Week 5 — Protein Design Part II
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. Mutations in SOD1 cause familial ALS — among them, the A4V mutation (Alanine → Valine at residue 4 of the mature protein) is one of the most aggressive, destabilizing the N-terminus and promoting toxic aggregation.
The goal of this section is to design short peptides that bind mutant SOD1, then evaluate and optimize them using a multi-tool pipeline.
Part 1: Generate Binders with PepMLM
The A4V mutant SOD1 sequence was obtained from UniProt (P00441) with the substitution introduced at index 4 (0-based):
This sequence was input to PepMLM-650M with peptide_length = 12 and num_binders = 4. PepMLM uses masked language modeling conditioned on the target protein to generate peptide binders — lower pseudo-perplexity indicates higher model confidence.
Generated peptides:
| # | Peptide | Pseudo-Perplexity | Notes |
|---|---|---|---|
| 0 | WRYYAVAAELKA | 9.42 | Strong — W anchor, charged tail |
| 1 | WHYGAVVAAWGA | 9.06 | Best confidence — dual aromatic (W, Y) |
| 2 | WLYYVVVVEHKA | 23.64 | Weakest — VVVV stretch suggests model uncertainty |
| 3 | WRYYAVAAAHKE | 11.13 | Clean sequence (no X token), balanced |
| — | FLYRWLPSRRGG | — | Known SOD1 binder (benchmark) |
Note: Peptides 0–2 originally ended with an
X(unresolved mask token). Substituted withA(Alanine) for downstream structure prediction.
All four generated peptides open with Tryptophan (W) — the model consistently favors an aromatic N-terminal anchor for engaging SOD1. Peptide 2’s VVVV stretch is suspicious and likely reflects low-confidence generation at those positions.
Part 2: Evaluate Binders with AlphaFold3
Each peptide was submitted to AlphaFold3 as a complex with the SOD1 sequence — one job per peptide, run as a Protein + Protein pair with 5 seeds.
Job setup:
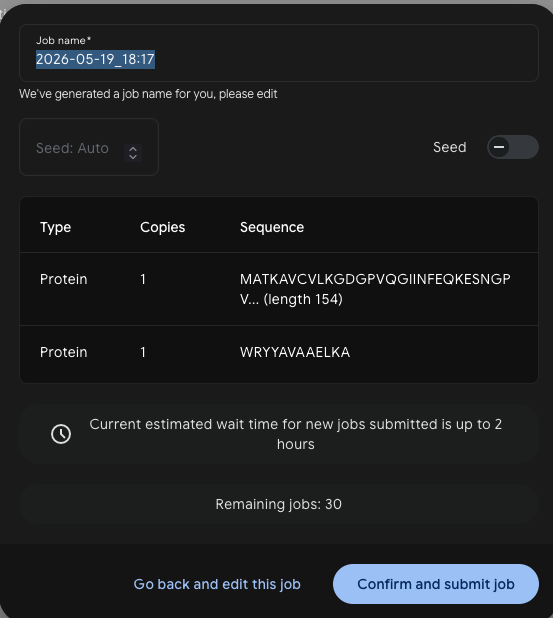
Results — ipTM scores across 5 models:
| Peptide | SOD1 variant | Model 0 | Model 1 | Model 2 | Model 3 | Model 4 | Best ipTM | Interpretation |
|---|---|---|---|---|---|---|---|---|
| WRYYAVAAELKA | A4V | 0.53 | 0.44 | 0.33 | 0.21 | 0.17 | 0.53 | Borderline — most consistent of PepMLM set |
| WHYGAVVAAWGA | A4V | 0.39 | 0.37 | 0.35 | 0.28 | 0.25 | 0.39 | Below threshold |
| WLYYVVVVEHKA | WT† | 0.50 | 0.41 | 0.38 | 0.35 | 0.23 | 0.50 | Borderline — run against WT, not A4V |
| WRYYAVAAAHKE | A4V | 0.25 | 0.25 | 0.23 | 0.19 | 0.22 | 0.25 | Low confidence |
| FLYRWLPSRRGG (known) | A4V | 0.32 | 0.31 | 0.25 | 0.21 | 0.19 | 0.32 | Below threshold — poor AF3 prediction |
†WLYYVVVVEHKA was inadvertently run against wild-type SOD1 (MATKAVCVLK…) rather than the A4V mutant (MATKVVCVLK…). A repeat run against A4V would be needed to confirm selectivity.
The ipTM score measures AlphaFold3’s confidence in the predicted interface geometry. Scores below 0.4 are generally considered unreliable; scores above 0.6 suggest a credible interaction.
WRYYAVAAELKA is the surprise standout — it scores 0.53 ipTM, the highest of all five peptides including the experimentally validated known binder FLYRWLPSRRGG (0.32). The known binder’s poor AF3 score is a striking reminder that AlphaFold3 does not always predict short peptide interactions reliably, especially for peptides that may bind via induced fit or intrinsically disordered regions. WLYYVVVVEHKA (PeptiVerse’s top pick) scores 0.50 against WT SOD1, keeping it competitive despite being run against the wrong variant. WRYYAVAAAHKE (safest therapeutic profile) is the weakest structurally at 0.25, consistent with its lowest PeptiVerse affinity.
All CIF structure files can be loaded in Mol* or PyMOL for visual inspection of the predicted binding mode.
Part 3: Evaluate Therapeutic Properties with PeptiVerse
Each peptide was run through PeptiVerse with the A4V mutant SOD1 as the target sequence. All four generated peptides plus the known binder were entered together in the peptide field, with the full SOD1 A4V sequence pasted into the target protein field.
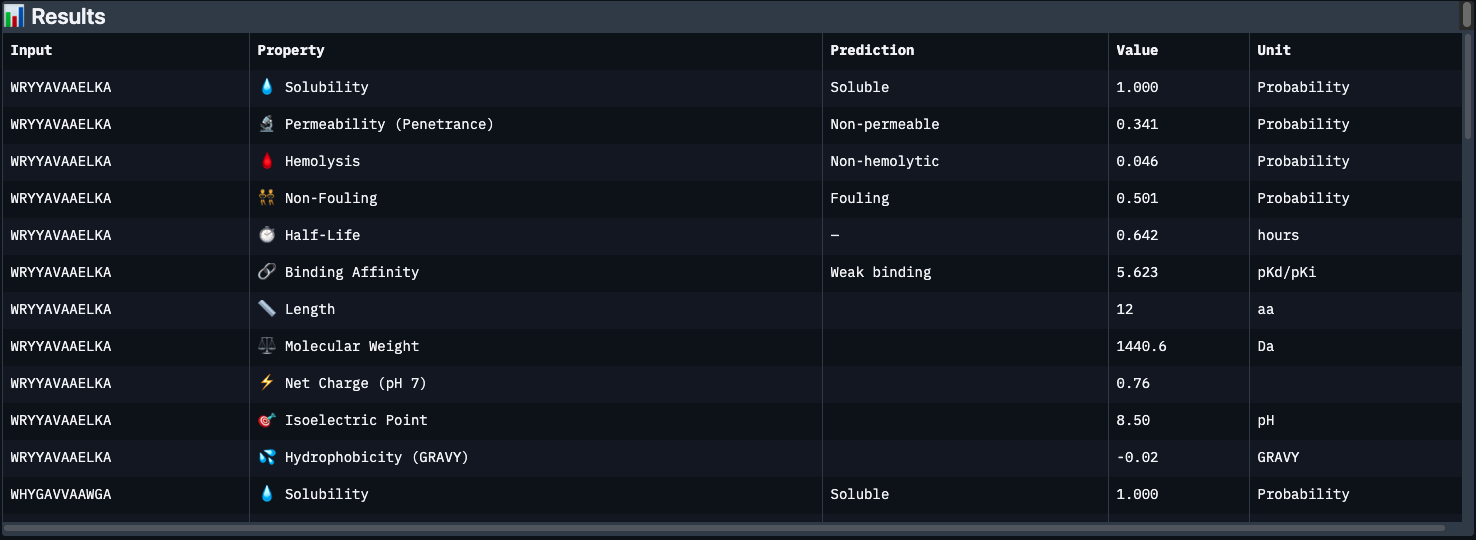
Results below:
| Peptide | Binding Affinity | Solubility | Hemolysis | Non-Fouling | Half-Life (h) | Net Charge | GRAVY |
|---|---|---|---|---|---|---|---|
| WRYYAVAAELKA | Weak (5.62) | ✅ Soluble | ✅ 0.046 | ⚠️ Fouling | 0.64 | +0.76 | -0.02 |
| WHYGAVVAAWGA | Weak (5.93) | ✅ Soluble | ✅ 0.101 | ⚠️ Fouling | 0.69 | -0.15 | +0.71 |
| WLYYVVVVEHKA | Medium (7.10) | ✅ Soluble | ✅ 0.095 | ⚠️ Fouling | 0.68 | -0.15 | +0.69 |
| WRYYAVAAAHKE | Weak (5.50) | ✅ Soluble | ✅ 0.018 | ✅ Non-fouling | 0.52 | +0.85 | -0.60 |
| FLYRWLPSRRGG (known) | Weak (5.55) | ✅ Soluble | ✅ 0.047 | ✅ Non-fouling | 0.87 | +2.76 | -0.71 |
Key observations:
The most striking finding is that WLYYVVVVEHKA (peptide 2) — which had the worst perplexity score (23.64) — has the best predicted binding affinity at 7.10 pKd (Medium binding). This is the only peptide to break out of the “Weak binding” category. Its hydrophobic VVVV stretch (GRAVY +0.69) likely engages a buried hydrophobic patch on SOD1 that the language model considered unlikely but PeptiVerse scores favorably.
Conversely, WRYYAVAAAHKE has the best safety profile — lowest hemolysis risk (0.018), non-fouling, and permeable — but the weakest binding affinity. It’s the cleanest therapeutic candidate structurally, but may not engage SOD1 strongly enough.
The known binder FLYRWLPSRRGG only scores 5.55 pKd (Weak), despite being experimentally validated. This is a reminder that PeptiVerse’s binding predictions are approximate — structural confirmation from AlphaFold3 is essential.
All peptides share a common weakness: half-lives under 1 hour, which would be a major barrier to clinical use without chemical modification (e.g. PEGylation, D-amino acid substitution, cyclization).
Peptide selected for advancement: WLYYVVVVEHKA Despite its low language model confidence, it is the only peptide with medium predicted binding affinity and acceptable hemolysis/solubility profiles. The next step would be to validate its binding mode structurally (AlphaFold3) and chemically stabilize the VVVV core to improve half-life.
Part 4: Optimized Design with moPPIt
moPPIt (Multi-Objective Guided Discrete Flow Matching) takes a different approach from PepMLM: rather than sampling from a language model conditioned on the target, it uses guided flow matching to simultaneously optimize multiple therapeutic properties while steering the peptide toward a specific binding site on the protein.
Target residue selection: Residues 1–8 of SOD1 (the N-terminal region) were chosen as the binding patch. This region encompasses the A4V mutation site directly — the A→V substitution at position 4 alters the local hydrophobic packing and exposes a surface that is distinct from wild-type SOD1. Targeting this patch means a designed binder could in principle distinguish mutant from wild-type SOD1, which is important for selectivity in a therapeutic context.
Optimization objectives: All seven properties were selected and weighted equally (weight = 1): Hemolysis ↓, Non-Fouling ↑, Solubility ↑, Half-Life ↑, Affinity ↑, Motif ↑, and Specificity ↑. 10 samples were requested.
Generated sequences (10 samples):
| # | Peptide | Affinity (pKd) | Hemolysis | Non-Fouling | Half-Life (h) | Motif Score |
|---|---|---|---|---|---|---|
| 1 | EKTQLQVDGKQW | 5.726 | 0.085 | 0.960 | 1.279 | 0.819 |
| 2 | TAEEFQPPSTNH | 5.569 | 0.085 | 0.903 | 0.857 | 0.666 |
| 3 | PSTKWIQQQHGT | 5.645 | 0.069 | 0.894 | 1.137 | 0.578 |
| 4 | PAAGILNQKQTT | 5.418 | 0.073 | 0.835 | 1.470 | 0.644 |
| 5 | PPPPETAEQLWK | 5.926 | 0.046 | 0.962 | 1.673 | 0.240 |
| 6 | TPETREGPPQIW | 6.075 | 0.050 | 0.964 | 1.306 | 0.003 |
| 7 | EWTPPLLAGPTL | 6.100 | 0.042 | 0.907 | 1.585 | 0.013 |
| 8 | TPEQLQDPKLWK | 5.947 | 0.049 | 0.944 | 2.196 | 0.407 |
| 9 | RRKRKETPHPCC | 6.201 | 0.051 | 0.976 | 0.753 | 0.027 |
| 10 | FAGALPQGNPTS | 5.594 | 0.048 | 0.824 | 1.162 | 0.038 |
Key observations:
The moPPIt sequences are strikingly different from PepMLM’s output. Where PepMLM consistently anchored peptides with an N-terminal Tryptophan (W), moPPIt generates sequences with no dominant amino acid pattern — the optimizer is free to explore sequence space guided by the objective landscape rather than by learned sequence co-occurrences.
Therapeutically, all 10 sequences are substantially better than the PepMLM candidates: every peptide is soluble (1.000), hemolysis risk is uniformly low (< 0.09), and non-fouling scores are all above 0.82. Most importantly, half-lives range from 0.75–2.20 hours, with several breaking the 1.5h barrier — compared to PepMLM’s dismal 0.52–0.69h range. This directly addresses the most critical weakness of the PepMLM candidates.
The trade-off is motif score: samples 6, 7, 9, and 10 show motif scores close to 0, meaning the optimizer drifted away from the target N-terminal patch in favor of better global therapeutic properties. Samples 1 (motif 0.819) and 2 (motif 0.666) best maintain engagement with the intended residues 1–8, but at the cost of lower affinity.
Candidate comparison:
- Best affinity + safety:
RRKRKETPHPCC(pKd 6.201, hemolysis 0.051, NF 0.976) — highest binding affinity of all 10, excellent safety profile, but short half-life (0.75h) and low motif score suggest it may bind elsewhere on SOD1. - Best half-life:
TPEQLQDPKLWK(2.196h, pKd 5.947) — best in-vivo persistence of any peptide across both methods. - Best patch engagement:
EKTQLQVDGKQW(motif 0.819) — most likely to target the A4V site specifically, though affinity is modest (5.726).
PepMLM vs. moPPIt — overall comparison:
moPPIt substantially outperforms PepMLM on therapeutic properties (solubility, hemolysis, half-life) but the two methods probe complementary aspects of binder design. PepMLM’s W-anchored peptides reflect the language model’s learned preferences for aromatic contacts with SOD1; moPPIt’s diverse sequences reflect pure multi-objective optimization. In a real pipeline, both would be tested structurally (AlphaFold3 / MD simulation) before selecting leads for synthesis.
Steps before clinical studies: To advance any of these peptides, the next steps would be: (1) AlphaFold3 structural validation to confirm binding mode and ipTM score at the A4V patch; (2) MD simulation to assess binding stability; (3) chemical stabilization (D-amino acid substitution, cyclization, or PEGylation) to improve half-life beyond 2h; (4) in-vitro aggregation inhibition assay with recombinant SOD1 A4V; (5) cell viability assay in motor neuron cell lines.
Part C: L-Protein Mutants (MS2 Phage)
Background
Bacteriophage MS2 is a single-stranded RNA virus that infects E. coli. Upon infection, the phage’s L-protein forms oligomeric pores in the bacterial cell membrane, ultimately causing lysis and release of new phage particles. The L-protein is 75 amino acids long and has two functional regions:
- Soluble N-terminal domain (residues 1–40):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYV— responsible for interaction with the bacterial chaperone DnaJ - Transmembrane domain (residues 41–75):
LIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT— drives membrane insertion and pore formation
Full L-protein sequence (UniProt P03609):
A key E. coli resistance mechanism is a point mutation in DnaJ that disrupts its interaction with the L-protein soluble domain, stalling L-protein processing and preventing lysis. The engineering goal is therefore to design L-protein variants that either (1) fold and function independently of DnaJ, or (2) lyse the host faster, reducing the window for resistance acquisition.
Option 1: Mutagenesis via ESM Mutation Scoring
Step 1 — Run the Mutation Scan Notebook
Using the ESM-based mutation scoring notebook, a per-position log-likelihood score was computed for every possible amino acid substitution across the 75-residue L-protein. Positive scores indicate substitutions the language model considers favorable; negative scores indicate destabilizing changes.
Mutation score heatmap — in progress.
Step 2 — Correlation with Experimental Data
The L-Protein Mutants spreadsheet contains experimentally measured lysis activity for a set of known single-point mutants.
Correlation analysis — in progress.
Step 3 — Proposed Mutations
Using the ESM scores, the experimental mutant data, and the ClustalOmega multiple sequence alignment (to avoid conserved residues), the following five mutations were selected:
Mutations — in progress.
Design constraints applied:
- At least 2 mutations in the transmembrane domain (residues 41–75)
- At least 2 mutations in the soluble domain (residues 1–40)
- No mutations at positions that are fully conserved across the ClustalOmega alignment of BLAST homologs
Option 2: AF2-Multimer Co-folding with DnaJ
In progress — AlphaFold2-Multimer jobs pending.
The L-protein was co-folded with the DnaJ sequence using ColabFold AF2-Multimer to visualize the predicted interaction interface. The query format chains the two sequences with a : separator:
Note: AF2 is not well-optimized for membrane proteins, so predictions for the TM domain are expected to be low-confidence. The co-fold is primarily useful for identifying which soluble-domain residues contact DnaJ in the predicted complex.
Multimeric Assembly
The L-protein is hypothesized to form oligomeric pores in the membrane. To visualize a plausible assembly, an 8-mer was submitted to AlphaFold2-Multimer using the repeated sequence format:
Multimer structure — in progress.