HW 1 First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
A biological engineering application I am interested in developing is biological haptic actuators. I envision a future where one can fabricate haptic systems driven by living cells to mimic touch sensations. Through some external stimulus, this device could output vibrations to mimic touch. The scenario I am presenting replaces electromechanical systems with biologically powered interfaces.
Part 1: Benchling & In-Silico Gel Art Using Benchling, I imported the Lambda DNA sequence and simulated restriction enzyme digestions with EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. The goal was to design a gel art pattern inspired by Paul Vanouse’s Latent Figure Protocol.
The design depicts a figure in a rocky victory pose with a crying face, using 5 lanes of selected enzyme combinations to form the image.
Assignment 1: Python Script for Opentrons Artwork I designed an Indonesian cloud pattern called Megamendung using the Opentrons pipetting robot. The pattern uses two colors — red (mRFP1) and green (Azurite) — arranged as interlocking arcs to replicate the traditional Javanese batik motif.
Simulated output of the Python script:
Python script:
Part 1: Protein Selection 1.1 Protein Choice I selected Titin (PDB: 1G1C) because it is a structural protein and acts as the “spring” in human muscle — it is the largest known protein in the human body and is responsible for the passive elasticity of muscle fibers.
Part A: SOD1 Binder Peptide Design Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. Mutations in SOD1 cause familial ALS — among them, the A4V mutation (Alanine → Valine at residue 4 of the mature protein) is one of the most aggressive, destabilizing the N-terminus and promoting toxic aggregation.
Part A: Pre-Lab Protocol Questions 1. Phusion High-Fidelity PCR Master Mix According to the NEB product page, the Phusion master mix is built around two key features of the Phusion polymerase itself:
A Pyrococcus-like polymerase core with a 3’→5’ proofreading exonuclease — catches and corrects misincorporated bases in real time, giving ~50x lower error rate than Taq. An Sso7d processivity domain fused to the polymerase — a DNA-binding clamp that keeps the enzyme on the template, increasing speed and fidelity together. The master mix also includes dNTPs, MgCl₂ (essential cofactor for polymerase activity), and a reaction buffer optimized for Phusion.
Part 1: Intracellular Artificial Neural Networks (IANNs) 1. Advantages of IANNs over Boolean Genetic Circuits Traditional genetic circuits implement Boolean logic — AND, OR, NOT gates — where every output is binary: a gene is either on or off. IANNs offer three key advantages over this:
General Homework Questions 1. Advantages of Cell-Free Protein Synthesis Cell-free protein synthesis offers several advantages over in vivo expression:
Speed — no transformation, culture growth, or induction cycle. Protein can be expressed in hours rather than days or weeks. Open system / debuggability — with no cell wall in the way, you can directly add, remove, or adjust components mid-reaction. Troubleshooting is hands-on: swap the energy system, add chaperones, tweak Mg²⁺ — all in real time. Biosafety — no live GMOs means no antibiotic resistance cassettes spreading in the environment and a lower containment burden overall. Toxic proteins — proteins that would kill a host cell can be expressed freely in lysate. Non-natural amino acids — easier to incorporate than in living cells where the genetic code is fixed. Two use cases where cell-free is preferred:
Final Project: Measurements Plan What to Measure The chimeric casein project has three distinct questions that each need a different measurement approach:
Stage 1 — Did the protein express? After inducing E. coli with IPTG and lysing the cells, I need to check whether the chimeric protein actually appeared. The primary tool for this is SDS-PAGE (gel electrophoresis for proteins) — the same ladder-based approach used for DNA, but with SDS added to unfold proteins and give them uniform charge so separation is purely by size. Running the pre- and post-induction lysate side by side, I’d look for a new band appearing at ~50 kDa (the expected molecular weight of the chimera). A Western blot using an anti-His antibody would then confirm the band is specifically my His-tagged chimera and not a coincidental band from the host cell.
Part A: Global Pixel Artwork In progress.
My contribution: I added a single red pixel late in the project to correct a gap in the pattern. A small fix, but satisfying to slot into place.
Subsections of Homework
Week 1 HW: Principles and Practices
HW 1
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
A biological engineering application I am interested in developing is biological haptic actuators. I envision a future where one can fabricate haptic systems driven by living cells to mimic touch sensations. Through some external stimulus, this device could output vibrations to mimic touch. The scenario I am presenting replaces electromechanical systems with biologically powered interfaces.
In current fabrication pipelines, the output is structural (eg. 3D Printing). To add interactivity, components need to be added at a later stage (microcontrollers + sensors + motors). This co-location of sensing, computation and actuation is built-in in biological systems. Furthermore, as devices are integrating with the user (we are currently at the wearable stage) current fabricated objects may be considered “clunky” whereas purely biological interfaces can be streamlined. Tools such as AlphaFold make it easier for users to author and design proteins and I am curious to see if it is possible to fabricate a biological actuator. If touch is not feasible I am open to exploring other output modalities such as color.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Sustainability, Safety and Agency are the biggest considerations regarding my selected application or tool. Current haptic devices are external objects to the user. Safety considerations should consider where the device is located such as handheld vs wearable. Constant interaction with the skin can potentially induce irritations to the user. Furthermore, once the device is no longer usable, policy considerations should also prioritize methods to safely dispose and recycle such systems. Throwing away objects that use biological systems to drive outputs can be hazardous to the ecosystem.
One trend in interfaces is the movement beyond wearables to systems that interface directly into the user. In the case of implantables/consumables, extra precaution is needed prior to deploying such systems at a larger scale. Haptic devices (through electro muscle stimulation) and AI systems introduce novel interaction paradigms where the user does not have agency over the outputs of the interaction. The introduction of biological systems will also have agency ramifications in the context of interaction.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Action 1: I will focus on biolab/makerspace leadership for Action 1. The action that this actor should take is rigorous safety onboarding for new members and ethical considerations when designing biological technologies. After onboarding, any new projects/prototypes need to undergo an IRB-like approval process. My proposal is to make a communication channel for all biolab spaces, in which for one to pursue a project in lab A, it must be approved by someone in an external lab B.
Purpose: As biolab spaces and computational tools such as AlphaFold are becoming more accessible to the general public it is important that the onboarding process communicates ethical considerations and the safety of handling biomaterials. Similar to personal fabrication (makerspaces etc.) users can fabricate objects ranging from fidget spinners to weapons without institutional/external approvals. Necessary governing policies need to be enacted that balances the need to rapid prototype and safety. Currently, regulation is done internally which may bias what gets built. Additionally a degree of familiarity with the staff can lead to less “scruntinization”.
Design: Bio makerspaces are unlike academia where there are no external auditors. For this to work, all multiple maker-spaces need to buy-in. Oftentimes these maker-spaces are run by volunteers and this approval process adds additional labor. The user interested in the project could pay a small fee (similar to college admissions) to incentivize the volunteers to read through the application.
Assumptions: I am assuming that the approval process is efficient and that the small fee is enough to incentivize the external reviewers to read through the project. I am also assuming that a penalizing mechanism is not necessary.
Action 2: Federal Regulators are my actor for Action 2 to tackle the issue of user agency. This action would be a law that enforces that bio-interfaces must be removable non-invasibly.
Purpose: The purpose of this action is to ensure that the user intent aligns with the output of the device and prevent unintended consequences. By allowing users to remove the device (similar to a wearable), the user can decide when they are ok with interacting with biological interfaces. This law would discourage implantable interfaces. Open-sourceness also prevents vendor lock-in and allows users to “debug” the circuitry.
Design: To make this work, regulators would need buy-in from other government agencies and the general population. Additionally, the corporations that manufacture the device must also comply with the law. Assumptions: I am assuming that the zeitgeist is in favor of using this bio-interface. Social norms will dictate whether or not the general population will agree with this action. If the utility of a discrete implantable system exceeds the risk and creates a significant advantage, then there may not be an incentive to pass this law.
Risks of Failure and Success: If the cost to implement the implant is lower than the wearable companies will push back on the way this law is written. Furthermore, by focusing this law to discourage implants, if this device is successful then there is a gap in how to dispose of the system.
Action 3: Academic researchers establish mandatory dual-use risk registries for biological interface projects.
Purpose: The purpose of this registry is for researchers to prevent researchers from developing bio-weapons. The registry serves purposes. The first is for the researcher to reflect through the design process. The second is to allow external audits.
Design: I envision an online portal where prior to starting a research project, the researcher will fill out a form. When designing bio-experiments, the researcher must disclose all funding sources and collaboration to ensure that no bad actors are involved. Additionally high-risk design choices (eg. non-conventional, unexplored) must be disclosed. In the context of haptic devices, actuation/vibration above some threshold should raise flags since the body can only perceive a certain amount.
Assumptions: The environment that the researchers operate in will be conducive to this change. Researchers will cooperate with each other (even in a world where funding is tight).
Risks of Failure and Success: In a high pressure publish or perish environment, this adds an additional bureaucratic layer they must go through. Researchers may underreport to perform experiments faster. If this is successful, external actors (non academic) may access the open source registry and reverse engineer successful designs for malicious use.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
X
• By helping respond
Foster Lab Safety
• By preventing incident
X
X
X
• By helping respond
X
Protect the environment
• By preventing incidents
X
X
• By helping respond
Other considerations
• Minimizing costs and burdens to stakeholders
X
X
• Feasibility?
X
X
• Not impede research
X
X
X
• Promote constructive applications
X
X
X
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
I would prioritize option 1. By making biohacker makerspaces more accessible, it has made it easier for any individual to prototype. Unlike academia, and corporations the people that utilize these spaces are hobbyists. It is difficult to regulate activities when people do it for “fun”. Furthermore, friendships are formed when frequenting a location. By adding a proposal step and asking approval from other makerspaces, an informal approval process is created. Even though this increases prototyping time, it ensures an unbiased review. The relevant audience would be other hackerspace leadership and small partnerships could be formed.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
I am new to biology (last time I did this was in highschool) so I am not familiar with the mechanisms and thus may have proposed something not feasible. One reflection I have is that my answers (and slides) reflect an anthropocentric view. The ethical perspective of non-humans and even the biological system that would realize these systems are not explored. If I had that perspective I would answer these questions differently. A governance action that I would have proposed is to compare the benefits of using biology as a substitute for an electromechanical system (at least from a government perspective). The would state law that that if an equal design works electromechanically then a biology-driven device should not be used.
AI Usage:
“Critique the below response: “
I then iteratively update my answer by adding details.
Assignment
Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error Rate: 1e-6. There is 3.2gbp (3.2e9). The error rate is much smaller (a factor of 1e17) compared to the length of the human genome. It is essentially near 0 error and that is how biology handles that discrepancy by doing a proofreading step.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The average human protein is 1036 bp. With 3bp in a codon for an amino acid this would yield around 345 amino acids. There are 4 possible sequences, so 4^{345} is the number of ways to code DNA.
Dr.LeProust
What’s the most commonly used method for oligo synthesis currently?
Coupling with phosphoramidite, Capping unreacted sites, Oxidation, Deblock. Repeat until done.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
I’m assuming error rates in the process? Error is fine but as you increase the length the rate of error can be “felt” more.
Why can’t you make a 2000bp gene via direct oligo synthesis?
The chip dimension is limited? It is also an imperfect process (there is an error rate) and deformations may result in a non-sharp clean peak.
George Church
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine
From Google, the lysine contingency is a reference to Jurassic Park. The dinosaurs at the Park are unable to produce lysine and thus need to consume lysine supplements to stay alive. This prevents the dinosaurs from escaping the park. Since it is an essential amino acid, it can not be synthesized fast enough and would need external supplements. The plan in Jurassic Park doesn’t work since all animals can not produce lysine fast enough and need to consume it.
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-Silico Gel Art
Using Benchling, I imported the Lambda DNA sequence and simulated restriction enzyme digestions with EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. The goal was to design a gel art pattern inspired by Paul Vanouse’s Latent Figure Protocol.
The design depicts a figure in a rocky victory pose with a crying face, using 5 lanes of selected enzyme combinations to form the image.
Full virtual digest — all 7 enzymes (reference):
Gel art design — victory pose with crying face:
The final design uses the following lanes:
Lane 1: Lambda_NEB - BamHI
Lane 2: Lambda_NEB - EcoRV
Lane 3: Lambda_NEB - KpnI
Lane 4: Lambda_NEB - EcoRV
Lane 5: Lambda_NEB - BamHI
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
We performed the wet-lab restriction digest and gel electrophoresis experiment designed in Part 1, following the Gel Art protocol. Photos of the gel were taken and will be uploaded shortly.
In progress — wet lab gel photos to be uploaded from Discord.
Part 3: DNA Design Challenge
3.1 Protein of Interest
I selected beta-casein (Bos taurus), a milk protein. Accession: UniProt P02666
Codon optimization translates a DNA sequence to match the codon preferences of a target organism. Beta-casein is naturally expressed in bovines (Bos taurus), but here I optimized for E. coli since it is a different organism with different preferred codon usage. By remapping the codons, we maximize translational efficiency and protein yield in the new host.
3.4 Protein Production Technologies
With the codon-optimized sequence, the protein can be produced via two approaches:
Cell-dependent: The codon-optimized DNA is introduced into E. coli cells (e.g. via a plasmid). The cell’s own transcription and translation machinery reads the sequence and produces the protein.
Cell-free: The necessary cellular components (ribosomes, tRNA, polymerases, etc.) are extracted and used in vitro to transcribe and translate the DNA into protein, without needing a living cell.
3.5 Transcriptional Protein Diversity (Optional)
Skipped.
Part 4: Twist DNA Synthesis Order
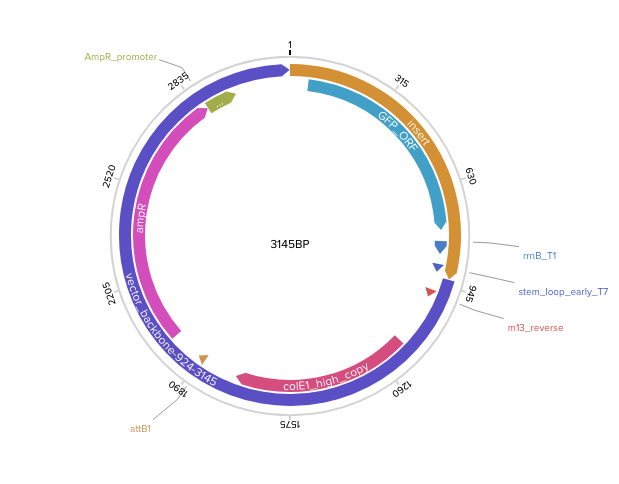
Following the Twist tutorial, I built an expression cassette in Benchling containing a promoter, RBS, start codon, codon-optimized coding sequence, 7x His tag, stop codon, and terminator. The linear sequence was then uploaded to Twist to prepare a clonal gene order, and the resulting construct was imported back into Benchling to verify the plasmid design.
Step 1 — Linear expression cassette in Benchling (924 bp):
Step 2 — Uploaded to Twist (clonal gene order):
Step 3 — Final circular plasmid verified in Benchling (3145 bp, pTwist Amp High Copy):
Part 5: DNA Read/Write/Edit
5.1 DNA Read
In progress.
5.2 DNA Write
In progress.
5.3 DNA Edit
In progress.
Week 3 HW: Opentrons & Lab Automation
Assignment 1: Python Script for Opentrons Artwork
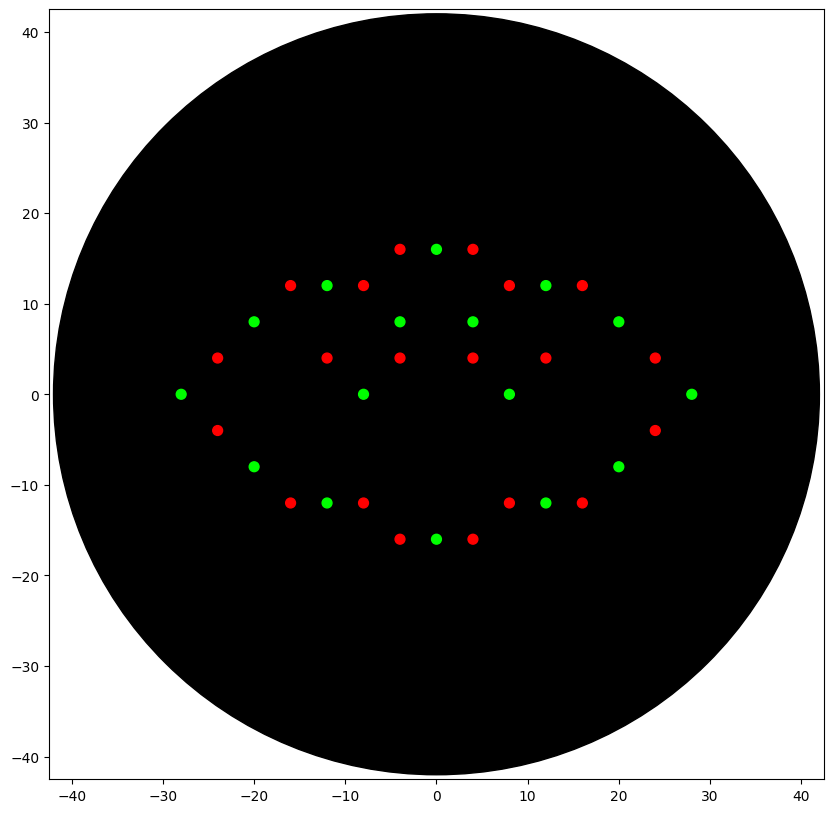
I designed an Indonesian cloud pattern called Megamendung using the Opentrons pipetting robot. The pattern uses two colors — red (mRFP1) and green (Azurite) — arranged as interlocking arcs to replicate the traditional Javanese batik motif.
Simulated output of the Python script:
Python script:
fromopentronsimporttypesmetadata={# see https://docs.opentrons.com/v2/tutorial.html#tutorial-metadata'author':'Aditya Retnanto','protocolName':'Megamendung','description':'Indonesian Cloud Pattern','source':'HTGAA 2026 Opentrons Lab','apiLevel':'2.20'}################################################################################# Robot deck setup constants - don't change these##############################################################################TIP_RACK_DECK_SLOT=9COLORS_DECK_SLOT=6AGAR_DECK_SLOT=5PIPETTE_STARTING_TIP_WELL='A1'well_colors={'A1':'Red','B1':'Green','C1':'Orange'}defrun(protocol):################################################################################# Load labware, modules and pipettes############################################################################### Tipstips_20ul=protocol.load_labware('opentrons_96_tiprack_20ul',TIP_RACK_DECK_SLOT,'Opentrons 20uL Tips')# Pipettespipette_20ul=protocol.load_instrument("p20_single_gen2","right",[tips_20ul])# Modulestemperature_module=protocol.load_module('temperature module gen2',COLORS_DECK_SLOT)# Temperature Module Platetemperature_plate=temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul','Cold Plate')# Choose where to take the colors fromcolor_plate=temperature_plate# Agar Plateagar_plate=protocol.load_labware('htgaa_agar_plate',AGAR_DECK_SLOT,'Agar Plate')## TA MUST CALIBRATE EACH PLATE!# Get the top-center of the plate, make sure the plate was calibrated before running thiscenter_location=agar_plate['A1'].top()pipette_20ul.starting_tip=tips_20ul.well(PIPETTE_STARTING_TIP_WELL)################################################################################# Patterning############################################################################### pass this e.g. 'Red' and get back a Location which can be passed to aspirate()deflocation_of_color(color_string):forwell,colorinwell_colors.items():ifcolor.lower()==color_string.lower():returncolor_plate[well]raiseValueError(f"No well found with color {color_string}")# For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_detach(pipette, 1, loc)defdispense_and_detach(pipette,volume,location):"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
"""assert(isinstance(volume,(int,float)))above_location=location.move(types.Point(z=location.point.z+5))pipette.move_to(above_location)pipette.dispense(volume,location)pipette.move_to(above_location)dispense_amount=0.5mrfp1_points=[(-4,16),(4,16),(-16,12),(-8,12),(8,12),(16,12),(-24,4),(-12,4),(-4,4),(4,4),(12,4),(24,4),(-24,-4),(24,-4),(-16,-12),(-8,-12),(8,-12),(16,-12),(-4,-16),(4,-16)]azurite_points=[(0,16),(-12,12),(12,12),(-20,8),(-4,8),(4,8),(20,8),(-28,0),(-8,0),(8,0),(28,0),(-20,-8),(20,-8),(-12,-12),(12,-12),(0,-16)]pipette_20ul.pick_up_tip()total_aspiration=len(mrfp1_points)*dispense_amountpipette_20ul.aspirate(total_aspiration,location_of_color('Red'))foriinrange(len(mrfp1_points)):adjusted_location=center_location.move(types.Point(x=mrfp1_points[i][0],y=mrfp1_points[i][1]))dispense_and_detach(pipette_20ul,dispense_amount,adjusted_location)pipette_20ul.drop_tip()pipette_20ul.pick_up_tip()total_aspiration=len(azurite_points)*dispense_amountpipette_20ul.aspirate(total_aspiration,location_of_color('Green'))foriinrange(len(azurite_points)):adjusted_location=center_location.move(types.Point(x=azurite_points[i][0],y=azurite_points[i][1]))dispense_and_detach(pipette_20ul,dispense_amount,adjusted_location)pipette_20ul.drop_tip()
Assignment 2: Post-Lab Questions
Published Paper on Lab Automation
Automation for Final Project
Assignment 3: Final Project Ideas
Week 4 — Protein Design Part I
Part 1: Protein Selection
1.1 Protein Choice
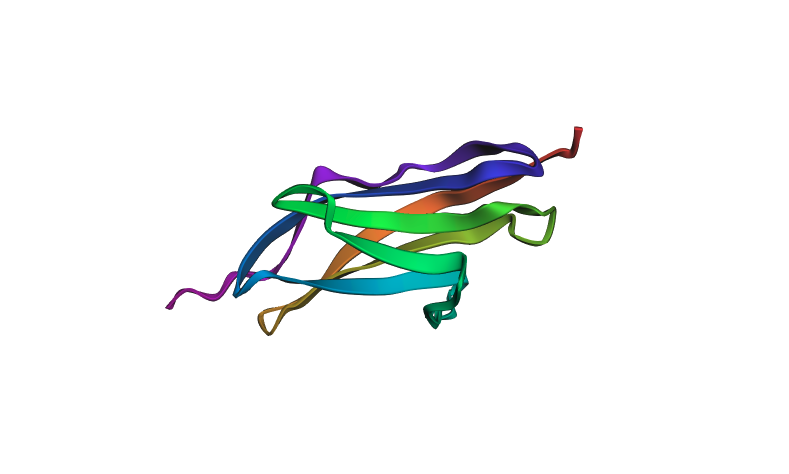
I selected Titin (PDB: 1G1C) because it is a structural protein and acts as the “spring” in human muscle — it is the largest known protein in the human body and is responsible for the passive elasticity of muscle fibers.
1.2 Sequence Properties
Sequence length: The 1G1C fragment of Titin is 146 amino acids.
Most frequent amino acid: V (Valine, 10 occurrences)
Protein family: Immunoglobulin (Ig) Superfamily
1.3 Structure Analysis (RCSB)
Deposition date: October 11, 2000
Resolution: 2.70 Å — considered a medium-quality structure (smaller resolution values are better)
Other molecules in structure: None — Titin is purely a structural protein and does not bind ligands or cofactors.
The protein was visualized in PyMOL in multiple representations.
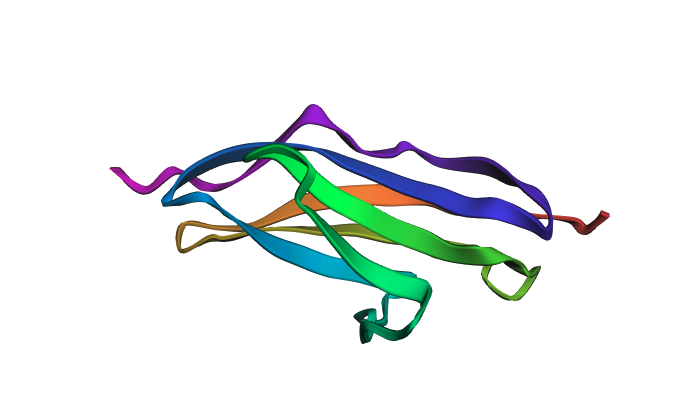
Cartoon view colored by secondary structure:
Ball-and-stick / surface view:
The structure is predominantly beta-sheet (beta-sandwich architecture), consistent with Titin’s role as a mechanical spring — the Ig domain folds into two antiparallel beta-sheets packed face-to-face. Hydrophobic residues are concentrated at the core of the sandwich, while hydrophilic residues are on the exterior surface. The protein does not have obvious binding pockets, as expected for a purely structural domain.
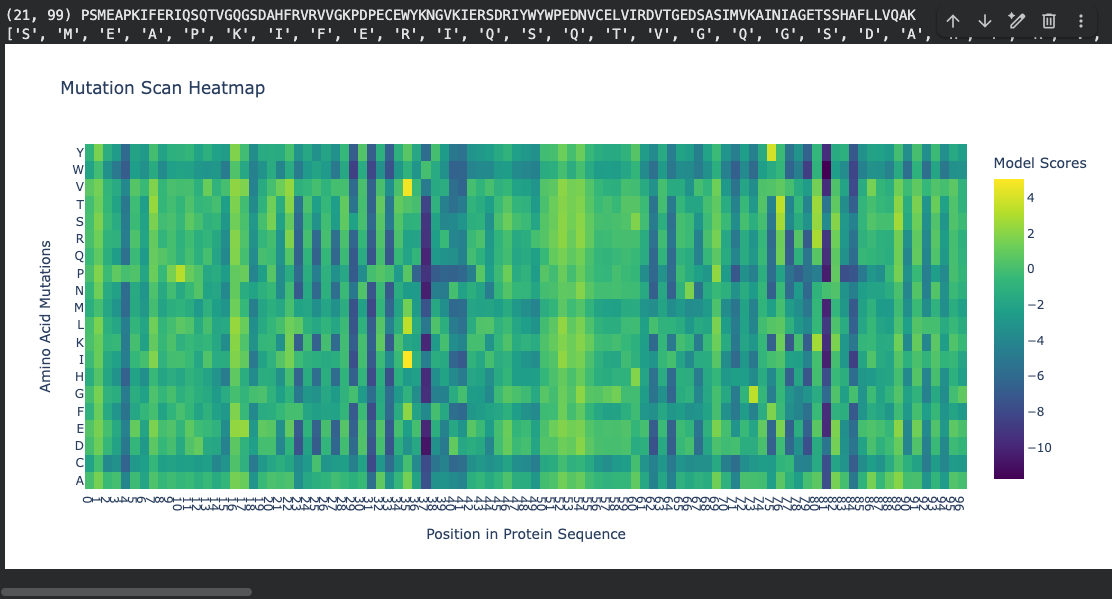
Part 2: Deep Mutational Scan
Using ESM2 (esm2_t6_8M_UR50D), I generated an unsupervised deep mutational scan by masking each position in the sequence and computing log-likelihood ratios (LLR) for every possible amino acid substitution.
Mutation scan heatmap (relative LLR scores):
Notable pattern: At positions ~37 and ~81, many amino acid substitutions show strongly negative log-likelihood scores (around −10). According to literature, these positions fall within the hydrophobic core of the beta-sandwich. The ESM2 language model correctly identifies that mutations at buried hydrophobic positions are highly unfavorable — substituting a core hydrophobic residue with a polar or charged amino acid would destabilize the fold. This is represented by the dark regions in the heatmap.
Part 3: Latent Space Analysis
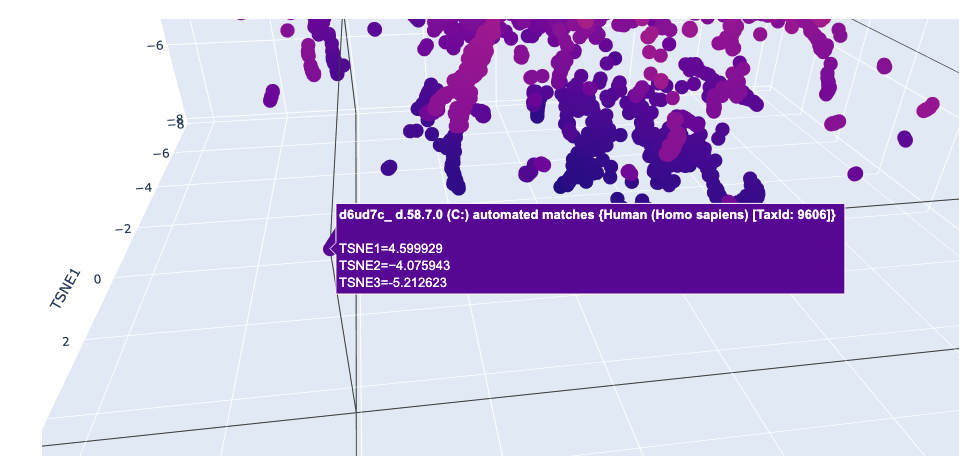
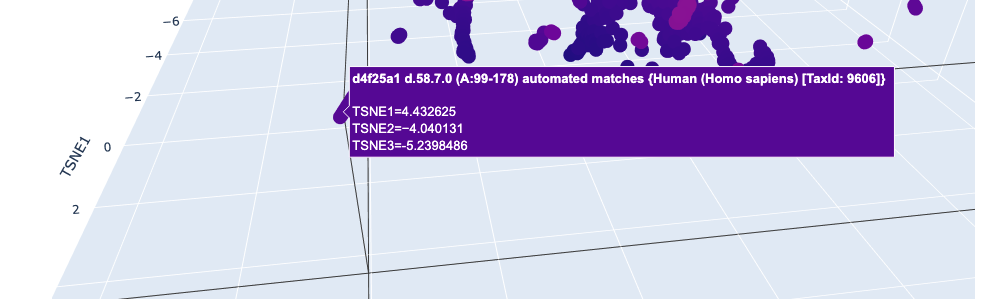
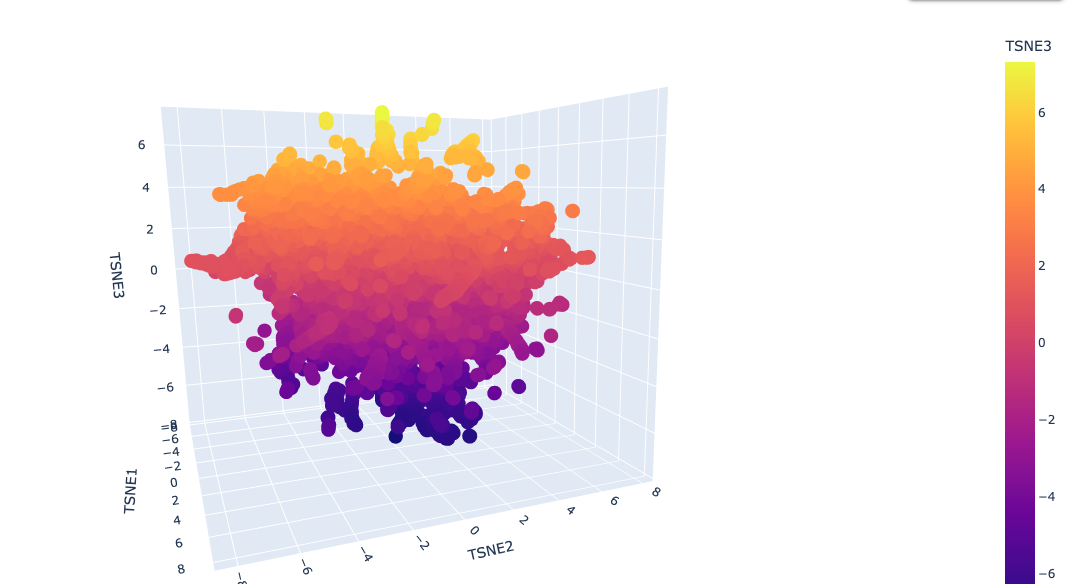
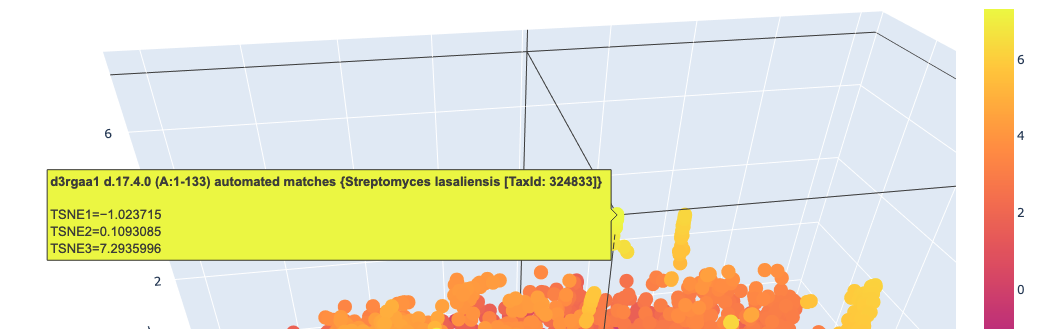
I used ESM2 to embed ~15,000 protein sequences from the SCOP 2.08 database (40% sequence identity cutoff) and reduced the dimensionality with 3D t-SNE for visualization.
Full latent space (t-SNE):
Nearby neighbors of Titin:
Distant proteins in latent space:
Analysis: The t-SNE map clusters proteins into distinct neighborhoods that broadly correspond to structural and functional families. Proteins near Titin in the latent space are predominantly other Ig-fold / beta-sandwich proteins, confirming that ESM2 embeddings capture structural similarity. Proteins in distant regions of the map tend to be alpha-helical or enzymatic proteins with very different sequence composition.
Part 4: Protein Folding with ESMFold
I folded the 1G1C sequence (146 residues) using ESMFold.
Folding results:
pTM score: 0.881 (high confidence in overall topology)
Mean pLDDT: 92.1 (high per-residue confidence)
Predicted structure (pre-mutation):
Predicted structure after mutations:
The predicted structure closely matches the experimentally determined 1G1C coordinates — the beta-sandwich topology is correctly recovered. When introducing point mutations (especially at surface-exposed residues), the predicted structure remains largely intact, showing that the Ig-fold is quite resilient to mutations at non-core positions. Larger segment substitutions begin to perturb the fold more noticeably.
Part 5: Inverse Folding with ProteinMPNN
Using the backbone of PDB 5MBA (chain A, 146 residues) as input to ProteinMPNN (v_48_020, noise level 0.2Å), I generated a new sequence candidate via inverse folding.
The model predicted sequence probabilities show high confidence at structurally constrained core positions (low entropy) and more variability at surface-exposed loops, which is consistent with the known structure-sequence relationship in Ig-fold proteins.
Comparison: Feeding the ProteinMPNN-generated sequence back into ESMFold yields a structure that closely matches the original 5MBA backbone (sequence recovery ~52% with a low sampling temperature of 0.1), demonstrating that ProteinMPNN successfully captured the backbone geometry even while proposing a distinct sequence.
Notebook
The full Colab notebook with all code and outputs is embedded below.
Week 5 — Protein Design Part II
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. Mutations in SOD1 cause familial ALS — among them, the A4V mutation (Alanine → Valine at residue 4 of the mature protein) is one of the most aggressive, destabilizing the N-terminus and promoting toxic aggregation.
The goal of this section is to design short peptides that bind mutant SOD1, then evaluate and optimize them using a multi-tool pipeline.
Part 1: Generate Binders with PepMLM
The A4V mutant SOD1 sequence was obtained from UniProt (P00441) with the substitution introduced at index 4 (0-based):
This sequence was input to PepMLM-650M with peptide_length = 12 and num_binders = 4. PepMLM uses masked language modeling conditioned on the target protein to generate peptide binders — lower pseudo-perplexity indicates higher model confidence.
Generated peptides:
#
Peptide
Pseudo-Perplexity
Notes
0
WRYYAVAAELKA
9.42
Strong — W anchor, charged tail
1
WHYGAVVAAWGA
9.06
Best confidence — dual aromatic (W, Y)
2
WLYYVVVVEHKA
23.64
Weakest — VVVV stretch suggests model uncertainty
3
WRYYAVAAAHKE
11.13
Clean sequence (no X token), balanced
—
FLYRWLPSRRGG
—
Known SOD1 binder (benchmark)
Note: Peptides 0–2 originally ended with an X (unresolved mask token). Substituted with A (Alanine) for downstream structure prediction.
All four generated peptides open with Tryptophan (W) — the model consistently favors an aromatic N-terminal anchor for engaging SOD1. Peptide 2’s VVVV stretch is suspicious and likely reflects low-confidence generation at those positions.
Part 2: Evaluate Binders with AlphaFold3
Each peptide was submitted to AlphaFold3 as a complex with the SOD1 sequence — one job per peptide, run as a Protein + Protein pair with 5 seeds.
Job setup:
Results — ipTM scores across 5 models:
Peptide
SOD1 variant
Model 0
Model 1
Model 2
Model 3
Model 4
Best ipTM
Interpretation
WRYYAVAAELKA
A4V
0.53
0.44
0.33
0.21
0.17
0.53
Borderline — most consistent of PepMLM set
WHYGAVVAAWGA
A4V
0.39
0.37
0.35
0.28
0.25
0.39
Below threshold
WLYYVVVVEHKA
WT†
0.50
0.41
0.38
0.35
0.23
0.50
Borderline — run against WT, not A4V
WRYYAVAAAHKE
A4V
0.25
0.25
0.23
0.19
0.22
0.25
Low confidence
FLYRWLPSRRGG (known)
A4V
0.32
0.31
0.25
0.21
0.19
0.32
Below threshold — poor AF3 prediction
†WLYYVVVVEHKA was inadvertently run against wild-type SOD1 (MATKAVCVLK…) rather than the A4V mutant (MATKVVCVLK…). A repeat run against A4V would be needed to confirm selectivity.
The ipTM score measures AlphaFold3’s confidence in the predicted interface geometry. Scores below 0.4 are generally considered unreliable; scores above 0.6 suggest a credible interaction.
WRYYAVAAELKA is the surprise standout — it scores 0.53 ipTM, the highest of all five peptides including the experimentally validated known binder FLYRWLPSRRGG (0.32). The known binder’s poor AF3 score is a striking reminder that AlphaFold3 does not always predict short peptide interactions reliably, especially for peptides that may bind via induced fit or intrinsically disordered regions. WLYYVVVVEHKA (PeptiVerse’s top pick) scores 0.50 against WT SOD1, keeping it competitive despite being run against the wrong variant. WRYYAVAAAHKE (safest therapeutic profile) is the weakest structurally at 0.25, consistent with its lowest PeptiVerse affinity.
All CIF structure files can be loaded in Mol* or PyMOL for visual inspection of the predicted binding mode.
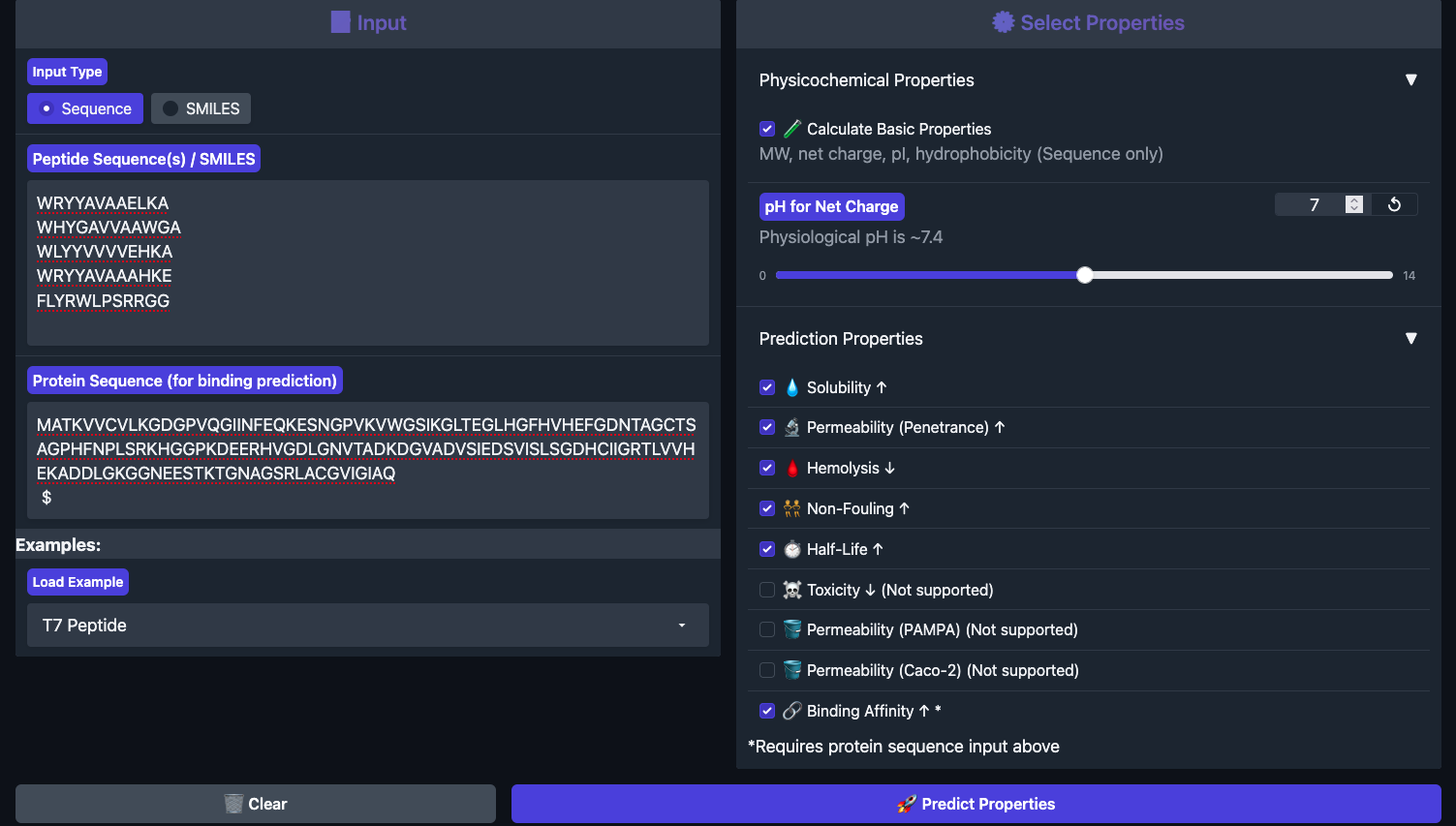
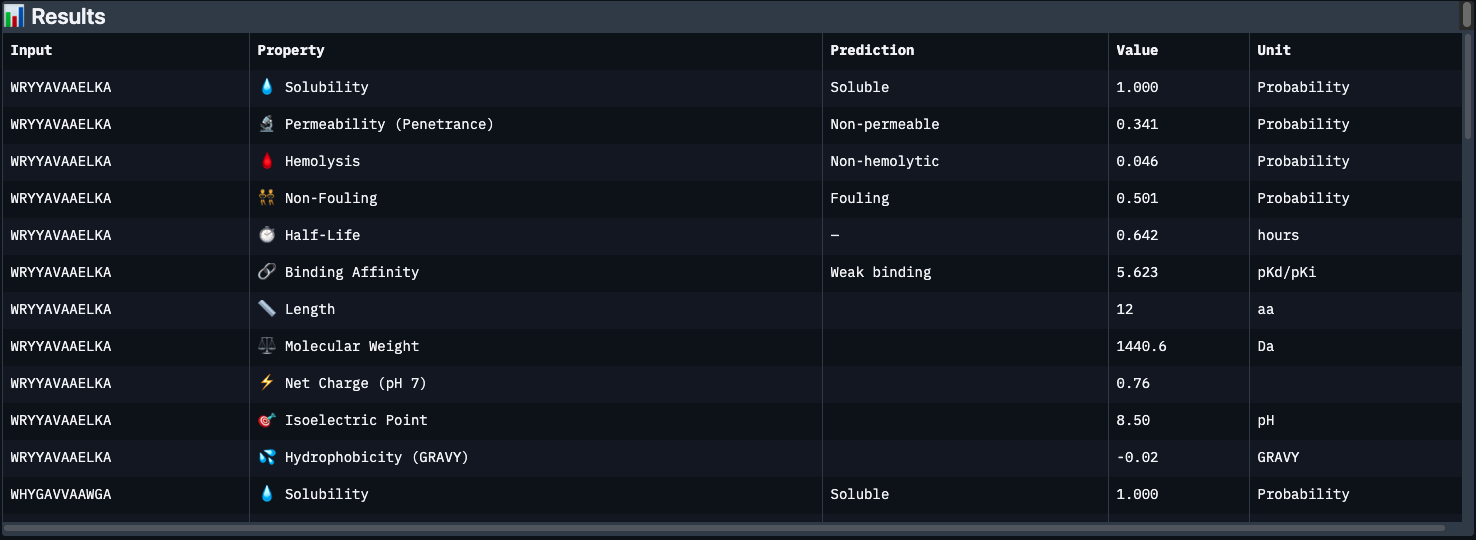
Part 3: Evaluate Therapeutic Properties with PeptiVerse
Each peptide was run through PeptiVerse with the A4V mutant SOD1 as the target sequence. All four generated peptides plus the known binder were entered together in the peptide field, with the full SOD1 A4V sequence pasted into the target protein field.
Results below:
Peptide
Binding Affinity
Solubility
Hemolysis
Non-Fouling
Half-Life (h)
Net Charge
GRAVY
WRYYAVAAELKA
Weak (5.62)
✅ Soluble
✅ 0.046
⚠️ Fouling
0.64
+0.76
-0.02
WHYGAVVAAWGA
Weak (5.93)
✅ Soluble
✅ 0.101
⚠️ Fouling
0.69
-0.15
+0.71
WLYYVVVVEHKA
Medium (7.10)
✅ Soluble
✅ 0.095
⚠️ Fouling
0.68
-0.15
+0.69
WRYYAVAAAHKE
Weak (5.50)
✅ Soluble
✅ 0.018
✅ Non-fouling
0.52
+0.85
-0.60
FLYRWLPSRRGG (known)
Weak (5.55)
✅ Soluble
✅ 0.047
✅ Non-fouling
0.87
+2.76
-0.71
Key observations:
The most striking finding is that WLYYVVVVEHKA (peptide 2) — which had the worst perplexity score (23.64) — has the best predicted binding affinity at 7.10 pKd (Medium binding). This is the only peptide to break out of the “Weak binding” category. Its hydrophobic VVVV stretch (GRAVY +0.69) likely engages a buried hydrophobic patch on SOD1 that the language model considered unlikely but PeptiVerse scores favorably.
Conversely, WRYYAVAAAHKE has the best safety profile — lowest hemolysis risk (0.018), non-fouling, and permeable — but the weakest binding affinity. It’s the cleanest therapeutic candidate structurally, but may not engage SOD1 strongly enough.
The known binder FLYRWLPSRRGG only scores 5.55 pKd (Weak), despite being experimentally validated. This is a reminder that PeptiVerse’s binding predictions are approximate — structural confirmation from AlphaFold3 is essential.
All peptides share a common weakness: half-lives under 1 hour, which would be a major barrier to clinical use without chemical modification (e.g. PEGylation, D-amino acid substitution, cyclization).
Peptide selected for advancement: WLYYVVVVEHKA
Despite its low language model confidence, it is the only peptide with medium predicted binding affinity and acceptable hemolysis/solubility profiles. The next step would be to validate its binding mode structurally (AlphaFold3) and chemically stabilize the VVVV core to improve half-life.
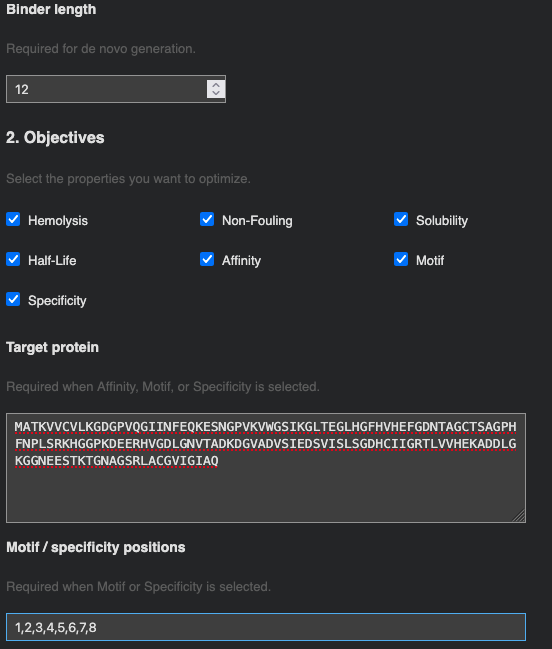
Part 4: Optimized Design with moPPIt
moPPIt (Multi-Objective Guided Discrete Flow Matching) takes a different approach from PepMLM: rather than sampling from a language model conditioned on the target, it uses guided flow matching to simultaneously optimize multiple therapeutic properties while steering the peptide toward a specific binding site on the protein.
Target residue selection: Residues 1–8 of SOD1 (the N-terminal region) were chosen as the binding patch. This region encompasses the A4V mutation site directly — the A→V substitution at position 4 alters the local hydrophobic packing and exposes a surface that is distinct from wild-type SOD1. Targeting this patch means a designed binder could in principle distinguish mutant from wild-type SOD1, which is important for selectivity in a therapeutic context.
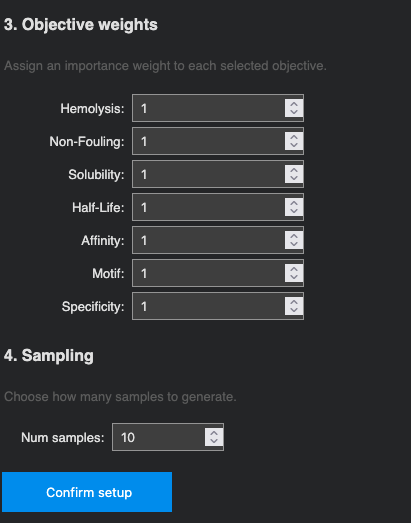
Optimization objectives: All seven properties were selected and weighted equally (weight = 1): Hemolysis ↓, Non-Fouling ↑, Solubility ↑, Half-Life ↑, Affinity ↑, Motif ↑, and Specificity ↑. 10 samples were requested.
Generated sequences (10 samples):
#
Peptide
Affinity (pKd)
Hemolysis
Non-Fouling
Half-Life (h)
Motif Score
1
EKTQLQVDGKQW
5.726
0.085
0.960
1.279
0.819
2
TAEEFQPPSTNH
5.569
0.085
0.903
0.857
0.666
3
PSTKWIQQQHGT
5.645
0.069
0.894
1.137
0.578
4
PAAGILNQKQTT
5.418
0.073
0.835
1.470
0.644
5
PPPPETAEQLWK
5.926
0.046
0.962
1.673
0.240
6
TPETREGPPQIW
6.075
0.050
0.964
1.306
0.003
7
EWTPPLLAGPTL
6.100
0.042
0.907
1.585
0.013
8
TPEQLQDPKLWK
5.947
0.049
0.944
2.196
0.407
9
RRKRKETPHPCC
6.201
0.051
0.976
0.753
0.027
10
FAGALPQGNPTS
5.594
0.048
0.824
1.162
0.038
Key observations:
The moPPIt sequences are strikingly different from PepMLM’s output. Where PepMLM consistently anchored peptides with an N-terminal Tryptophan (W), moPPIt generates sequences with no dominant amino acid pattern — the optimizer is free to explore sequence space guided by the objective landscape rather than by learned sequence co-occurrences.
Therapeutically, all 10 sequences are substantially better than the PepMLM candidates: every peptide is soluble (1.000), hemolysis risk is uniformly low (< 0.09), and non-fouling scores are all above 0.82. Most importantly, half-lives range from 0.75–2.20 hours, with several breaking the 1.5h barrier — compared to PepMLM’s dismal 0.52–0.69h range. This directly addresses the most critical weakness of the PepMLM candidates.
The trade-off is motif score: samples 6, 7, 9, and 10 show motif scores close to 0, meaning the optimizer drifted away from the target N-terminal patch in favor of better global therapeutic properties. Samples 1 (motif 0.819) and 2 (motif 0.666) best maintain engagement with the intended residues 1–8, but at the cost of lower affinity.
Candidate comparison:
Best affinity + safety:RRKRKETPHPCC (pKd 6.201, hemolysis 0.051, NF 0.976) — highest binding affinity of all 10, excellent safety profile, but short half-life (0.75h) and low motif score suggest it may bind elsewhere on SOD1.
Best half-life:TPEQLQDPKLWK (2.196h, pKd 5.947) — best in-vivo persistence of any peptide across both methods.
Best patch engagement:EKTQLQVDGKQW (motif 0.819) — most likely to target the A4V site specifically, though affinity is modest (5.726).
PepMLM vs. moPPIt — overall comparison:
moPPIt substantially outperforms PepMLM on therapeutic properties (solubility, hemolysis, half-life) but the two methods probe complementary aspects of binder design. PepMLM’s W-anchored peptides reflect the language model’s learned preferences for aromatic contacts with SOD1; moPPIt’s diverse sequences reflect pure multi-objective optimization. In a real pipeline, both would be tested structurally (AlphaFold3 / MD simulation) before selecting leads for synthesis.
Steps before clinical studies: To advance any of these peptides, the next steps would be: (1) AlphaFold3 structural validation to confirm binding mode and ipTM score at the A4V patch; (2) MD simulation to assess binding stability; (3) chemical stabilization (D-amino acid substitution, cyclization, or PEGylation) to improve half-life beyond 2h; (4) in-vitro aggregation inhibition assay with recombinant SOD1 A4V; (5) cell viability assay in motor neuron cell lines.
Part C: L-Protein Mutants (MS2 Phage)
Background
Bacteriophage MS2 is a single-stranded RNA virus that infects E. coli. Upon infection, the phage’s L-protein forms oligomeric pores in the bacterial cell membrane, ultimately causing lysis and release of new phage particles. The L-protein is 75 amino acids long and has two functional regions:
Soluble N-terminal domain (residues 1–40):METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYV — responsible for interaction with the bacterial chaperone DnaJ
A key E. coli resistance mechanism is a point mutation in DnaJ that disrupts its interaction with the L-protein soluble domain, stalling L-protein processing and preventing lysis. The engineering goal is therefore to design L-protein variants that either (1) fold and function independently of DnaJ, or (2) lyse the host faster, reducing the window for resistance acquisition.
Option 1: Mutagenesis via ESM Mutation Scoring
Step 1 — Run the Mutation Scan Notebook
Using the ESM-based mutation scoring notebook, a per-position log-likelihood score was computed for every possible amino acid substitution across the 75-residue L-protein. Positive scores indicate substitutions the language model considers favorable; negative scores indicate destabilizing changes.
Mutation score heatmap — in progress.
Step 2 — Correlation with Experimental Data
The L-Protein Mutants spreadsheet contains experimentally measured lysis activity for a set of known single-point mutants.
Correlation analysis — in progress.
Step 3 — Proposed Mutations
Using the ESM scores, the experimental mutant data, and the ClustalOmega multiple sequence alignment (to avoid conserved residues), the following five mutations were selected:
Mutations — in progress.
Design constraints applied:
At least 2 mutations in the transmembrane domain (residues 41–75)
At least 2 mutations in the soluble domain (residues 1–40)
No mutations at positions that are fully conserved across the ClustalOmega alignment of BLAST homologs
Option 2: AF2-Multimer Co-folding with DnaJ
In progress — AlphaFold2-Multimer jobs pending.
The L-protein was co-folded with the DnaJ sequence using ColabFold AF2-Multimer to visualize the predicted interaction interface. The query format chains the two sequences with a : separator:
Note: AF2 is not well-optimized for membrane proteins, so predictions for the TM domain are expected to be low-confidence. The co-fold is primarily useful for identifying which soluble-domain residues contact DnaJ in the predicted complex.
Multimeric Assembly
The L-protein is hypothesized to form oligomeric pores in the membrane. To visualize a plausible assembly, an 8-mer was submitted to AlphaFold2-Multimer using the repeated sequence format:
According to the NEB product page, the Phusion master mix is built around two key features of the Phusion polymerase itself:
A Pyrococcus-like polymerase core with a 3’→5’ proofreading exonuclease — catches and corrects misincorporated bases in real time, giving ~50x lower error rate than Taq.
An Sso7d processivity domain fused to the polymerase — a DNA-binding clamp that keeps the enzyme on the template, increasing speed and fidelity together.
The master mix also includes dNTPs, MgCl₂ (essential cofactor for polymerase activity), and a reaction buffer optimized for Phusion.
2. Factors That Determine Primer Annealing Temperature
The annealing temperature is set based on the primer’s melting temperature (Tm) — the temperature at which half the primer-template duplexes dissociate. Two main factors drive Tm:
GC content — G-C base pairs form 3 hydrogen bonds vs. 2 for A-T pairs, so GC-rich primers require more energy to dissociate and have a higher Tm.
Primer length — longer primers have more total bonds to the template, raising Tm.
Salt concentration also plays a minor role — higher salt stabilizes the duplex and raises Tm slightly.
In practice, Tm is calculated using the NEB Tm Calculator with Phusion selected as the polymerase. NEB recommends setting the annealing step at Tm or Tm +3°C for Phusion, which is higher than for Taq due to Phusion’s greater processivity. In a standard PCR cycle this sits between the denaturation step (94–96°C) and extension (72°C). The ideal annealing temperature in practice is 52–58°C — going above 65°C risks secondary annealing, where primers bind non-specifically to off-target sites.
3. PCR vs. Restriction Enzyme Digests
This is essentially the distinction between scarred and scarless cloning approaches.
Restriction enzyme digests cut at specific recognition sequences and create sticky-end overhangs. The recognition sequence remains in the final construct — leaving a scar at the junction. Simple and fast when compatible sites already exist on the insert and vector, but you’re constrained by where those sites naturally appear.
PCR lets you directly extract any region regardless of restriction sites, and primer tails can add any overhang, mutation, or homology region needed. This is what makes scarless assembly methods like Gibson possible — the overlap is designed directly into the primers, with no recognition sequence left behind.
Prefer RE digests when compatible sites already exist and the workflow needs to be simple. Prefer PCR when no convenient sites exist, when precise scarless junctions are needed, or when building multi-fragment assemblies.
4. Ensuring Sequences are Compatible for Gibson Cloning
Two things need to be in place before Gibson Assembly will work:
Overlaps — Gibson requires identical sequence at every junction between adjacent fragments. These overlaps don’t exist naturally, so they’re designed into the PCR primers: each primer has two regions — an 18–22 bp binding region that anneals to the template during PCR, followed by a 20–22 bp overhang matching the end of the adjacent fragment. The PCR product therefore comes out with the overlap already attached. The Gibson mix’s 5’ exonuclease chews back those ends to expose single-stranded overhangs that anneal, get filled, and get ligated into a seamless join. Overlaps should be verified in Benchling or SnapGene before ordering — align fragments and confirm homology at each junction, avoiding repetitive sequences or secondary structures in the overlap region.
Codon optimization — when codon-optimizing the insert sequence, the algorithm can freely swap synonymous codons and may accidentally introduce restriction enzyme recognition sequences inside the coding sequence. While this doesn’t affect Gibson directly, it can cause problems if restriction enzymes are used elsewhere in the workflow. Tools like IDT and Twist allow you to blacklist specific restriction sites during optimization, which is worth doing as a precaution before ordering.
5. How Does Plasmid DNA Enter E. coli During Transformation?
In the lab we used chemical transformation (heat shock). The process works in three stages:
Ice — competent cells (prepared with CaCl₂, which partially destabilises the membrane) are mixed with plasmid DNA and kept on ice. The cold temperature keeps everything stable and allows the negatively charged DNA to associate with the outer membrane surface.
Heat bath (42°C, ~45 sec) — the brief temperature spike creates transient pores in the membrane, through which the DNA enters the cell.
Back to ice — immediately halts the heat shock and stabilises the cells before recovery in SOC medium at 37°C. Cells are then pelleted by centrifuge and plated on selective agar.
An alternative method is electroporation, where a brief high-voltage pulse creates temporary membrane pores. It offers higher transformation efficiency and works well for large plasmids or low DNA concentrations, but requires specialised cuvettes and equipment.
6. Golden Gate Assembly
Two useful analogies help distinguish Gibson and Golden Gate:
Gibson Assembly is like a zipper. PCR primers add long overlapping sequences (~25 bp) to each fragment end. An exonuclease then rips back one side, exposing a single-stranded tail — which finds its complement on the adjacent fragment and zips together. It works because the sequences are identical mirrors of each other.
Golden Gate is like melodies blending seamlessly. Rather than overlapping, each fragment ends with a short 4-note motif (a 4-nt sticky end) that hands off directly into the opening motif of the next fragment. One musical phrase resolves into the next — no repeated section, no scar. The BsaI recognition site is like an intro riff that gets cut away before the song properly starts: it does its job generating the sticky end, then disappears from the final construct entirely.
Because each 4-nt overhang is unique, every fragment knows exactly which neighbour to find — making it possible to assemble many fragments simultaneously in a single pot reaction, with high efficiency and no scars at junctions.
Part B: Asimov Kernel
Note: Part B uses the Asimov Kernel platform. As a Global Committed Listener I did not have access to Asimov during the course — this section is left incomplete.
The Repressilator
The Repressilator is a synthetic genetic oscillator built from three transcriptional repressors wired in a loop: LacI represses tetR, TetR represses cI, and CI represses lacI. This negative feedback loop creates sustained oscillations in protein levels.
Repressilator construct — in progress.
Custom Constructs
Construct 1
In progress.
Construct 2
In progress.
Construct 3
In progress.
Week 7 — IANNs, Fungal Materials & DNA Design
Part 1: Intracellular Artificial Neural Networks (IANNs)
1. Advantages of IANNs over Boolean Genetic Circuits
Traditional genetic circuits implement Boolean logic — AND, OR, NOT gates — where every output is binary: a gene is either on or off. IANNs offer three key advantages over this:
Custom activation functions — Boolean logic is just a special case of an IANN. By choosing an activation function (e.g. a sharp step = Boolean; ReLU or tanh = analog), you can implement Boolean circuits as a subset or go beyond them entirely without redesigning the underlying architecture.
Non-linearity — non-linear activation functions allow the network to separate input combinations that a linear system can’t distinguish, enabling richer input-output mappings from the same number of genetic components.
Graded, continuous inputs — real biological signals (transcription factor concentrations, metabolite levels) exist on a spectrum, not just “present” or “absent.” IANNs integrate these natively without thresholding, preserving signal information that Boolean circuits discard.
A knitted, soft-robotic wearable that autonomously regulates body temperature using two biological sensors and an IANN to integrate them:
Input X1: sweat biosensor — detects sweat concentration (NaCl, lactate) as a continuous electrochemical signal
Input X2: mechanosensor — detects muscle vibration frequency as a proxy for shivering
The IANN hidden layer integrates both graded signals with learned weights. The key insight is that neither input alone is sufficient: a wearer can be slightly sweaty and slightly shivering simultaneously (e.g. after exercise in cold weather), and the correct actuator response is neither fully open nor fully contracted. A Boolean circuit would have to threshold each signal independently and lose that nuance; the IANN interpolates a response proportional to both.
Output: soft pneumatic or shape-memory actuators woven into the fabric — high sweat + no shiver → structure opens (ventilates, cools); low sweat + high shiver frequency → structure contracts (insulates, retains heat)
Limitations: protein-based signal transduction is orders of magnitude slower than electronic sensing; biological noise and signal crosstalk between the two inputs could cause erratic actuation; and the IANN weights would need to be tuned to individual wearers’ sweat profiles and shiver thresholds.
3. Diagram: Intracellular Multilayer Perceptron
The assignment provides a single-layer perceptron where:
X1 = DNA encoding Csy4 endoribonuclease
X2 = DNA encoding a fluorescent protein whose mRNA is regulated by Csy4
A multilayer perceptron adds a hidden layer between input and output. In the intracellular context, layer 1 would express an endoribonuclease that post-transcriptionally regulates layer 2 outputs.
Part 2: Fungal Materials
1. Existing Fungal Materials
The most compelling recent work on fungal materials comes from Jasmine Lu, whose research uses living mycelium as an interactive interface — essentially a biological tamagotchi. The fungus responds to care (light, humidity, touch) and the project deliberately questions what kind of relationship users develop with a living material versus a digital one. It’s a provocation as much as a technology: if your interface is alive, do you feel responsible for it? This framing is particularly relevant for wearables and soft robotics, where the boundary between tool and organism starts to blur.
At the infrastructure level, mycelium has also been shown to transmit electrical signals along hyphal networks — Andrew Adamatzky’s work on “fungal computers” demonstrated measurable voltage spikes in response to chemical and physical stimuli, opening the door to mycelium as a biological sensing and signaling substrate.
On the commercial side, several fungal materials have reached market:
Mycelium composites (Ecovative) — mycelium grown on agricultural waste forms a lightweight, biodegradable foam used for packaging and insulation. Produces far less CO₂ than styrofoam; limitation is moisture sensitivity and lower mechanical strength.
Fungal leather (Bolt Threads Mylo) — mycelium-based leather alternative used by Stella McCartney. Sustainable alternative to animal hide; scaling and long-term durability remain challenges.
Mycoprotein / Quorn — Fusarium venenatum fermented into a high-protein meat substitute. Established at scale; some allergenic potential in sensitive individuals.
Chitin — extracted from fungal cell walls, used in wound dressings and sutures. Biodegradable, biocompatible, and naturally antimicrobial.
Mycoremediation — species like Pleurotus ostreatus (oyster mushroom) can degrade petroleum, heavy metals, and some plastics. Paul Stamets has championed this as a low-cost environmental remediation tool.
Koji / Tempeh — Aspergillus oryzae and Rhizopus species underpin miso, sake, and soy sauce fermentation, and are now being explored as direct protein sources in their own right.
2. Genetic Engineering of Fungi
A useful way to frame fungal synthetic biology is to contrast it with bacterial engineering. In a project like synthesising casein from E. coli, bacteria are the fabricator — they express and secrete the protein, but the material itself is what they produce, not what they are. Mycelium flips this: the organism is the material. You’re not harvesting what it makes, you’re growing the structure itself.
This distinction shapes what genetic engineering of fungi looks like. Rather than inserting a production pathway, you’d engineer the fungus’s own structural properties — chitin composition, hyphal branching density, surface chemistry — to tune the material directly. For example:
Mechanical tuning — alter chitin synthase expression to make mycelium stiffer or more flexible on demand, useful for structural composites or soft robotics
Electrical conductivity — engineer metalloprotein networks or conductive surface coatings into hyphae for embedded sensing
Growth control — tune branching patterns to direct self-assembly into specific geometries without a mold
Fungi also offer practical advantages over bacteria for material applications: they self-assemble into complex 3D networks with no bioreactor required; their eukaryotic machinery supports post-translational modifications (glycosylation, disulfide bonds) needed for mammalian proteins; they grow on agricultural waste with minimal inputs; and many species are GRAS, lowering the regulatory barrier for consumer products.
Part 3: DNA Twist Order
Final Project DNA Design
In progress.
DNA Twist order submitted. Sequences and vector design finalized and sent for synthesis.
Week 9 — Cell-Free Protein Synthesis
General Homework Questions
1. Advantages of Cell-Free Protein Synthesis
Cell-free protein synthesis offers several advantages over in vivo expression:
Speed — no transformation, culture growth, or induction cycle. Protein can be expressed in hours rather than days or weeks.
Open system / debuggability — with no cell wall in the way, you can directly add, remove, or adjust components mid-reaction. Troubleshooting is hands-on: swap the energy system, add chaperones, tweak Mg²⁺ — all in real time.
Biosafety — no live GMOs means no antibiotic resistance cassettes spreading in the environment and a lower containment burden overall.
Toxic proteins — proteins that would kill a host cell can be expressed freely in lysate.
Non-natural amino acids — easier to incorporate than in living cells where the genetic code is fixed.
Two use cases where cell-free is preferred:
Biosafety-sensitive contexts — e.g. designing and testing antimicrobial peptides or toxins without engineering live bacteria that carry resistance genes.
Rapid prototyping — throughput per reaction is lower than in vivo, but the cycle time collapses from weeks to hours. Testing whether a protein variant folds or a genetic circuit fires becomes feasible at a pace that in vivo expression simply can’t match.
2. Components of a Cell-Free Expression System
A cell-free system is best understood as a build-your-own PC versus a sealed Mac. A living cell is like a Mac — you can give it inputs and read outputs, but everything happens inside a locked enclosure you can’t open. Cell-free lysate cracks that enclosure open and lays all the components on the table, letting you tinker directly with the machinery.
The key components and their roles:
Cell lysate — the burst contents of E. coli cells; contains all the hardware needed for transcription and translation: ribosomes, tRNA, elongation factors, RNA polymerase, and chaperones. This is the motherboard everything else plugs into.
DNA template — the software. Can be circular (plasmid) or linear (PCR product) — unlike in vivo expression, you don’t need to clone into a vector first. Swap in a different sequence and you get a different protein from the same hardware.
NTPs (ATP, GTP, CTP, UTP) — nucleotide building blocks for transcribing the DNA into mRNA.
Amino acids — the raw materials for translating mRNA into protein; all 20 must be present.
Energy regeneration system — keeps refuelling ATP as it gets consumed. Like a car that needs a continuous fuel supply, not a single tank fill: phosphoenolpyruvate (PEP) donates phosphate to ADP → ATP continuously throughout the reaction.
Mg²⁺ and K⁺ salts — ionic environment that ribosomes and polymerases require to function correctly.
RNase inhibitors — protect the mRNA from degradation so translation can proceed.
3. Energy Provision and ATP Regeneration
In progress.
4. Prokaryotic vs. Eukaryotic Cell-Free Systems
The choice between prokaryotic and eukaryotic lysate comes down to what protein you’re making and how much of it you need.
E. coli is a single-cell organism — no coordination overhead, no compartments to worry about. Prokaryotic lysate is cheaper and faster to prepare, and because each reaction is essentially independent, you can scale simply by running more reactions in parallel. It’s the right choice for enzymes, screening, and proteins that don’t require eukaryotic post-translational modifications.
Mammalian proteins are a different story. They evolved in a coordinated multi-system environment — glycosylation, signal peptides, disulfide bond formation — and a prokaryotic lysate lacks the machinery to faithfully reproduce that. If you’re targeting a protein that will eventually work in a mammalian context, you should express it in a mammalian (or at minimum eukaryotic) lysate from the start: wheat germ, rabbit reticulocyte, or HeLa-derived extracts. The trade-off is cost and complexity — eukaryotic systems are harder to prepare and less scalable — but for proteins that require correct folding and modification, there’s no shortcut.
Example: GFP or a simple enzyme → E. coli lysate. A glycosylated cytokine like erythropoietin (EPO) → mammalian lysate.
5. Optimizing Cell-Free Expression of a Membrane Protein
In progress.
6. Troubleshooting Low Yield
Three failure modes cover most low-yield scenarios:
Insufficient energy — translation is expensive: every peptide bond costs ~4 ATP. If the energy regeneration system runs dry mid-reaction, the ribosome stalls. Think of it like a car that stops not because the engine is broken but because the fuel ran out. Fix: switch from PEP to a longer-lasting energy source like maltodextrin, which can sustain reactions for over 12 hours.
mRNA degradation (signal integrity loss) — in a cell, mRNA is protected and chaperoned along optimal pathways. Outside that enclosure, RNases in the lysate chew up the transcript before translation can complete. The signal exists at the start but loses integrity before it reaches the output — like a message sent over a noisy channel with too much resistance. Fix: add RNase inhibitors to the reaction and use 5’ UTR sequences known to stabilize mRNA.
Protein misfolding and aggregation — without the cell’s full complement of chaperones guiding co-translational folding, hydrophobic domains that should be buried can stick to each other and aggregate. The protein is produced but can’t be separated into a usable form — it precipitates out of solution. Fix: supplement with chaperones (GroEL/GroES, DnaK), lower the reaction temperature to slow folding kinetics, and reduce Mg²⁺ concentration.
Kate Adamala: Design a Synthetic Minimal Cell
In progress.
Function chosen:In progress.
Membrane composition:In progress.
Encapsulated components:In progress.
Tx/Tl system:In progress.
Measurement:In progress.
Peter Nguyen: Cell-Free Materials Application
In progress.
Domain:In progress.
Pitch:In progress.
Ally Huang: Genes in Space Proposal
In progress.
Week 10 — Mass Spectrometry & Final Project Measurements
Final Project: Measurements Plan
What to Measure
The chimeric casein project has three distinct questions that each need a different measurement approach:
Stage 1 — Did the protein express?
After inducing E. coli with IPTG and lysing the cells, I need to check whether the chimeric protein actually appeared. The primary tool for this is SDS-PAGE (gel electrophoresis for proteins) — the same ladder-based approach used for DNA, but with SDS added to unfold proteins and give them uniform charge so separation is purely by size. Running the pre- and post-induction lysate side by side, I’d look for a new band appearing at ~50 kDa (the expected molecular weight of the chimera). A Western blot using an anti-His antibody would then confirm the band is specifically my His-tagged chimera and not a coincidental band from the host cell.
Stage 2 — Is it the right sequence?
SDS-PAGE confirms size but not sequence. Mass spectrometry (peptide mapping) goes further: the protein is digested with trypsin into small fragments, each fragment’s mass is measured precisely, and those masses are matched against the predicted tryptic peptides of the chimeric sequence. This confirms that the resilin and keratin inserts are present and correctly incorporated — not just that a ~50 kDa protein exists.
Stage 3 — Does it actually behave hygroscopically?
This is the most important question for the project, and it’s a macro-level measurement. The confirmed chimeric protein would be cast into a bioplastic film (using the same washing soda + glycerol protocol as standard casein plastic) and tested against a control film made from unmodified casein:
Curvature response — how many degrees does the film curl when exposed to a humidity change? A larger curl angle than unmodified casein confirms the inserts are adding hygroscopic actuation.
Response time — how many seconds or minutes does it take to reach full curl? Faster response would suggest better water uptake kinetics from the resilin and keratin domains.
Gravimetric water uptake — weigh the dry film, expose to a defined humidity for a fixed time, weigh again. The percentage weight gain directly measures hygroscopic capacity and can be compared to the +48% water-binding potential predicted computationally.
Measurement Technologies
Technology
What it confirms
SDS-PAGE (gel electrophoresis)
Protein expressed at correct molecular weight (~50 kDa)
Western blot (anti-His antibody)
Band is specifically the His-tagged chimera
Mass spectrometry (peptide mapping)
Correct primary sequence including resilin and keratin inserts
Gravimetric assay
Hygroscopic water uptake vs. unmodified casein baseline
Curvature measurement
Actuation response (degrees of curl) at defined humidity
Response time measurement
Kinetics of hygroscopic actuation
Note: The Waters Parts I–V below are in-person lab exercises. As a Global Committed Listener I participated remotely and did not have access to the lab instruments or figures — these sections are left incomplete.
Calculated MW (via ExPASy compute_pi): in progress.
2. Adjacent Charge State MW Calculation
In progress.
3. Charge State of Zoomed-In Peak
In progress.
Waters Part II: Native vs. Denatured eGFP (Optional)
1. Native vs. Denatured Conformations
In progress.
2. Charge State of Peak at ~2800 m/z
In progress.
Waters Part III: Peptide Mapping
1. Lysines and Arginines in eGFP
In progress.
2. Predicted Tryptic Peptides (PeptideMass Tool)
In progress.
3–7. Peptide Map Analysis
In progress.
Waters Part IV: KLH Oligomers (CDMS)
In progress.
Waters Part V: Did I Make GFP?
In progress.
Week 11 — Cloud Labs & Cell-Free Protein Optimization
Part A: Global Pixel Artwork
In progress.
My contribution: I added a single red pixel late in the project to correct a gap in the pattern. A small fix, but satisfying to slot into place.
What I liked: The collaborative nature immediately reminded me of Reddit’s r/Place — the emergent coordination between strangers to build something coherent out of individual dots is genuinely fun to watch. That same energy translates well here, especially with a class-sized group where you can actually track who did what.
Suggestions for improvement: Taking end-of-day screenshots to document how the image evolves over time would add a lot — it’d be interesting to watch the artwork form progressively rather than just seeing the final state. A timelapse of the build would be a nice archive to keep.
Part B: Cell-Free Reagents
Component Roles in the Cell-Free Reaction
In progress.
Differences: 1-Hour PEP-NTP vs. 20-Hour NMP-Ribose-Glucose