabolite diffusion. Deep into those 8 pockets is where the magnesium ions are located, which coordinate the substrates and are necessary for the reaction to proceed.
Deep Mutational Scans
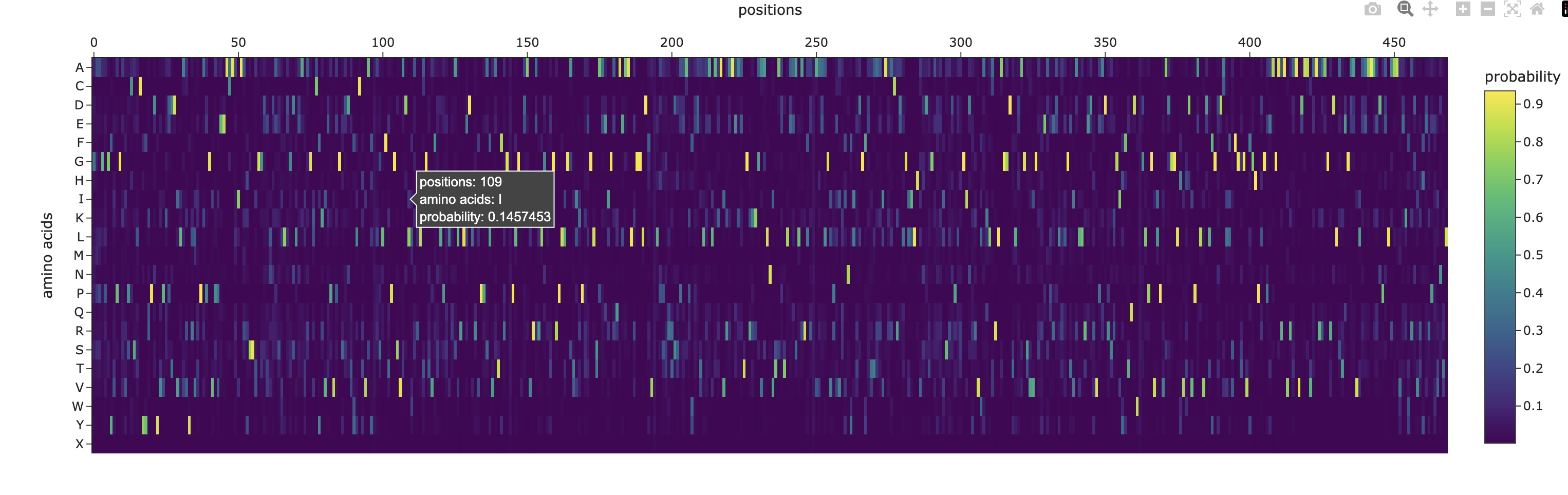
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
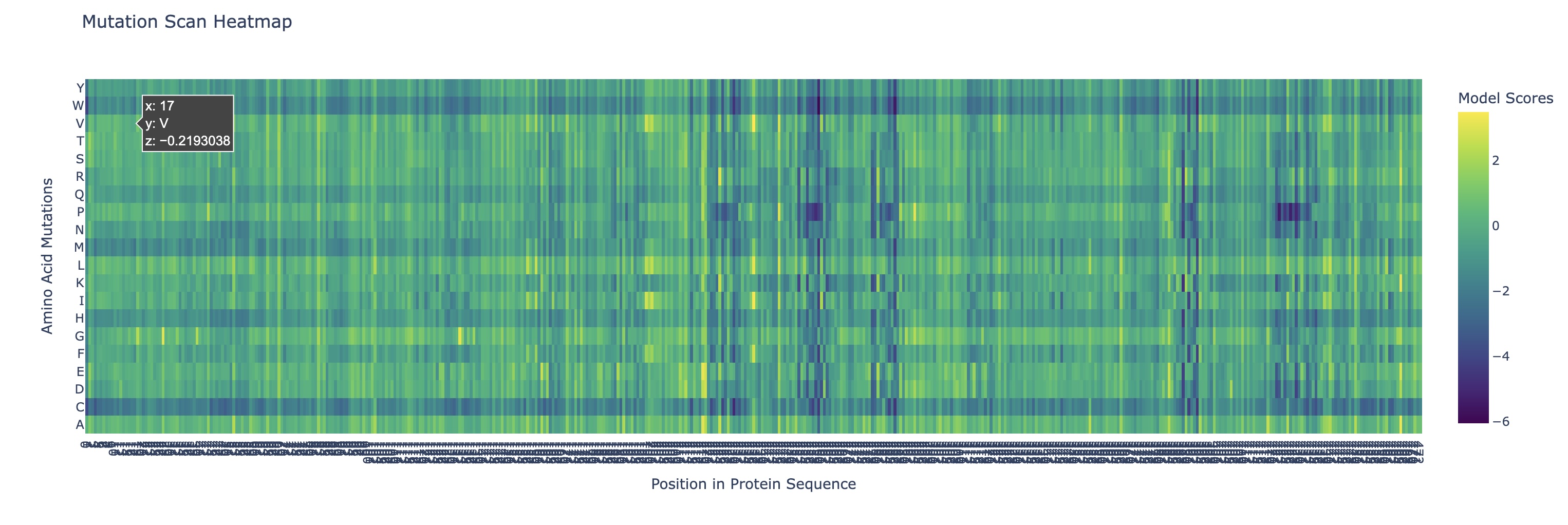
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
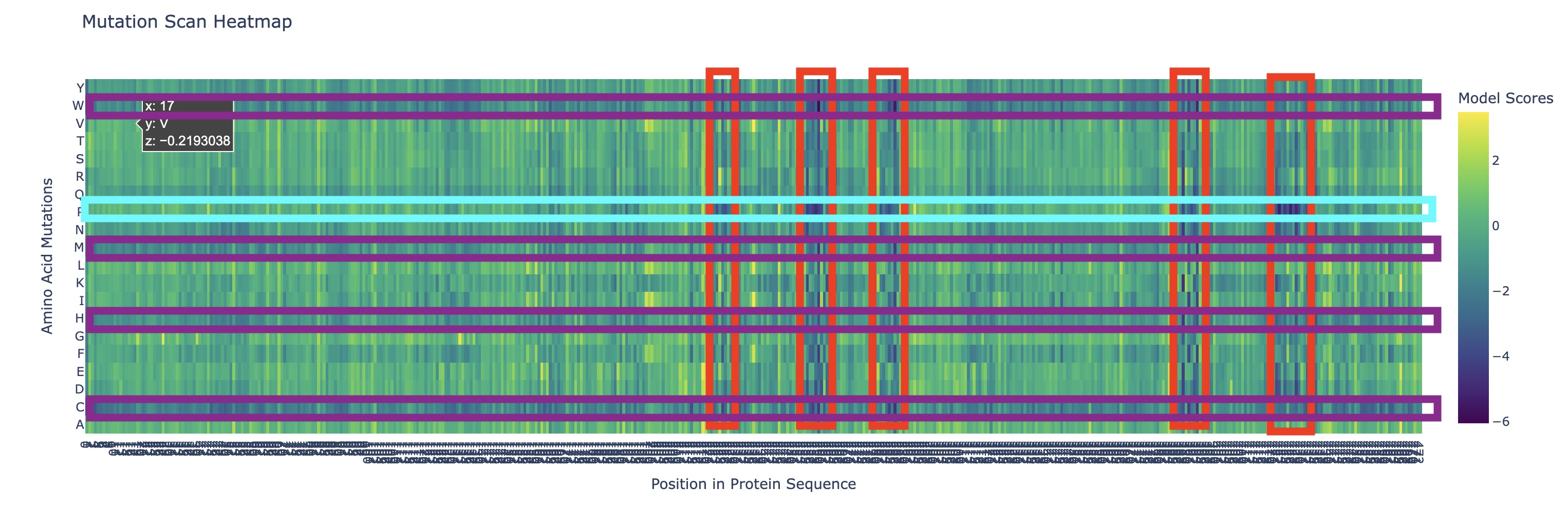
There are a couple of trends I’ve noticed from this mutational scan. There seem to be 5 main evolutionarily conserved sequences that result in catastrophic damage to the protein if substituted (these are the vertical lines I’ve highlighted in red). Additionally, there are many faintly dark vertical bands, indicating that Rubisco is generally highly conserved which I think is strongly supported by the fact that it’s the most abundant protein on earth (the sequence works and thus has resisted change over a long time scale).
Additionally, there are a couple main horizontal bands of negative change, mainly in amino acids W, C, M, H, Q (outlined in purple). This is actually not that surprising in my opinion. Methionine (M) and Cysteine (C) are unique in that they are the only two that contain sulfur atoms which could cause negative effects in a multitude of ways (sulfur is huge so steric hindrance is likely, it’s very polarizable so it makes an excellent nucleophile with higher reactivity, and its redox affinity allows for structural organizations like disulfide bridges to occur). Tryptophan (W) is a gigantic amino acid with its aromatic ring that again causes a lot of conformational changes due to sterics. Histidine (H) is one of three positively charged amino acid residues and altering electrostatic character (acidity/basicity) usually has significant effects. And Glutamine (Q) is a polar amino acid, again introducing a tangible change to binding character.
What I found interesting was that a substitution of Proline (P) (teal) across the protein was predicted to increase efficiency except at the highly conserved regions I highlighted earlier, where a substitution to proline was significantly more negatively impactful than other amino acids which I found a bit strange.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
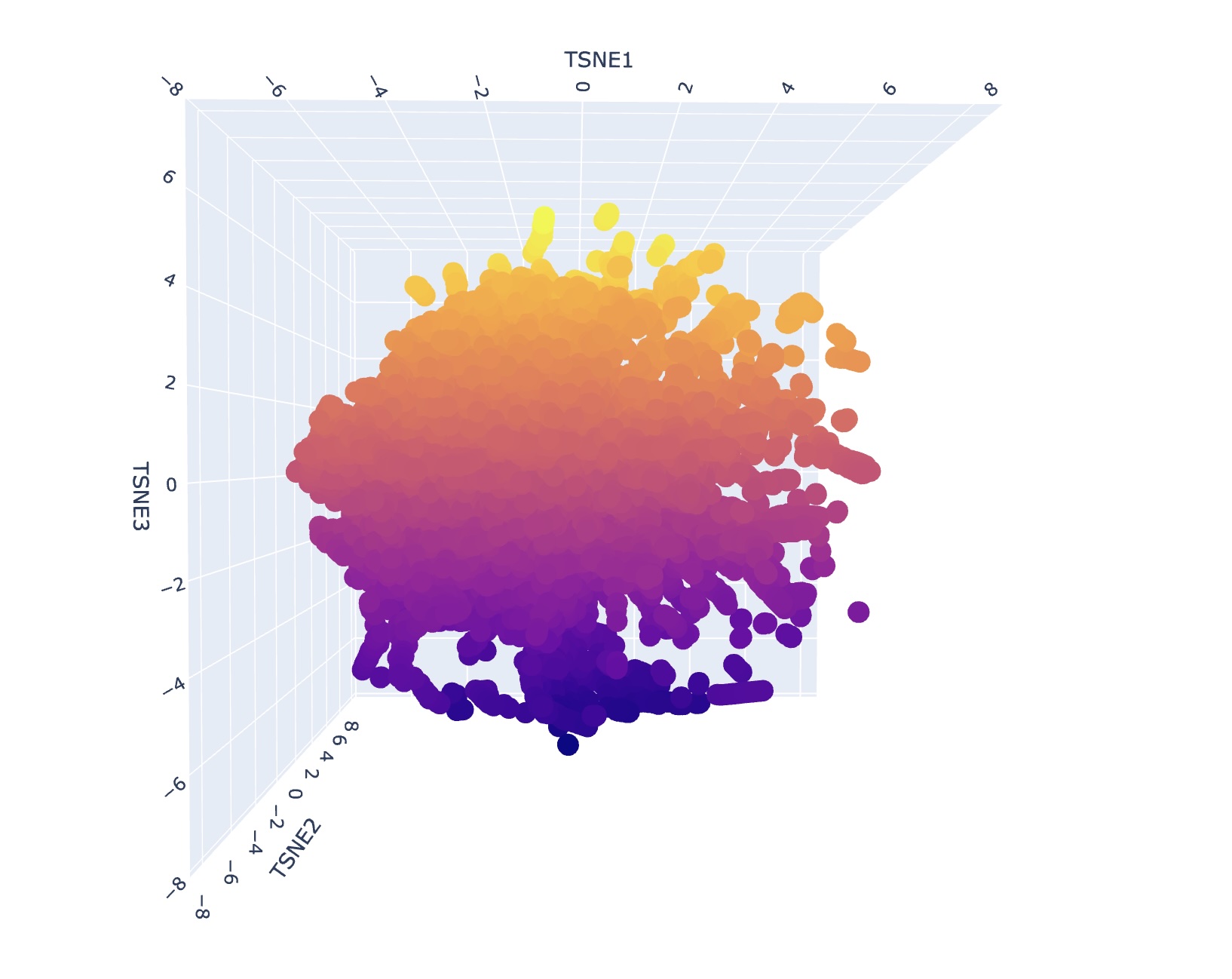
Analyze the different formed neighborhoods: do they approximate similar proteins? I generally could not find super consistent themes or narratives when looking at different neighborhood clusters. I think this is reflective of how protein structure might appear similar but depending on the environment can have significantly different functional attributes. However, upon a second search, I saw that some families contained only bacterial proteins/prokaryotes. Some families also contained only human proteins or proteins of mammals.
Folding a Protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure? The predicted coordinates are similar to my original structure but not a perfect match.
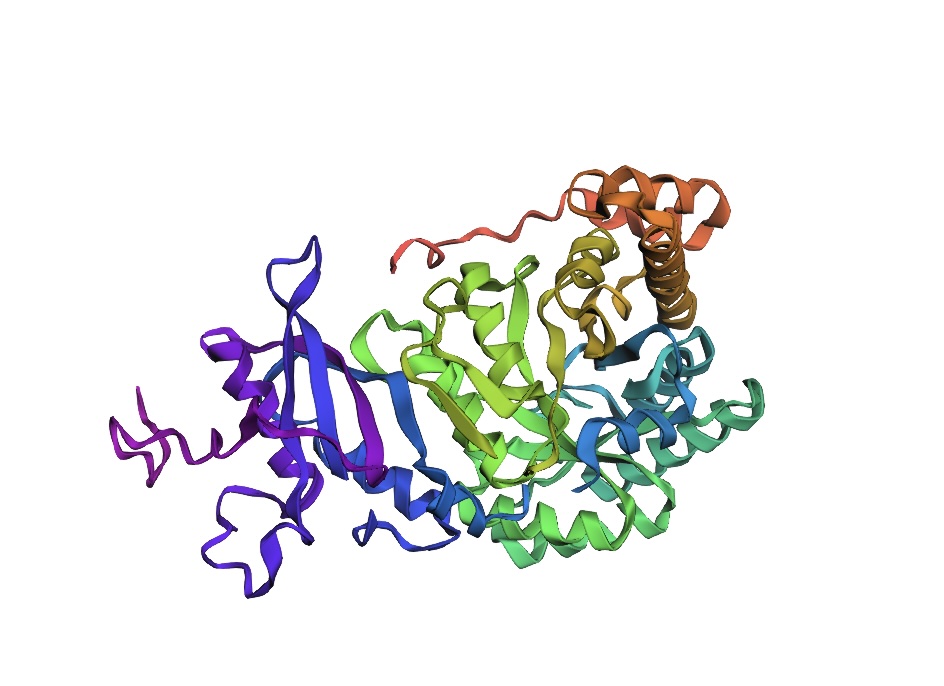
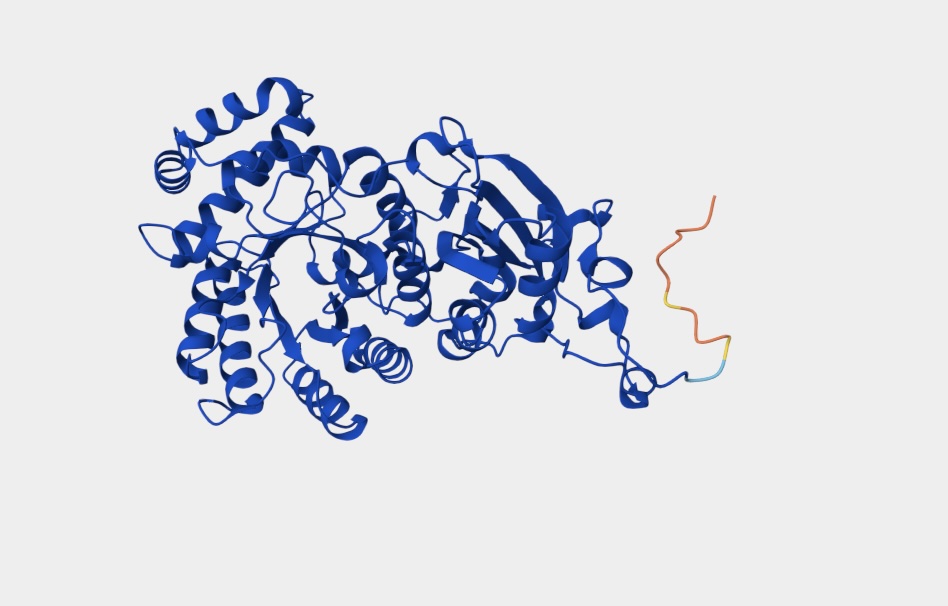
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
The protein structure is not resilient to mutations featuring distinct changes.
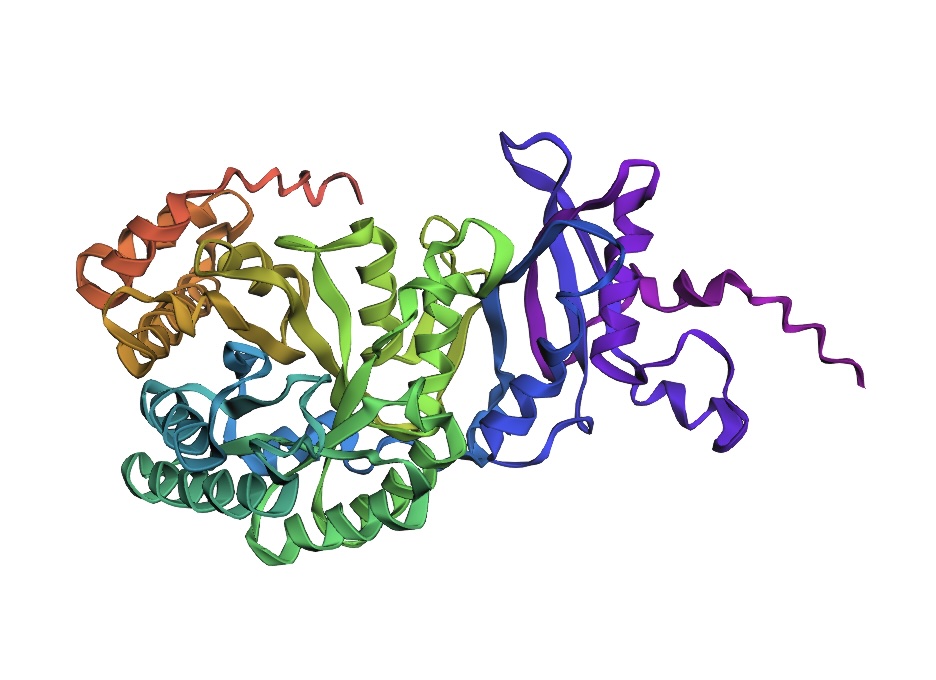
Inverse-Folding a Protein
Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN.
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
>1gk8, score=1.5831, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 TKAGAGFKAGVKDYRLTYYTPDYVVRDTDILAAFRMTPQPGVPPEECGAAVAAESSTGTWTTVWTDGLTSLDRYKGRCYDIEPVPGEDNQYIAYVAYXIDLFEEGSVTNMFTSIVGNVFGFKALRALRLEDLRIPPAYVKTFVGXPHGIQVERDKLNKYGRGLLGCTIKPKLGLSAKNYGRAVYECLRGGLDFTXDDENVNSQPFMRWRDRFLFVAEAIYKAQAETGEVKGHYLNATAGTCEEMMKRAVXAKELGVPIIMHDYLTGGFTANTSLAIYCRDNGLLLHIHRAMHAVIDRQRNHGIHFRVLAKALRMSGGDHLHSGTVVGKLEGEREVTLGFVDLMRDDYVEKDRSRGIYFTQDWXSMPGVMPVASGGIHVWHMPALVEIFGDDACLQFGGGTLGHPWGNAPGAAANRVALEACTQARNEGRDLAREGGDVIRSACKWSPELAAACEVWKEIKFEFDTIDKL
>T=0.1, sample=0, score=0.7630, seq_recovery=0.4440 GAAGPGYVPGATPPSLRYYQPDYVPKDDDVLAAYLVTPAPGVPPEEACALIARYSSIGGPENDASILLGDPEKTRGICYKVEPVPGSSDRYLCYIAYXLSLFTPGSVADIWAKLCGTVFTLPELKALKLLDIRFPRALVATFRGXPLGIAAVRARLGIEGRPLLGATLKPPLGLSAAELGEQARAALVGGLDFVXLHSSQRSSPAAPWADTLAALAEAVRAAEAETGRPKGAALNVTADTLEAALARLDXAAAAGLRVVRINFITLGFDAAAAIAAACRDRGIILHASDTGIRKYSLNPDSGIDYRVWAKLARLTGADTLHAGSVVGRFEAPEALLKGTIALLREDYVEKDESKGIYFTQDWXGLPGVLPVAGGGLHVHDVPALVALFGDDCIISFGTGVFGHPLGPRAGARAVRTAVDAAVAAARAGRDLRTEGAAVVAEAAARDPELAAAVERWRDVRYERPWVNTL
New Sequence:
GAAGPGYVPGATPPSLRYYQPDYVPKDDDVLAAYLVTPAPGVPPEEACALIARYSSIGGPENDASILLGDPEKTRGICYKVEPVPGSSDRYLCYIAYXLSLFTPGSVADIWAKLCGTVFTLPELKALKLLDIRFPRALVATFRGXPLGIAAVRARLGIEGRPLLGATLKPPLGLSAAELGEQARAALVGGLDFVXLHSSQRSSPAAPWADTLAALAEAVRAAEAETGRPKGAALNVTADTLEAALARLDXAAAAGLRVVRINFITLGFDAAAAIAAACRDRGIILHASDTGIRKYSLNPDSGIDYRVWAKLARLTGADTLHAGSVVGRFEAPEALLKGTIALLREDYVEKDESKGIYFTQDWXGLPGVLPVAGGGLHVHDVPALVALFGDDCIISFGTGVFGHPLGPRAGARAVRTAVDAAVAAARAGRDLRTEGAAVVAEAAARDPELAAAVERWRDVRYERPWVNTL
Input this sequence into ESMFold and compare the predicted structure to your original.
The predicted structure is somewhat similar to the original structure indicating the viability of ESMFold readout for protein identification, modification, and discovery.