Week 4 — Protein Design Part I
Part 1: Protein Selection
1.1 Protein Choice
I selected Titin (PDB: 1G1C) because it is a structural protein and acts as the “spring” in human muscle — it is the largest known protein in the human body and is responsible for the passive elasticity of muscle fibers.
1.2 Sequence Properties
- Sequence length: The 1G1C fragment of Titin is 146 amino acids.
- Most frequent amino acid: V (Valine, 10 occurrences)
- Protein family: Immunoglobulin (Ig) Superfamily
1.3 Structure Analysis (RCSB)
- Deposition date: October 11, 2000
- Resolution: 2.70 Å — considered a medium-quality structure (smaller resolution values are better)
- Other molecules in structure: None — Titin is purely a structural protein and does not bind ligands or cofactors.
- Structure classification: Immunoglobulin-like beta-sandwich fold (SCOP)
1.4 3D Visualization in PyMOL
The protein was visualized in PyMOL in multiple representations.
Cartoon view colored by secondary structure:
Ball-and-stick / surface view:
The structure is predominantly beta-sheet (beta-sandwich architecture), consistent with Titin’s role as a mechanical spring — the Ig domain folds into two antiparallel beta-sheets packed face-to-face. Hydrophobic residues are concentrated at the core of the sandwich, while hydrophilic residues are on the exterior surface. The protein does not have obvious binding pockets, as expected for a purely structural domain.
Part 2: Deep Mutational Scan
Using ESM2 (esm2_t6_8M_UR50D), I generated an unsupervised deep mutational scan by masking each position in the sequence and computing log-likelihood ratios (LLR) for every possible amino acid substitution.
Mutation scan heatmap (relative LLR scores):
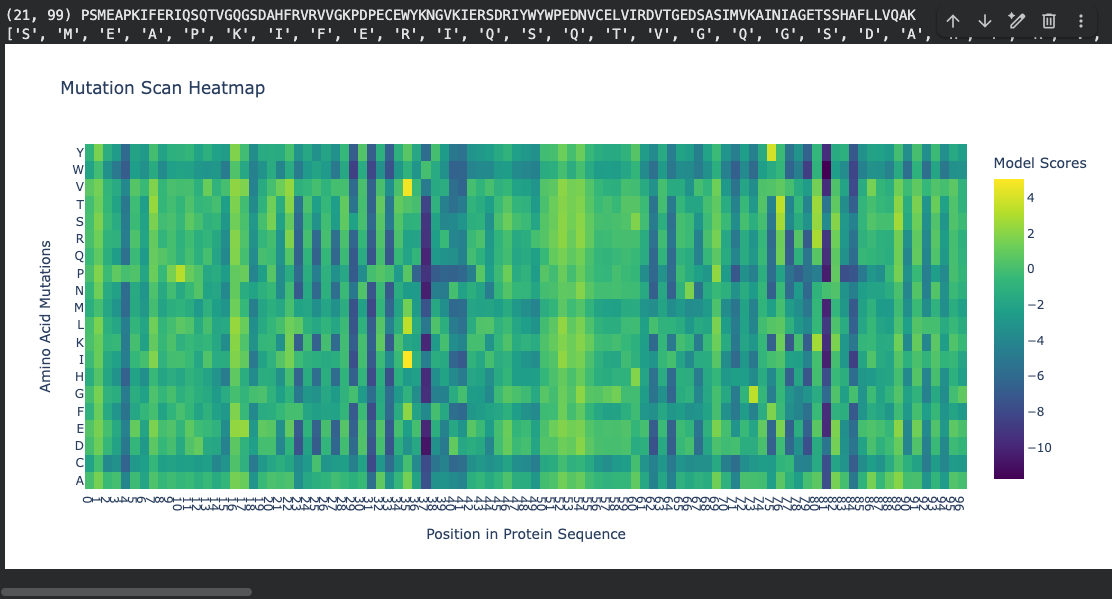
Notable pattern: At positions ~37 and ~81, many amino acid substitutions show strongly negative log-likelihood scores (around −10). According to literature, these positions fall within the hydrophobic core of the beta-sandwich. The ESM2 language model correctly identifies that mutations at buried hydrophobic positions are highly unfavorable — substituting a core hydrophobic residue with a polar or charged amino acid would destabilize the fold. This is represented by the dark regions in the heatmap.
Part 3: Latent Space Analysis
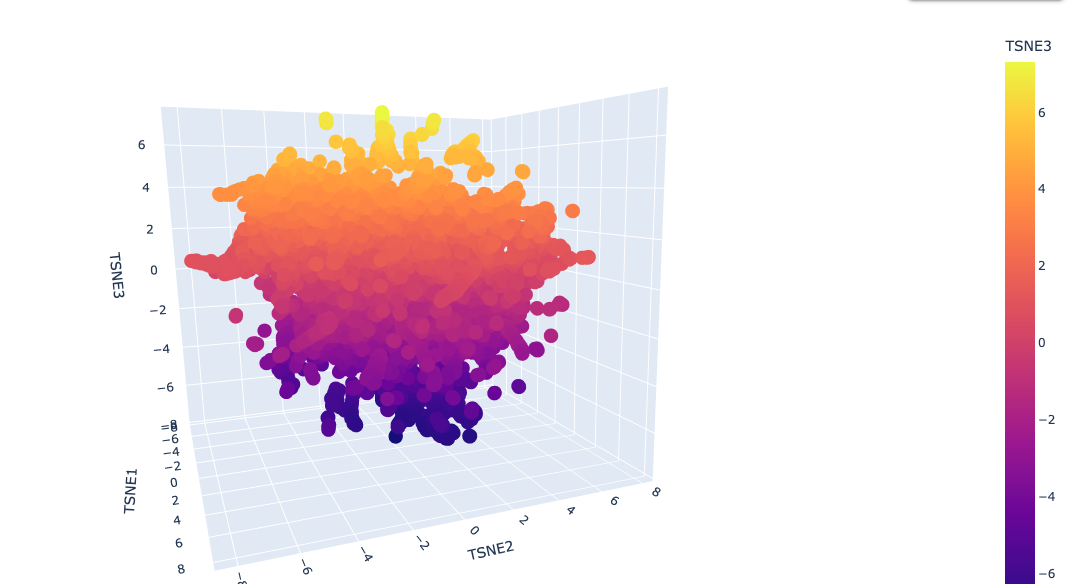
I used ESM2 to embed ~15,000 protein sequences from the SCOP 2.08 database (40% sequence identity cutoff) and reduced the dimensionality with 3D t-SNE for visualization.
Full latent space (t-SNE):
Nearby neighbors of Titin:

Distant proteins in latent space:
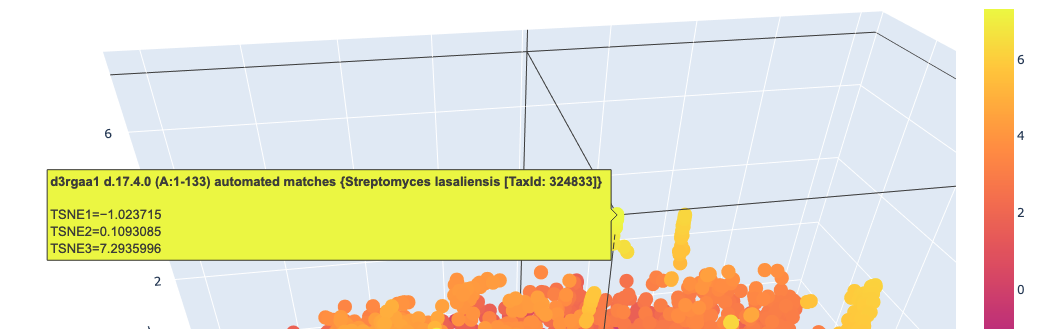
Analysis: The t-SNE map clusters proteins into distinct neighborhoods that broadly correspond to structural and functional families. Proteins near Titin in the latent space are predominantly other Ig-fold / beta-sandwich proteins, confirming that ESM2 embeddings capture structural similarity. Proteins in distant regions of the map tend to be alpha-helical or enzymatic proteins with very different sequence composition.
Part 4: Protein Folding with ESMFold
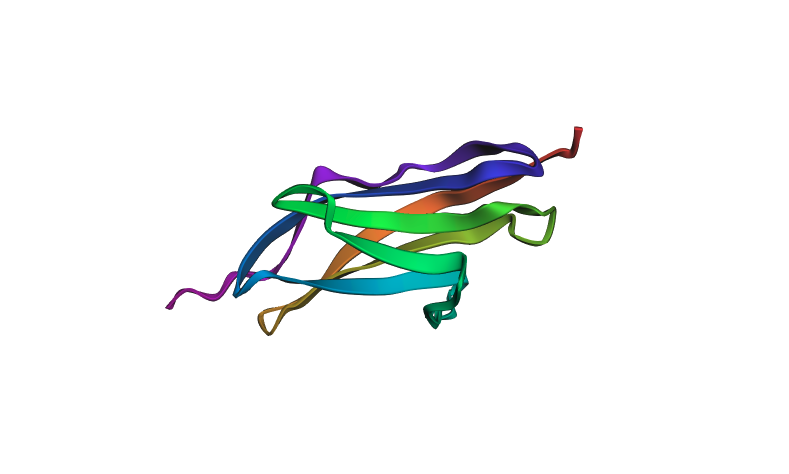
I folded the 1G1C sequence (146 residues) using ESMFold.
Folding results:
- pTM score: 0.881 (high confidence in overall topology)
- Mean pLDDT: 92.1 (high per-residue confidence)
Predicted structure (pre-mutation):
Predicted structure after mutations:
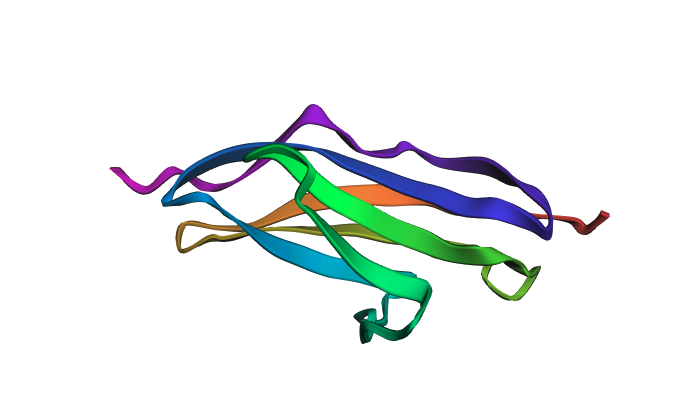
The predicted structure closely matches the experimentally determined 1G1C coordinates — the beta-sandwich topology is correctly recovered. When introducing point mutations (especially at surface-exposed residues), the predicted structure remains largely intact, showing that the Ig-fold is quite resilient to mutations at non-core positions. Larger segment substitutions begin to perturb the fold more noticeably.
Part 5: Inverse Folding with ProteinMPNN
Using the backbone of PDB 5MBA (chain A, 146 residues) as input to ProteinMPNN (v_48_020, noise level 0.2Å), I generated a new sequence candidate via inverse folding.
Native sequence (5MBA Chain A):
Native score (−log prob): 1.3374
ProteinMPNN generated sequence (T=0.1, seq_recovery=52%):
Sampled score: 0.8087
Amino acid probability heatmap (ProteinMPNN):
The model predicted sequence probabilities show high confidence at structurally constrained core positions (low entropy) and more variability at surface-exposed loops, which is consistent with the known structure-sequence relationship in Ig-fold proteins.
Comparison: Feeding the ProteinMPNN-generated sequence back into ESMFold yields a structure that closely matches the original 5MBA backbone (sequence recovery ~52% with a low sampling temperature of 0.1), demonstrating that ProteinMPNN successfully captured the backbone geometry even while proposing a distinct sequence.
Notebook
The full Colab notebook with all code and outputs is embedded below.