Governance means different things to different people, but what truly matters to me is building systems that share power rather than seize it. I imagine resilient structures that adapt, nurture well-being in many forms, and thrive on transparency and shared commitment.
First, describe a biological engineering application or tool you want to develop and why
Plants can be reciprocal and help each other. For example, ectomycorrhizal (ECM) fungi, which form sheaths around roots, help restore soil and make plants more resilient, especially in the face of climate change. I want to create a system inspired by the ectomycorrhizal (ECM) fungi living model of resilience. By mimicking their metabolism and distribution, we could recover and manage resources more wisely, whether in organizations, food systems, or conservation efforts. Imagine a biological pump that naturally generates, transports, and processes information, strengthening our collective resilience.
Here is the reference of the assignment: https://2026a.htgaa.org/2026a/course-pages/weeks/week-02/lab/index.html
Basic Understanding DNA Gel, restriction enzymes, Benchling intro, Twist intro (▶️Recording | 💻Slides)
easy content in Spanish: ¿Cómo hacer EDICIÓN GENÉTICA con CRISPR? https://www.youtube.com/watch?v=UaxrYWCyLdY&t=1s
As a committed listener in distance, I can only make the benching :
First step: https://www.benchling.com/, make an account. Second Step: choose a DNA sequence: https://www.neb.com/en-gb/tools-and-resources/interactive-tools/dna-sequences-and-maps-tool Begin by importing your DNA sequence and use the Digests tool to test the effects of different restriction enzyme(s). Export your final design as a png and compare with your lab results on your Notion page. See the images below for where to find the Digests tool, selecting the “NEB 2-log” ladder in the Virtual Digest tab, and how to have multiple Digests appear in the same Virtual Digest.
basic Concepts
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script that draws your design using the Opentrons. I took the Elephant as a starting point for my art in OpenTrons. This was an experience we complemented by understanding th step by step how to set up the Opentrons machine
Basics
Questions
Amino Acids, Protein Structure, and β-Sheets 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? It depends on the type and cut of meat, but a reasonable estimate is about 20–26 g of protein per 100 g of beef. Therefore, 500 g of meat would provide approximately 100–130 g of protein.
Still not really sure what I am doing :-)!
Basics
Design short peptides that bind mutant SOD1. “Design short peptides that bind mutant SOD1” means creating small, synthetic chains of amino acids (peptides) specifically engineered to attach to a deformed version of the Superoxide Dismutase 1 (SOD1) protein. This is a therapeutic strategy aimed at treating SOD1-related Amyotrophic Lateral Sclerosis (ALS).
Mutant SOD1: A faulty version of the SOD1 enzyme produced due to genetic mutations. Unlike healthy SOD1, mutant SOD1 misfolds, becomes unstable, and aggregates (clumps together), leading to toxicity in motor neurons.
basic Concepts
This week we learn core molecular biology tools and techniques for processing and assembling DNA, including PCR and Gibson Assembly.
Components of Phusion High-Fidelity PCR Master Mix Phusion Master Mix contains several key components:
Phusion Hot Start II DNA Polymerase — A high-fidelity polymerase with a proofreading (3’→5’ exonuclease) domain that corrects misincorporated bases, resulting in ~50× lower error rates than Taq. It also has a processivity-enhancing domain that speeds up elongation. dNTPs (dATP, dCTP, dGTP, dTTP) — The nucleotide building blocks incorporated during strand synthesis. MgCl₂ — Magnesium ions are an essential cofactor for DNA polymerase activity and also stabilize the dNTP substrates. Optimized reaction buffer — Maintains proper pH and ionic conditions for polymerase activity and primer/template annealing. Stabilizers/additives — Help maintain enzyme stability and can improve yield on difficult templates (e.g., GC-rich regions). 2. Factors Determining Primer Annealing Temperature Primer GC content — G·C pairs form 3 hydrogen bonds vs. 2 for A·T, so higher GC content raises the melting temperature (Tm). A rough formula is Tm = 4(G+C) + 2(A+T). Primer length — Longer primers have higher Tm values because more base-pair interactions must be disrupted. Salt/ion concentration — Higher Mg²⁺ or monovalent salt concentrations stabilize the DNA duplex and raise Tm. Primer secondary structure — Hairpins or self-dimers can reduce effective annealing efficiency. Template secondary structure — Highly structured templates may require higher annealing temperatures or additives like DMSO. Mismatches — Deliberate mismatches (e.g., for mutagenesis) lower Tm and require adjusted annealing temperatures. Annealing temperature rule of thumb — Typically set 5°C below the lower Tm of the two primers used. 3. PCR vs. Restriction Enzyme Digests Feature PCR Restriction Enzyme Digest Input template Any DNA (plasmid, genomic, cDNA) Usually plasmid or purified DNA Output Amplified, defined fragment Fragment(s) cut at specific recognition sites End type Blunt (Phusion) or 3’ A-overhang (Taq) Blunt or sticky (cohesive) ends depending on enzyme Precision Defined by primer design; any sequence Defined by restriction site locations in DNA Flexibility Very high — you design the fragment Limited to where restriction sites naturally exist Time ~1–3 hours ~1–2 hours Error risk Polymerase errors possible (mitigated by HiFi) No sequence errors; only wrong cut possible Requires sequence knowledge? Yes, for primer design Yes, to identify restriction sites When to prefer PCR You need to amplify a fragment from a complex mixture (e.g., genomic DNA). You want to add sequences (overhangs, restriction sites, Gibson overlaps) to the ends of a fragment. No convenient restriction sites flank your gene of interest. You are introducing a point mutation or modifying a sequence. When to prefer restriction enzyme digest You are sub-cloning between two vectors that already have compatible restriction sites. You need sticky ends for directional cloning. You want to cut a vector backbone without amplifying it (avoids PCR errors in the vector). Speed and simplicity are priorities when restriction sites are already present. 4. Ensuring Compatibility with Gibson Assembly Gibson Assembly requires fragments with overlapping homologous sequences (~15–30 bp) at their ends. To ensure compatibility:
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs have several advantages over traditional Boolean genetic circuits.
First, they can process continuous and graded inputs rather than only treating signals as ON or OFF. This is important because many biological signals, such as metabolite concentrations, transcription factor levels, or signaling gradients, are not binary.
Second, IANNs can perform weighted integration of multiple inputs. Instead of responding only when a rigid logical condition is met, they can combine signals with different strengths, similar to how neurons sum inputs.
Cell-Free Protein Synthesis: Questions and Answers 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis, or CFPS, produces proteins outside living cells using the molecular machinery extracted from cells. Compared with traditional in vivo expression, where proteins are produced inside organisms such as E. coli, yeast, or mammalian cells, CFPS offers more flexibility and experimental control.
Measurement Plan for Final Project: Piezoelectric Tone Modulation Project Context My final project explores a concept called Piezoelectric Tone Modulation, where a biologically produced peptide or protein-based scaffold, called here PiezoTone, could be integrated into a soft robotic wearable system for muscle tone modulation or rehabilitation support. The project combines synthetic biology, biomaterials, and wearable soft robotics.
HTGAA 1536 Pixel Artwork Canvas – Collective Bioart Experiment As part of Week 11, I participated in the HTGAA 1536 Pixel Artwork Canvas, a collective bioart experiment where each participant could contribute at least one pixel to a shared global artwork. The artwork was connected to cell-free reaction compositions, where each pixel represented a small contribution to a larger collaborative biological and visual system.
Subsections of Homework
Week 1 HW: Principles and Practices
Governance
means different things to different people, but what truly matters to me is building systems that share power rather than seize it. I imagine resilient structures that adapt, nurture well-being in many forms, and thrive on transparency and shared commitment.
First, describe a biological engineering application or tool you want to develop and why
Plants can be reciprocal and help each other. For example, ectomycorrhizal (ECM) fungi, which form sheaths around roots, help restore soil and make plants more resilient, especially in the face of climate change.
I want to create a system inspired by the ectomycorrhizal (ECM) fungi living model of resilience. By mimicking their metabolism and distribution, we could recover and manage resources more wisely, whether in organizations, food systems, or conservation efforts. Imagine a biological pump that naturally generates, transports, and processes information, strengthening our collective resilience.
Arbuscular mycorrhizae (AM), a type of endomycorrhizal fungi, associate with 80% of terrestrial plants. AM supports soil stabilisation, nutrient cycling, microbial diversity, and soil organic matter (SOM) formation.
Figure 1. “Diagram showing the updated conceptual framework of arbuscular mycorrhizal (AM) fungi-mediated soil organic matter (SOM) dynamics. Plants fix carbon through photosynthesis, which is then delivered to AM fungi. Arbuscular mycorrhizal fungi influence SOM dynamics through four pathways classified as (1) generating, (2) reprocessing, (3) reorganizing, and (4) stabilizing” Wu et al. 2024. (Image credits: https://doi.org/10.1111/nph.19178)reference: https://cid-inc.com/blog/2024-research-insights-how-mycorrhizal-fungi-benefits-agriculture/
Wu et al. (2024) introduced a framework explaining AM’s role in soil organic matter formation, considering factors such as organic compound diversity, mineral weathering, chemical interactions, and hyphosphere microbial contributions. They identify four pathways: SOM generation, reprocessing, reorganisation, and stabilisation (see Figure 1).
Generating: AM fungi produce exudates, metabolites, mucilage, and necromass. Wu et al. refer to the diversity, composition, and properties of these compounds as chemodiversity. They argue that chemodiversity, rather than individual compounds, is key to SOM composition, microbial biodegradation rates, and persistence.
Reprocessing: AM fungi attract specific microbes to their hyphosphere, the soil area influenced by AM hyphal exudates. These microbes drive soil biochemistry by decomposing SOM components that AM fungi cannot process. Through internal and extracellular pathways, they break down and assimilate SOM, contributing to chemodiversity, persistence, and SOM resynthesis. This process is known as the hyphosphere “microbial carbon pump.”
Reorganising: The fungi’s mycelial growth, expansion, and colonisation change the soil’s physical porosity and hydraulic properties. While AM stabilises macro soil aggregates, the mycelial dynamics increase micro aggregate turnover, water infiltration, soil-water retention capacity, hydraulic conductivity, and redistribution of AM exudates. The changing soil conditions due to mycelial expansion also change nutrient availability, temperature, and oxygen. It results in SOM redistribution and transformation.
Stabilising: AM fungi cause mineral weathering and alter interactions that influence SOM formation and stabilisation. AM rock mineral weathering makes nitrogen, phosphorus, potassium, and magnesium available in soils that form secondary compounds with different sizes, surfaces, and reactivity. This can alter mineral absorption, catalysis, and oxidation of SOM. These processes are called the “soil mineral carbon pump”.
Takeaway: The new concept can explain AM’s role in small- to large-scale SOM dynamics, which can help develop mycorrhiza-based technologies to enhance soil health.
I am facinating of the concept of carbon cycling and the major processes and mechanisms involved in this process, and how it can create positive changes in the ecosystem:
Figure 2. The solubility carbon pump (SCP) is driven by the difference of CO2 partial pressures between the atmosphere and surface waters; exchanges of CO2 occur through dissolution into water or release into the air. Generally, the SCP refers to the pumping of CO2 from the atmosphere into the ocean driven by abiotic processes such as lowering temperature and downward mixing. The biological carbon pump (BCP) refers to a series of biogeochemical processes that transport organic carbon (mainly particulate organic carbon (POC)) from the surface to the ocean interior
Figure 3. Microorganism–dissolved organic matter (DOM) complex networks consist of two types of nodes: microbial and DOM. Connections are made between nodes based on correlations of data sets. Microbial diversity can be analysed using 16S rRNA amplicons, metagenomics, metatranscriptomics, and metaproteomics, as described in Jiao, N., Luo, T., Chen, Q. et al. The microbial carbon pump and climate change. Nat Rev Microbiol 22, 408–419 (2024). https://doi.org/10.1038/s41579-024-01018-0
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals
Restoring governance in our ecosystems, especially within food systems and conservation, is crucial for protecting key species. By studying these organisms, we can build a deeper, more collaborative knowledge base and move toward distributed, systemic governance.
Purpose: Transform the soil by introducing this plant, aiming to regenerate and sustain a thriving, healthy food environment.
Design: Build partnerships among small businesses, farmers, and policymakers to expand these practices across diverse ecosystems.
Assumptions: it is not a law or regutlation is ian a mediation with the ecosystmen
Risks of Failure & Success: Outcomes depend on the ecosystem, but the ultimate goal is to design interventions that measure plant survival and resilience as the true indicators of success.
Although I am not entirely certain how the process functions, I believe in the potential of replicating plant-based or bioinspired systems to support regeneration and address deforestation within food systems.
First, implementing plant-based systems can address the irrigation and soil nutrition needs of various sectors. For example, cacao plantations, which are highly sensitive to extreme temperatures, could benefit from such approaches and support regenerative agriculture. Monitoring these plants could facilitate the development of sensing systems and enable the implementation of more complex networks for reforestation efforts.
Idea 2
My family’s cacao farm in Colombia has always sparked my curiosity about our crop’s roots and how we might make it more sustainable. For years, we have wondered about the true quality of our cacao and whether it belongs to the prized Criollo variety, since its genetics remain a mystery. Now, I am exploring how synthetic biology could unlock new possibilities for our farm, strengthen local leadership, and add value by revealing and celebrating the unique DNA of our cacao. hire a vision with a longer term
Step 1: Identify and document ancestral cacao varieties, which can be the first stage of a HTGAA
Map existing Criollo cacao varieties and their genetic diversity.
Record sensory profiles, cultural significance, and local cultivation knowledge. Connect with the idea 1 to produce regenerative practices
Collaborate with farmers, researchers, and local organisations. to contribut into a ecosytem DNA picture
Develop genetic repositories combining scientific methods with community participation.
Ensure fair access, ethical data governance, and benefit sharing with producer communities.
Integrate sustainable agricultural policies and biodiversity frameworks.
Step 3: Strengthen biodiversity conservation
Preserve endangered cacao genetic resources.
Promote agroecological cultivation models that maintain ecosystem health.
Support climate resilience through genetic diversity.
Step 4: Empower local farming communities
Recognise farmers as co-stewards of genetic heritage.
Provide training, technical support, and participatory decision-making spaces.
Foster economic opportunities linked to high-quality heritage cacao.
Step 5: Support sustainable certification and traceability
Use genetic data to strengthen transparency in cacao supply chains.
Enable certification schemes that reflect biodiversity conservation and ethical production.
Improve governance mechanisms linking agriculture, sustainability, and cultural heritage.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
x
• By helping respond
Foster Lab Safety
• By preventing incident
• By helping respond
Protect the environment
x
• By preventing incidents
• By helping respond
x
Other considerations
• Minimizing costs and burdens to stakeholders
• Feasibility?
• Not impede research
x
• Promote constructive applications
x
Bio Questions Week 1 <3 !
-> Question by J. Jacobson´s Presentation <-
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
As all concepts are new for me, I might need a bit more explanation of all concepts
A polymerase is a type of enzyme, such as DNA or RNA polymerase, that synthesises long chains of nucleic acids by adding nucleotides to a template strand. These enzymes are essential for critical biological processes, including DNA replication, repair, and transcription. Polymerases are also vital in laboratory applications, most notably in the polymerase chain reaction (PCR) for amplifying DNA. https://en.wikipedia.org/wiki/Polymerase
Function: DNA polymerases catalyze the synthesis of DNA by adding nucleoside triphosphates, creating two identical DNA duplexes from one.
Types: DNA polymerase (replicates DNA) and RNA polymerase (transcribes DNA into RNA) are the primary types, found in all living organisms.
Mechanism: They require a primer and a template strand to function, adding nucleotides in a
to
direction.
Accuracy: DNA polymerases often have built-in proofreading abilities (
to
exonuclease activity) to ensure high-fidelity replication.
PCR Application: Thermostable polymerases, such as Taq polymerase, are used in PCR to automate DNA copying, allowing for billions of copies to be made in a few hours.
Structural Variation: Polymerases range from simple single proteins to complex, multi-subunit assemblies.
What is the error rate of polymerase?
Throughput Error Rate Product Differential: ~10⁹
based on a consensus DNA polymerase is extremely accurate, with typical error rates around 1 mistake per 10⁵–10⁷ bases before cellular repair, and ~10⁹–10¹¹ per base per replication after all proofreading and repair.
How does this compare to the length of the human genome?
3.2 Gbp
How does biology deal with that discrepancy?
By error correcton MutS Repair System
by consensus: Biology layers multiple error‑correction systems to shrink polymerase’s raw error rate down to ~1 mutation per genome copy.
How many different ways are there to code (DNA nucleotide code) for an average human protein?
I am not sure, but in this paper they present a method for constructing complex and diverse DNA sequences using DNA three-way junctions. Theoretically, because of genetic code degeneracy, an “average” human protein can be encoded by an astronomically large number of DNA sequences.
In practice, what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
I guess because the fragmentation :S
-> Questions by LEPROUST´s Presentation <-
What’s the most commonly used method for oligo synthesis currently?
Oligonucleotide Synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis?
I didn´t find the aswer but I found this: phosphoramidite synthesis struggles beyond ~150–200 nt because stepwise inefficiency and side reactions make long, error‑free chains very rare. Filges, S., Mouhanna, P., & Ståhlberg, A. (2021). Digital Quantification of Chemical Oligonucleotide Synthesis Errors. Clinical chemistry. https://doi.org/10.1093/clinchem/hvab136.
Why can’t you make a 2000bp gene via direct oligo synthesis?
If I understand correctly, it is more efficient to work based on the Twist Silicon Platform, which can produce 9,600 genes. but not sure
-> Question by George Church´s Presentation <-
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Across vertebrates, most mammals (including humans) require nine essential amino acids:
histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine
Biochemically, a “lysine contingency” (as in Jurassic Park) is plausible only in the trivial sense that any essential amino acid could serve as a single‑point nutritional dependency. Lysine is important and often limiting, but it is not uniquely essential compared with the rest of the conserved EAA set, and species differ enough that no single “10‑EAA rule” applies universally. Mccann, J., & Rawls, J. (2023). Essential Amino Acid Metabolites as Chemical Mediators of Host-Microbe Interaction in the Gut.. Annual review of microbiology. https://doi.org/10.1146/annurev-micro-032421-111819.
As a committed listener in distance, I can only make the benching :
First step: https://www.benchling.com/, make an account. Second Step: choose a DNA sequence: https://www.neb.com/en-gb/tools-and-resources/interactive-tools/dna-sequences-and-maps-tool Begin by importing your DNA sequence and use the Digests tool to test the effects of different restriction enzyme(s). Export your final design as a png and compare with your lab results on your Notion page. See the images below for where to find the Digests tool, selecting the “NEB 2-log” ladder in the Virtual Digest tab, and how to have multiple Digests appear in the same Virtual Digest.
Part 0: Basics of Gel Electrophoresis
We did this call nicely online!
Thanks to the introduction from Digby Usher. Here are a couple of pics of the experience:
The work is saved in the following link: https://benchling.com/s/seq-33Yxt01UY8AwvjfPOIbd?m=slm-XZXh4GbXIU8Dh07RBlDZ
Attend or watch all lecture and recitation videos. Optionally watch bootcamp.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis ( just the play with the gel art interface)
Part of the assignment was to create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
What was very useful for understanding the second part, at least the preparation for the lab, was Ronan’s website (Ronan’s website, a helpful tool for quickly iterating on designs!
For this interface, I play with the Enzimes:
EcoRI, EcoRV,Sall and SacI
Restriction Digest Parameters
37°C for 60 minutes (incubation)
80°C for 20 minutes (heat inactivation) (optional)
DNA Gel Electrophoresis
Hand-Cast Gel
Digest
14.7 μL Water
3.3 μL Loading Dye
2 μL Digest
Which protein have you chosen and why?
I selected the Protein Titin, which is related to muscle function and movement; I used UniProt to look up information about the protein and found fascinating functions.
Cell color indicative of number of GO terms
Aspect Term
Molecular Function actin bindingSource:UniProtKB-KW
Molecular Function alpha-actinin bindingSource:MGI1 publication
Molecular Function axon guidance receptor activitySource:GO_Central
Molecular Function structural constituent of muscleSource:ProtInc1 publication
Biological Process homophilic cell adhesion via plasma membrane adhesion moleculesSource:GO_Central
Biological Process muscle contractionSource:ProtInc1 publication
Biological Process synapse organizationSource:GO_Central
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
This is the Exercise: The Central Dogma discussed in class and recitation describes the process by which the DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
LOCUS NW_023416316 74216526 bp DNA linear CON 30-SEP-2020
DEFINITION Myotis myotis isolate mMyoMyo1 unplaced genomic scaffold,
mMyoMyo1.p scaffold_m19_p_12, whole genome shotgun sequence.
ACCESSION NW_023416316
VERSION NW_023416316.1
DBLINK BioProject: PRJNA665501
BioSample: SAMN14734277
Assembly: GCF_014108235.1
KEYWORDS WGS; RefSeq.
SOURCE Myotis myotis
ORGANISM Myotis myotis
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Laurasiatheria; Chiroptera; Yangochiroptera;
Vespertilionidae; Myotis.
REFERENCE 1 (bases 1 to 74216526)
AUTHORS Jebb,D., Huang,Z., Pippel,M., Hughes,G.M., Lavrichenko,K.,
Devanna,P., Winkler,S., Jermiin,L.S., Skirmuntt,E.C.,
Katzourakis,A., Burkitt-Gray,L., Ray,D.A., Sullivan,K.A.M.,
Roscito,J.G., Kirilenko,B.M., Davalos,L.M., Corthals,A.P.,
Power,M.L., Jones,G., Ransome,R.D., Dechmann,D.K.N.,
Locatelli,A.G., Puechmaille,S.J., Fedrigo,O., Jarvis,E.D.,
Hiller,M., Vernes,S.C., Myers,E.W. and Teeling,E.C.
TITLE Six reference-quality genomes reveal evolution of bat adaptations
JOURNAL Nature 583 (7817), 578-584 (2020)
PUBMED 32699395
COMMENT REFSEQ INFORMATION: The reference sequence is identical to
JABWUV010000012.1.
Assembly name: mMyoMyo1.p
The genomic sequence for this RefSeq record is from the
whole-genome assembly released by the Bat1K on 2020/08/07. The
original whole-genome shotgun project has the accession
JABWUV000000000.1.
##Genome-Assembly-Data-START##
Assembly Provider :: Bat1K
Assembly Date :: 16-APR-2019
Assembly Method :: DAmar v. APRIL-2019; Bionano Solve DLS v.
3.3; Salsa2 HiC v. git commit: e3ae7d8;
GenomicConsensus v. git commit: 038de5c;
longranger align v. 2.2.0; Freebayes v.
1.2.0; HiGlass manual curation v. 1.5
Assembly Name :: mMyoMyo1.p
Genome Representation :: Full
Expected Final Version :: No
Genome Coverage :: 90.9x
Sequencing Technology :: PacBio Sequel CLR; 10X Genomics chromium
linked reads; Bionano Genomics; Phase
Genomics HiC; PacBio Sequel IsoSeq
##Genome-Assembly-Data-END##
##Genome-Annotation-Data-START##
Annotation Provider :: NCBI
Annotation Status :: Full annotation
Annotation Name :: Myotis myotis Annotation Release 100
Annotation Version :: 100
Annotation Pipeline :: NCBI eukaryotic genome annotation
pipeline
Annotation Software Version :: 8.5
Annotation Method :: Best-placed RefSeq; Gnomon
Features Annotated :: Gene; mRNA; CDS; ncRNA
##Genome-Annotation-Data-END##
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Lysis protein DNA sequence with Codon-Optimization ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
Week 3 Automation
basic Concepts
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script that draws your design using the Opentrons.
I took the Elephant as a starting point for my art in OpenTrons. This was an experience we complemented by understanding th step by step how to set up the Opentrons machine
I adapted the colors for the ones of the LifeFabs: purple Pink and Blue, which is actually a great Fluorescent
After exporting the code from the website, I ran the code in the Colab notebook as shown in the following picture
As our protocols didn’t work because we still had some issues in our programming, we did another practical part inoculating bacteria of the diferent colors, which we could set up in the future for the OpenTrons work.
Here is the Python code, and in the lab session you can find the pictures showing how to set up the OpenTrons software.
‘# -- coding: utf-8 --
“““Untitled0.ipynb
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
It depends on the type and cut of meat, but a reasonable estimate is about 20–26 g of protein per 100 g of beef. Therefore, 500 g of meat would provide approximately 100–130 g of protein.
If we assume an average amino acid residue has a mass of ~100 g/mol, this corresponds to about 1.0–1.3 moles of amino acid residues.
Since 1 mole = 6.022 × 10²³ molecules, this means:
1.0 mole ≈ 6 × 10²³ amino acid residues
1.3 moles ≈ 8 × 10²³ amino acid residues
So, eating 500 g of meat gives you on the order of 6 × 10²³ to 8 × 10²³ amino acid units.
2. Why do humans eat beef but do not become cows, eat fish but do not become fish?
Humans do not become the organisms they eat because food is first digested. Proteins from beef or fish are broken down into amino acids and small peptides in the digestive system. These small molecules are then absorbed and reused by the body to build human proteins, following the instructions encoded in human DNA.
In other words, the body does not copy the identity of the food organism. It only reuses its chemical building blocks.
3. Why are there only 20 natural amino acids?
There are not literally only 20 amino acids in nature, but there are 20 standard amino acids that are universally encoded by the genetic code in most proteins.
These 20 were likely selected during early evolution because they provide:
a broad range of chemical properties
good structural diversity
compatibility with the ribosome
efficient use in the genetic code
They include hydrophobic, polar, charged, aromatic, small, and flexible side chains, which together allow proteins to fold and function in many different ways.
There are also rare exceptions, such as selenocysteine and pyrrolysine, but the core set remains the same.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, scientists can make non-natural or non-canonical amino acids. These are useful in chemistry, protein engineering, and synthetic biology.
Examples of designed amino acids
Fluoro-leucine Similar to leucine, but with a fluorine atom added to the side chain. This could change hydrophobicity and stability.
Photo-switch amino acid An amino acid with an azobenzene group in its side chain, allowing it to change shape when exposed to light.
Metal-binding amino acid An amino acid containing a bipyridine-like side chain that can bind metal ions such as copper or zinc.
Redox amino acid An amino acid with a quinone or ferrocene-like group that could participate in electron transfer.
Click-ready amino acid An amino acid containing an azide or alkyne group for bioorthogonal “click” chemistry.
These new amino acids could give proteins new properties such as:
light responsiveness
selective chemical reactivity
conductivity
catalytic activity
metal binding
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life began, amino acids likely formed through prebiotic chemistry. This means they were produced by natural chemical reactions without enzymes or living cells.
Possible sources include:
reactions in the early Earth atmosphere
hydrothermal systems
lightning or UV-driven chemistry
meteorites and extraterrestrial delivery
This suggests that amino acids may have existed before life and later became incorporated into the first biological systems. Enzymes appeared later and made these processes faster and more controlled.
6. If you make an α-helix using D-amino acids, what handedness would you expect?
A normal α-helix made from L-amino acids is usually right-handed.
If the helix were made from D-amino acids, it would be expected to form a left-handed α-helix, which is the mirror image of the normal structure.
7. Can you discover additional helices in proteins?
Yes. Besides the classical α-helix, proteins and peptides can adopt other helical forms.
Examples include:
3₁₀-helices
π-helices
left-handed helices in special contexts
synthetic helical structures designed in peptides and foldamers
It is possible to discover or design additional helices by studying unusual protein structures, computational modeling, and synthetic peptide chemistry.
8. Why are most molecular helices right-handed?
In biology, most helices are right-handed because proteins are built mainly from L-amino acids. The stereochemistry of L-amino acids favors the formation of right-handed α-helices.
So the preference is not random: it arises from the chirality of the molecular building blocks.
9. Why do β-sheets tend to aggregate?
β-sheets tend to aggregate because their peptide backbones can form extensive hydrogen-bonding networks between neighboring strands. These interactions are repetitive and highly stabilizing.
Also, β-strands often expose side chains in an alternating pattern, which makes them good at packing together into larger assemblies such as fibrils.
10. What is the driving force for β-sheet aggregation?
The main driving forces are:
hydrogen bonding between peptide backbones
hydrophobic interactions between side chains
release of water molecules from the interface, which increases solvent entropy
Together, these effects make β-sheet assemblies, especially cross-β structures, very stable.
11. Why do many amyloid diseases form β-sheets?
Many amyloid diseases involve proteins that misfold and then assemble into β-sheet-rich fibrils. The cross-β structure is very stable and can grow by recruiting additional misfolded protein molecules.
This makes β-sheet aggregation a common structural feature in diseases such as:
Alzheimer’s disease
Parkinson’s disease
Huntington’s disease
other protein misfolding disorders
12. Can you use amyloid β-sheets as materials?
Yes. Amyloid β-sheet assemblies can be used as functional biomaterials because they are often:
strong
stable
self-assembling
nanoscale and highly ordered
Potential applications include:
tissue engineering scaffolds
nanomaterials
functional coatings
drug delivery systems
bio-inspired structural materials
So although amyloids are linked to disease, they can also be useful when carefully designed and controlled.
13. Design a β-sheet motif that forms a well-ordered structure.
A good β-sheet design should encourage:
β-strand formation
regular side-chain patterning
controlled intermolecular interactions
reduced disorder at the ends
Example 1: Amphipathic β-strand peptide
Sequence: Ac–Val-Lys-Val-Glu-Val-Lys-Val-Glu–NH2
Why this may work
Val promotes β-strand structure and creates a hydrophobic face.
Lys and Glu create a charged face.
Oppositely charged residues can form salt bridges.
The alternating arrangement supports ordered packing.
N-terminal acetylation and C-terminal amidation reduce end effects.
Example 2: More aggregation-prone fibril-forming motif
Sequence: Ac–Phe-Val-Phe-Val-Lys-Glu-Phe-Val–NH2
Why this may work
Phe and Val strongly favor packing and aggregation.
Aromatic residues may strengthen intermolecular interactions.
Lys/Glu improve some balance between solubility and assembly.
This sequence may form fibrils more easily, but it also carries a higher risk of uncontrolled aggregation.
Example 3: β-hairpin motif with defined turn
Sequence: RGKWTWQ–DPro-Gly–QWTVKGR
Why this may work
The DPro-Gly pair promotes a defined hairpin turn.
The strands can align in a controlled intramolecular β-sheet.
Aromatic and charged residues can help stabilize folding and packing.
This design is often more controlled than open-ended fibril-forming strands.
Briefly describe the protein you selected and why you selected it.
Identify the amino acid sequence of your protein.
I am interested in Proteins that can enable movement or have realtionship in designing soft robotics, so I was interested in the following proteins:
Silk-Elastin-Like Proteins (SELPs)
Silk-Elastin-Like Proteins (SELPs) are a class of genetically engineered, chimeric biopolymers that combine the structural, mechanical properties of silk (specifically Bombyx mori silk fibroin) with the elasticity and thermo-responsiveness of elastin. By leveraging recombinant DNA technology, these proteins can be precisely tailored for applications in biomedical engineering, drug delivery, and tissue engineering.
Silk-Elastin-Like Proteins (SELPs) are engineered block copolymers comprising repeating amino acid sequences of silk, typically GAGAGS (Gly-Ala-Gly-Ala-Gly-Ser), and elastin, often GVGVP (Val-Pro-Gly-Val-Gly). A common, highly studied monomer unit is, one that combines elastic and structural properties
Composition: SELPs are block copolymers, consisting of alternating silk-like motifs (typically GAGAGS) and elastin-like motifs (typically GVGVP).
Self-Assembly: In aqueous solutions, SELPs form micellar-like nanoparticles, with the hydrophobic silk blocks forming the core and the hydrophilic elastin blocks forming the corona.
Stimuli-Responsiveness: SELPs are “smart” materials that respond to environmental triggers, most notably temperature, but also pH, ionic strength, and light.
Mechanical Properties: The silk-to-elastin ratio determines the mechanical behavior. Higher silk content increases beta-sheet formation, resulting in stiffer materials, while higher elastin content increases flexibility.
Production: Produced through E. coli expression systems, allowing for high control over sequence, molecular weight, and monodispersity, which improves reproducibility compared to natural materials
Chambre L, Martín-Moldes Z, Parker RN, Kaplan DL. Bioengineered elastin- and silk-biomaterials for drug and gene delivery. Adv Drug Deliv Rev. 2020;160:186-198. doi: 10.1016/j.addr.2020.10.008. Epub 2020 Oct 17. PMID: 33080258; PMCID: PMC7736173.
Collagen
Collagen’s primary amino acid sequence is characterized by a repeating, unique motif, most commonly Glycine-Proline-X or Glycine-X-Hydroxyproline, where Glycine appears every third residue. These ~1,000 amino acid-long chains form a triple helix, rich in glycine, proline, and hydroxyproline.
Repeating Units: The primary sequence is defined by
repeats, making up a significant portion of the chain.
Glycine (Gly): Occurs at every third position, essential for the tight packing of the triple helix.
Proline (Pro) & Hydroxyproline (Hyp): The ‘X’ and ‘Y’ positions are frequently occupied by Proline (approx. 28%) and Hydroxyproline (approx. 38%).
Hydroxyproline and Hydroxylysine: These modified amino acids are crucial for stabilizing the triple helix structure via hydrogen bonding.
Structure: Three left-handed polyproline II helices intertwine to create a right-handed superhelical triple helix, known as tropocollagen.
Actin and myosin are highly conserved, complex proteins, with actin typically comprising 374-376 amino acids and myosin (specifically the heavy chain) being a much larger molecule (~2000+ residues). Due to their size and various isoforms, they are generally identified by their full sequences in protein databases (like UniProt) rather than a single short string.
Below are the key details regarding their amino acid sequences based on rabbit skeletal muscle, which is the standard reference:
Actin Amino Acid Sequence (Rabbit Skeletal Muscle)
Actin is a 374-residue protein with a highly conserved sequence. It includes a unique-methyl histidine residue.
Key Features: High proportion of proline and glycine.
Sequence Data Source: The complete sequence was first determined by Elzinga et al. (1973).
Isoforms: While highly conserved, differences occur between skeletal, cardiac, and cytoplasmic isoforms (e.g., about 25 amino acid differences between skeletal and cytoplasmic actin).
Myosin Amino Acid Sequence (Heavy Chain/S1 Fragment)
Myosin is a large motor protein (Hexamer: 2 heavy chains, 4 light chains). The functional motor domain is the S1 fragment (globular head).
Active Site Sequence: A key 20-residue peptide containing the active site in Acanthamoeba and rabbit skeletal myosin has been identified, with sequences such as Thr-Glu-Asn-Thr-Me2Lys-Lys.
Fragment Identification: A 92-residue fragment containing SH-1 and SH-2 groups in the globular head was identified by Maita et al..
Motor Domain: The motor domain of myosin II comprises approximately 700-800 amino acids at the N-terminus of the heavy chain.
Key Structural Sites (Interaction Points)
The interaction between actin and myosin involves specific binding sites on both proteins:
Actin Binding Site on Myosin: Located on the S1 head, this area involves multiple hinged segments that change shape to facilitate contraction.
Myosin Binding Site on Actin: The interaction involves specific residues that can be mapped using peptide fragments.
Loop 4/CM Loop: Specific loops on the myosin head are critical for binding to actin.
For the full, exact sequence, searching for “Rabbit skeletal muscle actin UniProt” or “Human Beta-Myosin Heavy Chain UniProt” in scientific databases is required.
Some images from :
Myotilin Monomer
AF-Q9UBF9-2-F1-v6
Protein: Myotilin
Gene: MYOT
Source organism: Homo sapiens search this organism
UniProt: Q9UBF9-2 go to UniProt
Experimental structures: 2 PDB structures for Q9UBF9-2go to PDBe-KB
Global quality average pLDDT 77.06 (High)
Sequence length 314
week 05 protein design part 2
Still not really sure what I am doing :-)!
Basics
Design short peptides that bind mutant SOD1.
“Design short peptides that bind mutant SOD1” means creating small, synthetic chains of amino acids (peptides) specifically engineered to attach to a deformed version of the Superoxide Dismutase 1 (SOD1) protein. This is a therapeutic strategy aimed at treating SOD1-related Amyotrophic Lateral Sclerosis (ALS).
Mutant SOD1: A faulty version of the SOD1 enzyme produced due to genetic mutations. Unlike healthy SOD1, mutant SOD1 misfolds, becomes unstable, and aggregates (clumps together), leading to toxicity in motor neurons.
Short Peptides: Small molecules, often consisting of only a few amino acids, designed to act as targeted “decoys.”
Bind: The peptides are engineered to stick to specific, exposed, or misfolded areas of the mutant SOD1 protein.
Goal: The binding stops the mutant SOD1 from interacting with, and damaging, vital parts of the cell—such as mitochondrial outer membranes (e.g., VDAC1) or Bcl-2 proteins—thereby preventing cell death and slowing the progression of ALS
Why this approach?
Mutant SOD1 binds to mitochondria, disrupting energy production and causing toxicity. Designed short peptides can mimic the normal binding partners of the mitochondria (like the VDAC1 N-terminus), acting as a decoy to prevent the toxic mutant protein from sticking to the mitochondria, as demonstrated in scientific studies where these peptides improved neuronal survival.
Then decide which ones are worth advancing toward therapy.
The goal of designing these peptides is typically to:
-> Block Aggregation: By binding to the “sticky” parts of the mutant protein, the peptides can prevent it from clumping into toxic aggregates.
-> Prevent Toxic Interactions: Mutant SOD1 often interferes with other vital cell parts, like mitochondria. Short “decoy” peptides can bind to the mutant protein first, blocking it from damaging these organelles.
-> Stabilize the Protein: Some peptides are designed to help the mutant protein keep its proper shape, making it less likely to become toxic.
In essence, “Design short peptides that bind mutant SOD1” means creating custom-made, small molecules to “clamped onto” the broken protein responsible for ALS to stop it from causing harm.
Would you like to see current examples of these peptides or learn more about how they are tested in the lab?
PART 1
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
PepMLM: target sequence-conditioned peptide generation via masked language modeling
This week we learn core molecular biology tools and techniques for processing and assembling DNA, including PCR and Gibson Assembly.
1. Components of Phusion High-Fidelity PCR Master Mix
Phusion Master Mix contains several key components:
Phusion Hot Start II DNA Polymerase — A high-fidelity polymerase with a proofreading (3’→5’ exonuclease) domain that corrects misincorporated bases, resulting in ~50× lower error rates than Taq. It also has a processivity-enhancing domain that speeds up elongation.
dNTPs (dATP, dCTP, dGTP, dTTP) — The nucleotide building blocks incorporated during strand synthesis.
MgCl₂ — Magnesium ions are an essential cofactor for DNA polymerase activity and also stabilize the dNTP substrates.
Optimized reaction buffer — Maintains proper pH and ionic conditions for polymerase activity and primer/template annealing.
Stabilizers/additives — Help maintain enzyme stability and can improve yield on difficult templates (e.g., GC-rich regions).
2. Factors Determining Primer Annealing Temperature
Primer GC content — G·C pairs form 3 hydrogen bonds vs. 2 for A·T, so higher GC content raises the melting temperature (Tm). A rough formula is Tm = 4(G+C) + 2(A+T).
Primer length — Longer primers have higher Tm values because more base-pair interactions must be disrupted.
Salt/ion concentration — Higher Mg²⁺ or monovalent salt concentrations stabilize the DNA duplex and raise Tm.
Primer secondary structure — Hairpins or self-dimers can reduce effective annealing efficiency.
Template secondary structure — Highly structured templates may require higher annealing temperatures or additives like DMSO.
Mismatches — Deliberate mismatches (e.g., for mutagenesis) lower Tm and require adjusted annealing temperatures.
Annealing temperature rule of thumb — Typically set 5°C below the lower Tm of the two primers used.
3. PCR vs. Restriction Enzyme Digests
Feature
PCR
Restriction Enzyme Digest
Input template
Any DNA (plasmid, genomic, cDNA)
Usually plasmid or purified DNA
Output
Amplified, defined fragment
Fragment(s) cut at specific recognition sites
End type
Blunt (Phusion) or 3’ A-overhang (Taq)
Blunt or sticky (cohesive) ends depending on enzyme
Precision
Defined by primer design; any sequence
Defined by restriction site locations in DNA
Flexibility
Very high — you design the fragment
Limited to where restriction sites naturally exist
Time
~1–3 hours
~1–2 hours
Error risk
Polymerase errors possible (mitigated by HiFi)
No sequence errors; only wrong cut possible
Requires sequence knowledge?
Yes, for primer design
Yes, to identify restriction sites
When to prefer PCR
You need to amplify a fragment from a complex mixture (e.g., genomic DNA).
You want to add sequences (overhangs, restriction sites, Gibson overlaps) to the ends of a fragment.
No convenient restriction sites flank your gene of interest.
You are introducing a point mutation or modifying a sequence.
When to prefer restriction enzyme digest
You are sub-cloning between two vectors that already have compatible restriction sites.
You need sticky ends for directional cloning.
You want to cut a vector backbone without amplifying it (avoids PCR errors in the vector).
Speed and simplicity are priorities when restriction sites are already present.
4. Ensuring Compatibility with Gibson Assembly
Gibson Assembly requires fragments with overlapping homologous sequences (~15–30 bp) at their ends. To ensure compatibility:
For PCR fragments: Design primers so that the 5’ overhang of each primer matches the end of the adjacent fragment. This way, after PCR, the amplified insert carries ~20–30 bp of sequence identical to the neighboring fragment or vector.
For restriction-digested fragments: After digestion, check that the sticky ends or blunt ends are located within the overlap region you plan to use — or add Gibson overlaps via a subsequent PCR step using primers that extend into the adjacent sequence.
Check orientation: Use Benchling or SnapGene to simulate the assembly and verify that all overlaps are in the correct orientation and reading frame.
Avoid internal repeat sequences in the overlap regions, as the exonuclease in Gibson mix can cause misannealing.
Ensure no unwanted restriction sites or stop codons are introduced at junctions.
Gel-purify or column-purify fragments after PCR or digest to remove enzymes, primers, and small fragments that could interfere.
5. How Plasmid DNA Enters E. coli During Transformation
The most common method in lab courses is heat-shock transformation of chemically competent cells:
Chemical competency preparation — Cells are treated with divalent cations (typically CaCl₂), which neutralize the negative charges on the LPS of the outer membrane and on the DNA, reducing electrostatic repulsion.
DNA binding — Plasmid DNA associates with the cell surface, facilitated by the Ca²⁺ ions.
Heat shock (42°C, ~30–45 sec) — The rapid temperature increase is thought to create a thermal imbalance that momentarily destabilizes the membrane and drives DNA into the cell, possibly through transient pores or membrane disruptions. The exact mechanism is still not fully understood.
Recovery on ice — Cells are rapidly cooled to stabilize the membrane after DNA entry.
Outgrowth in SOC/LB — Cells recover and begin expressing antibiotic resistance genes before plating on selective media.
Alternative method — Electroporation: A brief electrical pulse (~1.8–2.5 kV) creates transient pores in the membrane through which DNA passes. This is more efficient but requires electrocompetent cells and specialized equipment.
6. Golden Gate Assembly
6.1 Explanation in 5–7 sentences
Golden Gate Assembly is a DNA assembly technique that uses Type IIS restriction enzymes, such as BsaI or BsmBI, which cut outside of their recognition sites rather than within them. This makes it possible to design custom overhangs that determine the exact order in which DNA fragments join together. In a single reaction tube, the restriction enzyme cuts the DNA fragments and vector, and DNA ligase joins the matching overhangs. Because the recognition sites can be removed during the assembly process, the final DNA construct is often scarless, meaning no extra unwanted sequence remains at the junctions. Golden Gate Assembly is especially useful for assembling multiple DNA fragments in a defined order with high efficiency. It is widely used in modular cloning systems and synthetic biology workflows. Compared with Gibson Assembly, Golden Gate relies on restriction sites and short designed overhangs rather than long homologous overlaps.
NEB’s (New England Biolabs) explanation & protocols for Gibson Assembly®
General principle
Fragment 1 Fragment 2 Fragment 3
[BsaI] [BsaI] [BsaI]
| | |
v v v
Cut outside the recognition sequence to create custom overhangs
Overhangs designed as:
Fragment 1 ---> AATG
Fragment 2 ---> GCTT
Fragment 3 ---> CGGA
Matching overhangs guide ligation in the correct order:
Fragment 1 + Fragment 2 + Fragment 3
↓
Final assembled construct
1. Type IIS restriction enzyme cuts DNA outside its recognition site
2. Custom sticky ends are generated
3. Matching sticky ends anneal
4. DNA ligase seals the backbone
5. Final construct forms without the original restriction sites
week 7 genetic circuits part II
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs have several advantages over traditional Boolean genetic circuits.
First, they can process continuous and graded inputs rather than only treating signals as ON or OFF. This is important because many biological signals, such as metabolite concentrations, transcription factor levels, or signaling gradients, are not binary.
Second, IANNs can perform weighted integration of multiple inputs. Instead of responding only when a rigid logical condition is met, they can combine signals with different strengths, similar to how neurons sum inputs.
Third, they can generate nonlinear and more complex input-output behaviors, such as band-pass filters, threshold responses, or spatial patterns. This makes them more suitable for approximating real biological decision-making.
Fourth, multilayer IANNs can achieve greater design flexibility and generalization. By stacking regulatory layers, they can produce behaviors that would be difficult or inefficient to implement with simple Boolean gates alone.
Finally, IANNs are useful when the goal is not just logical control but also prediction, optimization, and adaptive design, especially when paired with AI-based modeling tools.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application for an IANN would be a smart therapeutic cell for liver disease detection and response.
Application idea
The engineered cell could sense several biomarkers associated with liver injury or inflammation and produce a therapeutic or reporter output only when a specific combination of signals is detected.
Inputs
The IANN could receive multiple intracellular or extracellular inputs, for example:
• X1: inflammatory cytokine level
• X2: oxidative stress signal
• X3: metabolite associated with liver dysfunction
• X4: hypoxia-related signal
Each of these inputs would not simply be present or absent, but could vary in concentration.
Output behavior
The output, Y, could be:
• expression of a fluorescent reporter for diagnosis, or
• release of a protective therapeutic protein
The IANN would integrate the four inputs using weighted biological regulation. For example:
• low inflammation alone would not activate the output
• moderate inflammation plus high oxidative stress might produce a medium output
• a specific disease-like combination of all four signals could trigger a strong output
• healthy or nonspecific combinations would remain below threshold
This would allow the system to distinguish a true pathological state from random fluctuations or isolated signals.
Why IANN is useful here
A Boolean circuit might require strict YES/NO cutoffs and could be too rigid. In contrast, an IANN could better handle noisy biological data and produce a more nuanced response.
Limitations
However, an IANN would face several limitations:
• biological noise: gene expression varies from cell to cell
• limited predictability: real cells may behave differently from the model
• cross-talk: regulators may unintentionally affect other components
• timing delays: transcription and translation are slower than electronic computation
• metabolic burden: large circuits can stress the cell
• safety and stability: long-term behavior may drift due to mutation or epigenetic changes
So while IANNs are powerful, achieving reliable therapeutic performance would require careful design, validation, and containment.
Week 9 Cell Free Systems
Cell-Free Protein Synthesis: Questions and Answers
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis, or CFPS, produces proteins outside living cells using the molecular machinery extracted from cells. Compared with traditional in vivo expression, where proteins are produced inside organisms such as E. coli, yeast, or mammalian cells, CFPS offers more flexibility and experimental control.
The main advantage is that the system is open. In living cells, the researcher cannot easily control everything inside the cell because metabolism, growth, toxicity, stress responses, and gene regulation all influence protein production. In a cell-free system, the researcher can directly add DNA, RNA, amino acids, cofactors, energy sources, salts, chaperones, detergents, liposomes, or other molecules. This makes it easier to test variables quickly and systematically.
CFPS is also useful because it avoids problems related to cell viability. Some proteins are toxic to living cells, difficult to fold inside cells, or interfere with the host metabolism. Since CFPS does not require cells to stay alive, it can produce proteins that would otherwise reduce cell growth or kill the host.
Two cases where cell-free expression is more beneficial than cell-based production are:
Toxic proteins For example, antimicrobial peptides, pore-forming proteins, or regulatory proteins that interfere with cell metabolism can be difficult to produce in E. coli. CFPS allows their production without harming a living host.
Membrane proteins or difficult-to-fold proteins Membrane proteins often aggregate or are poorly expressed in cells. In CFPS, detergents, nanodiscs, liposomes, or microsomes can be added directly to support proper folding and insertion.
Other useful cases include rapid prototyping of genetic circuits, testing many DNA designs quickly, producing proteins with non-natural amino acids, and screening enzyme variants.
Example of a Useful Synthetic Minimal Cell
Function
A useful synthetic minimal cell could be designed as a smart therapeutic microcell for localized inflammation detection and drug release.
The function of this synthetic cell would be to detect signs of inflammation in the body and respond by releasing an anti-inflammatory molecule only when needed. This would make treatment more precise and reduce side effects compared with systemic drug delivery.
For example, the synthetic minimal cell could be designed to sense inflammatory signals such as TNF-α, IL-6, or high levels of reactive oxygen species, which are often present in inflamed tissues.
What would the synthetic cell do?
The synthetic minimal cell would act like a small programmable therapeutic device. It would circulate or be placed near a target tissue, such as an inflamed joint, damaged muscle, or rehabilitation injury site.
When the synthetic cell detects inflammation, it activates an internal genetic or biochemical circuit. This circuit triggers the production or release of a therapeutic molecule, such as an anti-inflammatory peptide, cytokine inhibitor, or tissue-repair factor.
In simple terms, the synthetic cell would:
Sense a disease-related signal.
Process the information using a minimal genetic circuit.
Respond by producing or releasing a therapeutic output.
Stop responding when the inflammatory signal decreases.
Input and Output
Element
Description
Input
Inflammatory signals, such as TNF-α, IL-6, or reactive oxygen species
Processing system
Minimal gene circuit or synthetic receptor system that detects inflammation
Output
Controlled release of an anti-inflammatory protein, peptide, or repair-promoting molecule
Expected effect
Reduction of local inflammation and support of tissue healing
Example Scenario
A patient has chronic inflammation in a joint, muscle, or tendon. Instead of taking anti-inflammatory medicine that affects the whole body, synthetic minimal cells could be delivered locally.
When the cells detect high levels of inflammatory molecules, they release a therapeutic protein. When inflammation decreases, the synthetic cells reduce or stop production. This creates a feedback-controlled treatment system.
Why this is useful
This type of synthetic minimal cell could be useful because it allows localized, controlled, and responsive therapy. It could reduce the risk of side effects and avoid unnecessary drug exposure.
It could be especially valuable for:
Chronic inflammatory diseases
Arthritis
Muscle or tendon injuries
Rehabilitation after trauma
Smart biomaterials for wearable or implantable therapeutic systems
Summary
The synthetic minimal cell would function as a programmable inflammation-sensing therapeutic system.
Its input would be inflammatory biomarkers such as TNF-α, IL-6, or reactive oxygen species.
Its output would be the controlled release of an anti-inflammatory or tissue-repair molecule.
The goal would be to create a minimal biological system that can sense the body’s condition and respond only when treatment is needed.
2. Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system usually contains the following components:
DNA or mRNA template
This provides the genetic instructions for the protein. DNA templates include a promoter, ribosome binding site or translation initiation sequence, coding sequence, and terminator. In some systems, mRNA can be added directly.
Cell extract
The extract contains the biological machinery needed for transcription and translation. This includes ribosomes, tRNAs, aminoacyl-tRNA synthetases, translation factors, and sometimes RNA polymerases. The extract can come from E. coli, wheat germ, rabbit reticulocytes, insect cells, or mammalian cells.
Amino acids
These are the building blocks used to synthesize the protein.
Energy source
Protein synthesis requires energy, mainly ATP and GTP. The system needs an energy source such as phosphoenolpyruvate, creatine phosphate, glucose, maltodextrin, or other energy-regeneration molecules.
Nucleotides
NTPs such as ATP, GTP, CTP, and UTP are needed for transcription when DNA is used as the template.
Salts and ions
Magnesium, potassium, and other ions are essential for ribosome function, enzyme activity, and RNA stability. Their concentration strongly affects protein yield.
Cofactors and additives
Some proteins require cofactors such as heme, metals, flavins, or disulfide-bond-supporting reagents. Chaperones can also be added to help folding.
Optional components
Depending on the protein, the system may include detergents, liposomes, nanodiscs, microsomes, protease inhibitors, molecular chaperones, or non-natural amino acids.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision and regeneration are critical because transcription and translation consume large amounts of ATP and GTP. Without continuous energy regeneration, protein synthesis stops quickly because the system runs out of usable energy.
ATP is needed for many steps, including amino acid charging of tRNAs and general enzymatic activity. GTP is especially important during translation elongation and translocation. Since a cell-free reaction is not a living cell with full metabolism, the energy supply must be added externally and maintained during the experiment.
One method to ensure continuous ATP supply is to use an energy-regeneration system. For example:
Phosphoenolpyruvate system
Phosphoenolpyruvate, or PEP, can be added as a high-energy phosphate donor. Enzymes in the extract transfer phosphate groups to regenerate ATP from ADP. This helps maintain ATP levels during the reaction.
Another option is the creatine phosphate and creatine kinase system, where creatine phosphate regenerates ATP from ADP. More modern systems can use glucose, maltodextrin, or 3-phosphoglycerate because they can provide a more stable and less expensive energy supply.
For my own experiment, I would use a glucose or maltodextrin-based energy system if I wanted longer protein expression, because these systems can support more sustained ATP regeneration and are often more affordable.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic and eukaryotic CFPS systems differ mainly in their translation machinery, folding environment, post-translational modifications, and complexity.
Feature
Prokaryotic CFPS
Eukaryotic CFPS
Common extract
E. coli extract
Wheat germ, rabbit reticulocyte, insect, or mammalian extract
Human or eukaryotic proteins, proteins needing complex folding
Post-translational modifications
Limited
Better support for some eukaryotic modifications
Main limitation
Poor for complex eukaryotic proteins
Higher cost and sometimes lower yield
Protein example for prokaryotic CFPS
I would produce a small antimicrobial peptide or a bacterial enzyme in an E. coli CFPS system. For example, a designed peptide such as PiezoTone-His could be produced in this system because it is relatively small and does not require complex eukaryotic modifications. CFPS would also be useful if the peptide is toxic to living E. coli cells.
Protein example for eukaryotic CFPS
I would produce a human membrane receptor or a protein with disulfide bonds in a eukaryotic system. For example, a human G-protein-coupled receptor, or GPCR, would be better suited to a eukaryotic CFPS system supplemented with microsomes, liposomes, or nanodiscs. This is because GPCRs need proper membrane insertion and folding, which are difficult to achieve in a simple bacterial system.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To optimize the expression of a membrane protein in a cell-free system, I would design the experiment around both expression yield and correct folding.
Target protein
As an example, I would choose a membrane protein such as a GPCR or an ion channel. These proteins are challenging because they contain hydrophobic transmembrane domains that can aggregate if they are not inserted into a membrane-like environment.
Main challenges
The main challenges are:
Aggregation of hydrophobic regions Membrane proteins can misfold or form aggregates in aqueous solution.
Incorrect folding The protein may be produced but not adopt its functional structure.
Lack of membrane environment Many membrane proteins need lipids, detergents, nanodiscs, or microsomes during translation.
Low yield Membrane proteins are often expressed at lower levels than soluble proteins.
Experimental setup
I would use a eukaryotic or E. coli cell-free system depending on the protein. For a human membrane protein, I would choose a eukaryotic system or an E. coli system supplemented with membrane-mimicking structures.
I would test several conditions in parallel:
Variable
Optimization strategy
DNA concentration
Test low, medium, and high template concentrations
Temperature
Compare lower temperatures to improve folding
Magnesium and potassium
Optimize ion concentration for translation efficiency
Detergents
Test mild detergents that stabilize membrane proteins
Liposomes
Add artificial lipid vesicles for co-translational insertion
Nanodiscs
Use nanodiscs to provide a controlled membrane-like environment
Chaperones
Add folding helpers if needed
Reaction time
Compare short and long incubation times
The best setup would likely include co-translational insertion into liposomes or nanodiscs. This means the membrane protein is synthesized in the presence of a membrane-like structure, allowing the hydrophobic domains to enter the lipid environment as the protein is being produced.
To evaluate success, I would measure total protein yield, soluble fraction, correct size using SDS-PAGE or Western blot, and function using a ligand-binding or activity assay if available.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
If I observe low yield of my target protein in a cell-free system, I would consider at least three possible causes.
Possible reason
Explanation
Troubleshooting strategy
Poor DNA template design
The promoter may be weak, the ribosome binding site may not work efficiently, or the coding sequence may contain rare codons.
Redesign the construct with a stronger promoter, optimized RBS, and codon optimization for the chosen system. Check DNA quality and concentration.
Incorrect reaction conditions
CFPS is sensitive to magnesium, potassium, pH, DNA concentration, and temperature.
Run a small optimization matrix testing magnesium, potassium, DNA concentration, and temperature. Include a positive control such as GFP.
Protein instability or degradation
The protein may be unstable, degraded by proteases, or prone to aggregation.
Add protease inhibitors, reduce temperature, shorten reaction time, or add stabilizing agents, chaperones, detergents, liposomes, or nanodiscs.
Additional possible reasons
Other causes of low yield include poor mRNA stability, insufficient energy regeneration, missing cofactors, incorrect folding environment, or an overloaded reaction caused by too much DNA template.
A good troubleshooting workflow would be:
Test a positive control protein to confirm the CFPS system works.
Check DNA quality and template design.
Optimize salts, temperature, DNA concentration, and energy source.
Add folding aids, cofactors, or membrane-supporting components if needed.
Analyze both total protein and soluble/functional protein, because high expression does not always mean correct folding.
Implementation Strategy Analysis
1. Could this be realized by cell-free Tx/Tl alone, without encapsulation?
Partially, but not fully.
Aspect
Cell-Free Tx/Tl Capability
Limitation
Produce IL-10 or IL-1Ra
Yes
One-shot, finite yield
Sense cytokine input dynamically
No
No living feedback loop
Sustain production over time
No
Reaction degrades within hours
Respond repeatedly to signal
No
Cannot reset or re-trigger
Cell-free systems are open-loop. They can produce the protein once, but they cannot sense, decide, and respond autonomously. The sensor-actuator logic requires a living, persistent system.
Key insight: Cell-free would be useful for prototyping the genetic circuit before building the full synthetic cell — which is exactly its strength.
2. Could a genetically modified natural cell do this?
Yes, and this is the closest real-world precedent.
A natural cell such as a T cell or macrophage could be engineered with:
A cytokine-sensing promoter (e.g. NF-κB responsive) driving IL-10 expression
This is conceptually similar to CAR-T cell engineering, already in clinical use
Comparison: Synthetic Cell vs. Genetically Modified Natural Cell
Feature
Synthetic Cell
Genetically Modified Natural Cell
Control over function
High
Moderate
Own metabolism
Must be engineered
Already present
Host immune interference
Minimal (by design)
Possible
Gene regulation interference
Minimal
High — host regulation still active
Development complexity
High
Lower (builds on existing biology)
Clinical precedent
Low
High (e.g. CAR-T)
The natural cell brings its own metabolism, membrane, and longevity — but host gene regulation, immune responses, and survival pressures interfere with the engineered function.
3. Desired Outcome of Synthetic Cell Operation
The ideal outcome has three layers:
🎯 Therapeutic Outcome
Local inflammation is suppressed at the site of the flare, tissue damage is reduced, and systemic drug exposure is minimized compared to injected biologics.
⚙️ Operational Outcome
The cell reliably switches on above a defined cytokine threshold, produces a sufficient and bounded quantity of anti-inflammatory protein, and switches off when the signal resolves — avoiding chronic immunosuppression.
🔒 Safety Outcome
The cell does not proliferate uncontrollably, does not produce protein constitutively in the absence of signal, and can ideally be cleared or switched off externally if needed.
Conceptual Summary
The synthetic cell behaves like a biological thermostat:
[Cytokine signal rises]
↓
[Sensor promoter activates]
↓
[IL-10 / IL-1Ra produced and secreted]
↓
[Local inflammation suppressed]
↓
[Cytokine signal falls → cell returns to quiet state]
This closed-loop design is particularly relevant for chronic inflammatory conditions such as:
Rheumatoid arthritis
Inflammatory bowel disease (IBD)
Psoriasis
Where localized, on-demand anti-inflammatory delivery would significantly reduce the side effects associated with systemic biologic therapies.
Synthetic Cell: Full Component Design & Experimental Details
1. Cell Design Components
A. Membrane Composition
The membrane is a giant unilamellar vesicle (GUV) made of four lipids chosen to mimic a mammalian plasma membrane:
Lipid
Role
POPC
Main phospholipid backbone, fluid bilayer at 37°C
DOPE
Promotes negative curvature, supports membrane protein insertion
Bacterial (E. coli) CFPS would not work here for three reasons:
The NF-κB responsive promoter requires mammalian RNA Pol II and eukaryotic transcription factors — E. coli sigma factors cannot drive it
The Tet-ON system (rtTA3 + TRE3G) is designed for mammalian transactivation machinery
IL-10 and IL-1Ra are human proteins that benefit from a mammalian co-translational folding environment
D. Communication with the Environment
The lipid bilayer is largely impermeable to cytokines and proteins, so two mechanisms are required:
Direction
Mechanism
Gene
Input — sense TNF-α
Transmembrane receptor TNFR1 co-translationally inserted during GUV formation
TNFRSF1A
Input — sense IL-6
Receptor complex gp130 + IL-6Rα
IL6ST + IL6R
Output — secrete IL-10 / IL-1Ra
Alpha-hemolysin (α-HL) pore, a self-assembling heptameric channel (~2 nm lumen) that allows protein diffusion out
hla (from S. aureus)
2. Experimental Details
Full Gene List
Gene
Product
Purpose
TNFRSF1A
TNFR1
Senses extracellular TNF-α
IL6ST
gp130
IL-6 signal transducer
IL6R
IL-6Rα
IL-6 receptor alpha chain
RELA
p65 NF-κB
Transcriptional activator
NFKB1
p50 NF-κB
Dimerization partner of p65
IKBKB
IKKβ
Phosphorylates and releases IκBα
IL10
IL-10
Anti-inflammatory output
IL1RN
IL-1Ra
Anti-inflammatory output
EGFP
GFP
Fluorescent reporter of circuit activity
hla
α-hemolysin
Pore for protein secretion
How to Measure Function
Assay
What it measures
ELISA
IL-10 and IL-1Ra concentration in supernatant — primary functional readout
Fluorescence microscopy / flow cytometry
EGFP signal confirms circuit activation
Western blot
Total protein production and correct molecular weight
Confocal microscopy
Vesicle integrity, receptor localization, co-localization of output
DLS (dynamic light scattering)
Vesicle size distribution and stability over time
Luminex bead array
Multiplexed cytokine detection — IL-10, IL-1Ra, and any off-target cytokines simultaneously
Macrophage activation assay
Functional test — do secreted proteins suppress LPS-activated macrophages?
Note: The macrophage activation assay is the most critical readout. It tests whether the output is biologically active, not just present.
Summary: Signal Flow
[TNF-α / IL-6 in tissue]
↓
[TNFR1 / gp130 receptors on membrane surface]
↓
[NF-κB pathway activated: IKBKB → IκBα release → RELA/NFKB1 nuclear entry]
↓
[NF-κB promoter drives IL10 transcription]
[TRE3G promoter drives IL1RN + EGFP transcription]
↓
[Proteins produced by mammalian CFPS machinery]
↓
[IL-10 and IL-1Ra exit via α-hemolysin pore]
↓
[Local inflammation suppressed]
Bioreactive Architectural Wall Panel: Cell-Free Air Purification Surface
I really got inspired by this paper: and approach of bioinspiration:
Figure 1. Example diagram related to synthetic cells and bioengineering systems. Source: Frontiers in Bioengineering and Biotechnology.
Based on: Ho, Kubušová et al. (2023) — Multiscale design of cell-free biologically active architectural structures, Frontiers in Bioengineering and Biotechnology. https://doi.org/10.3389/fbioe.2023.1125156
Field: Architecture
One-sentence pitch
A 3D-printed silk fibroin indoor wall panel, built on the multiscale CFPS biopolymer platform demonstrated by Ho et al. (2023), that autonomously detects formaldehyde and VOC off-gassing from furniture and produces a laccase enzyme in situ to oxidatively degrade them — turning the building surface itself into a living air-purification membrane.
How it works
The paper by Ho et al. demonstrates that freeze-dried CFPS pellets can be mechanically attached into 3D-printed foldable fibrous biopolymer lattices — combining silk fibroin and sodium alginate matrices with cell-free transcription-translation machinery across three design scales: microscale expression within the biopolymer matrix, mesoscale variation of porosity and strength within printed lattices, and macroscale folded indoor surfaces at the meter scale. This proposal takes that exact platform and redirects it toward a functional air-quality application.
The wall panel is fabricated by the same extrusion-based additive manufacturing approach, with the biopolymer lattice designed at mesoscale to maximize surface-area-to-volume ratio and air contact. At the microscale, freeze-dried CFPS pellets carry two components:
A formaldehyde-responsive biosensor circuit — using the frmR repressor and PfrmA promoter from E. coli
A DNA template encoding fungal laccase — lcc2 from Trametes versicolor
Laccase is a copper-containing oxidoreductase that degrades formaldehyde, benzene, and other VOCs into non-toxic products. When indoor humidity contacts the biopolymer matrix — as naturally occurs in occupied spaces — it partially rehydrates the CFPS pellets and initiates transcription and translation. If formaldehyde is simultaneously present at the surface, the frmR repressor is inactivated, the PfrmA promoter opens, and laccase is expressed and diffuses outward through the porous lattice into the surrounding air layer.
Societal challenge addressed
Indoor air quality is a recognized public health problem: the EPA estimates that indoor VOC concentrations are routinely 2–5× higher than outdoor levels, driven primarily by off-gassing from furniture, flooring, adhesives, and paints. Formaldehyde is classified as a Group 1 carcinogen by IARC and is present in virtually every modern interior.
Current solutions — activated carbon filters, air purifiers, houseplants — are passive, require maintenance, consume energy, and do not scale to building surfaces. A biologically active architectural surface that responds proportionally to pollutant load and degrades contaminants without electricity or consumables addresses this gap directly, and aligns with the paper’s stated goals of supporting health, resource optimization, and reduction of energy use in the built environment.
Addressing cell-free limitations — grounded in the paper’s framework
Limitation
Strategy — based on Ho et al. approach
Water activation
The paper notes that biomaterials are excellent candidates to host CFPS due to their high water content. In an occupied interior, ambient humidity (40–60% RH) provides the moisture needed to partially solvate the silk fibroin matrix and rehydrate pellets — no liquid water required
Stability
The paper uses commercially available freeze-dried CFPS pellets embedded in the biopolymer matrix. Lyophilized CFPS in silk fibroin maintains activity over extended storage; trehalose co-lyophilization can extend shelf life to 1+ year at room temperature
One-time use
The paper proposes modular fabrication with biologically active zones, which directly enables a replacement strategy: individual lattice modules carrying CFPS pellets are designed as snap-in cartridges within the larger panel frame. Spent modules are composted — the silk fibroin and alginate matrix is fully biodegradable — and replaced seasonally
Expression levels
The paper acknowledges challenges to experimental setup affecting expression levels as an open problem. For a laccase application, the key insight is that enzymatic output accumulates and acts catalytically — even modest expression yields meaningful degradation activity, unlike stoichiometric therapeutic proteins
Scale
The paper’s three-scale framework (micro → meso → macro) directly solves this: CFPS activity is engineered at microscale, pore geometry for air diffusion at mesoscale, and structural integrity of the full wall surface at macroscale
Key differences from the paper’s GFP reporter proof-of-concept
The paper uses GFP as a reporter to confirm CFPS compatibility with the biopolymer matrix. This proposal takes the next step: replacing the reporter with a functional enzymatic output (lcc2 laccase) and adding an upstream inducible biosensor circuit (frmR/PfrmA) so that expression is conditional on pollutant detection rather than always-on.
This converts the platform from a demonstrator into a closed-loop sense-and-respond system, which is precisely the trajectory the paper describes as the future direction for bio-interactive architectural structures.
Multiscale design summary
Scale
Element
Design choice
Microscale
CFPS pellets in silk fibroin / sodium alginate matrix
High surface-area-to-volume ratio; pore size tuned for air diffusion and humidity uptake
Macroscale
Foldable wall panel, meter scale
Modular snap-in cartridge zones; structurally sound; fully compostable at end-of-life
Key genes and components
Component
Gene / molecule
Role
Biosensor repressor
frmR (E. coli)
Binds formaldehyde; releases promoter when formaldehyde detected
Inducible promoter
PfrmA (E. coli)
Drives transcription only when frmR is inactivated by formaldehyde
Enzymatic output
lcc2 (Trametes versicolor)
Laccase — oxidizes and degrades VOCs including formaldehyde and benzene
Reporter (validation)
EGFP
Confirms CFPS activity in biopolymer matrix (as per Ho et al.)
Matrix material
Silk fibroin + sodium alginate
Biopolymer host; high water content; 3D-printable; biodegradable
Cryoprotectant
Trehalose
Stabilizes freeze-dried CFPS pellets during storage
Copper cofactor
Cu²⁺ ions
Required for laccase activity; co-loaded into pellet
Signal flow
[Ambient humidity contacts silk fibroin matrix]
↓
[CFPS pellets rehydrate → transcription/translation activates]
↓
[Formaldehyde / VOC present at panel surface?]
↓ Yes
[frmR repressor inactivated → PfrmA promoter opens]
↓
[lcc2 laccase synthesized and secreted through lattice pores]
↓
[Laccase oxidizes VOCs → non-toxic products]
↓
[Indoor air quality improved — no electricity, no maintenance]
References
Ho, G., Kubušová, V., Irabien, C., Li, V., Weinstein, A., Chawla, S., Yeung, D., Mershin, A., Zolotovsky, K., & Mogas-Soldevila, L. (2023). Multiscale design of cell-free biologically active architectural structures. Frontiers in Bioengineering and Biotechnology, 11. https://doi.org/10.3389/fbioe.2023.1125156
Genes in Space — Mock Proposal
Title: Early detection of spaceflight-induced muscle atrophy using cell-free toehold switch biosensors
Tools used: BioBits® cell-free protein expression system · miniPCR® thermal cycler · P51 Molecular Fluorescence Viewer
1. Background
(100 words max)
Spaceflight causes rapid skeletal muscle atrophy — astronauts can lose up to 20% of muscle mass during a 6-month mission. This impairs performance, increases injury risk, and complicates post-flight recovery. Current monitoring relies on infrequent MRI or exercise tests, which cannot track molecular-level changes in real time. On long-duration missions to Mars, detecting early-stage atrophy at the molecular level would enable timely countermeasure adjustments before irreversible mass loss occurs. Understanding the molecular drivers of spaceflight-induced atrophy also informs treatment of age-related muscle loss on Earth, giving this research dual significance for space exploration and human health broadly.
2. Molecular target
(30 words max)
mRNA transcripts of atrogin-1 (FBXO32) and MuRF1 (TRIM63) — E3 ubiquitin ligase genes that are early transcriptional markers of skeletal muscle atrophy — detected in astronaut blood samples.
3. Target relevance
(100 words max)
Atrogin-1 and MuRF1 are the master regulators of the ubiquitin-proteasome pathway that drives muscle protein degradation. Both genes are transcriptionally upregulated within hours of muscle disuse or microgravity exposure, making their mRNA levels sensitive early indicators of atrophy onset — detectable before measurable mass loss occurs. Elevated transcript levels in blood reflect active muscle breakdown signaling. Monitoring these markers longitudinally during a mission would give flight surgeons a real-time molecular window into crew muscle health, enabling proactive rather than reactive adjustment of exercise countermeasures such as resistive training protocols.
4. Hypothesis and reasoning
(150 words max)
We hypothesize that toehold switch biosensors targeting atrogin-1 and MuRF1 mRNA, deployed in a BioBits® freeze-dried cell-free expression system, can detect upregulation of these atrophy markers in astronaut blood during spaceflight — providing an early warning of muscle degradation before clinically detectable mass loss occurs.
Toehold switches are programmable RNA sensors that trigger translation of a GFP reporter only when a complementary target mRNA sequence is present. By encoding atrogin-1- and MuRF1-specific toehold switches into a BioBits® reaction, we create a portable, single-use diagnostic requiring no living cells, no cold chain, and no specialized equipment. A positive GFP signal, read with the P51 Molecular Fluorescence Viewer, indicates active atrophy signaling in that sample. This approach is uniquely suited to spaceflight constraints: the entire assay fits in a small pouch, is stable at room temperature as a lyophilized pellet, and produces a result readable by a non-specialist crew member.
5. Experimental plan
(100 words max)
Samples: Weekly fingerprick blood draws from crew members throughout the mission.
Controls:
Positive control — synthetic atrogin-1 / MuRF1 mRNA added directly to BioBits® reaction
Negative control — BioBits® reaction with no RNA added
Protocol:
Extract total RNA from blood sample
Reverse-transcribe and amplify cDNA using miniPCR® with T7 promoter-tagged primers
Read GFP fluorescence with P51 Viewer
Measurements: Fluorescence intensity proportional to target mRNA abundance, tracked longitudinally across the mission to detect atrophy trajectory.
Primary atrophy marker; upregulated within hours of microgravity exposure
MuRF1 (TRIM63) mRNA
Secondary atrophy marker; co-upregulated with atrogin-1 during muscle degradation
Toehold switch constructs
RNA sensor sequences that unlock GFP translation only in the presence of target mRNA
BioBits® CFPS extract
Freeze-dried transcription-translation machinery; stable at room temperature
GFP reporter
Fluorescent output protein produced when toehold switch is triggered
miniPCR®
Amplifies and tags cDNA with T7 promoter for use in BioBits® reaction
P51 Viewer
Portable fluorescence reader; detects GFP signal without laboratory equipment
Why this works in space
Spaceflight constraint
How this proposal addresses it
No cold chain for reagents
BioBits® is freeze-dried and stable at room temperature
No laboratory equipment
miniPCR® and P51 are compact, low-power, purpose-built for field use
Crew time is limited
Entire assay requires minimal hands-on steps; result is a simple yes/no fluorescence signal
No trained laboratory personnel
P51 readout requires no interpretation beyond presence or absence of fluorescence
Resource scarcity
Single-use lyophilized pellets; minimal consumables per assay
No living cell cultures
BioBits® CFPS uses no living organisms; no containment risk
week 10 imaging-and-measurement
Measurement Plan for Final Project: Piezoelectric Tone Modulation
Project Context
My final project explores a concept called Piezoelectric Tone Modulation, where a biologically produced peptide or protein-based scaffold, called here PiezoTone, could be integrated into a soft robotic wearable system for muscle tone modulation or rehabilitation support. The project combines synthetic biology, biomaterials, and wearable soft robotics.
Because this project includes both a biological production phase and a functional wearable prototype phase, I would measure several aspects at different levels:
DNA/plasmid design and verification
Protein or peptide expression
Protein purification and identity
Material integration into a scaffold or film
Piezoelectric/mechanical response
Muscle-related sensing or tone modulation performance
Biocompatibility and safety, if the project advances toward biological or wearable testing
1. DNA / Plasmid Verification
What I would measure
The first element I would measure is whether the designed DNA construct is correct. The plasmid should contain the correct components for expression in E. coli:
Promoter
Ribosome binding site
PiezoTone peptide/protein coding sequence
His-tag for purification
Terminator
Antibiotic resistance marker
Origin of replication
Why this is important
Before producing the peptide or protein, I need to confirm that the genetic construct is correct. If the sequence has mutations, missing regions, or incorrect orientation, the expression may fail.
Technologies and methods
Agarose Gel Electrophoresis
I would use agarose gel electrophoresis to verify the size of the plasmid or DNA insert after digestion or PCR.
Procedure:
Prepare a sample of plasmid DNA.
Digest the plasmid with restriction enzymes or amplify the insert by PCR.
Load the DNA sample into an agarose gel.
Run the gel using an electric field.
Compare the DNA bands with a DNA ladder.
Confirm whether the band size matches the expected plasmid or insert size.
Expected result:
A DNA band corresponding to the expected size of the PiezoTone insert and/or complete plasmid.
Sanger Sequencing
I would use Sanger sequencing to confirm the exact nucleotide sequence of the PiezoTone coding region.
Procedure:
Send the plasmid DNA with specific sequencing primers.
Sequence across the promoter, coding sequence, His-tag, and terminator.
Compare the sequencing result with the designed sequence in Benchling.
Check for mutations, frame shifts, or incorrect orientation.
Expected result:
The sequence should match the designed PiezoTone construct with no unwanted mutations.
2. Protein / Peptide Expression
What I would measure
After confirming the plasmid, I would measure whether the PiezoTone peptide or protein is successfully expressed in E. coli or in a cell-free expression system.
The main measurable elements are:
Presence or absence of the PiezoTone protein
Approximate molecular weight
Expression level
Solubility of the protein
Difference between induced and non-induced samples
Why this is important
The project depends on producing the PiezoTone peptide/protein. Measuring expression allows me to know whether the biological system is producing the desired material.
Technologies and methods
SDS-PAGE
I would use SDS-PAGE to separate proteins by molecular weight and verify whether a new band appears at the expected size of the PiezoTone protein.
SDS-PAGE is a standard method used to analyze protein expression and purity. In protein expression workflows, SDS-PAGE is commonly used together with Western blotting to verify whether a recombinant protein has been produced. oai_citation:0‡PMC
Procedure:
Grow transformed E. coli cells containing the PiezoTone plasmid.
Induce expression, for example with IPTG if using an inducible promoter.
Collect samples before and after induction.
Lyse the cells.
Separate soluble and insoluble fractions.
Load samples on an SDS-PAGE gel.
Stain the gel with Coomassie Blue.
Compare the bands with a protein ladder.
Samples to compare:
Non-induced cells
Induced cells
Soluble fraction
Insoluble pellet
Purified protein fraction
Expected result:
A protein band should appear at the expected molecular weight after induction. A stronger band in the induced sample would suggest successful expression.
3. Protein Identity and Purification
What I would measure
After expression, I would measure whether the produced protein is really the PiezoTone protein and whether it can be purified.
If the PiezoTone construct includes a His-tag, I would purify it using Ni-NTA affinity chromatography. His-tagged proteins can bind to immobilized metal ions such as nickel, cobalt, or copper, which makes the tag useful for purification and detection. oai_citation:1‡thermofisher.com
Procedure:
Lyse the transformed E. coli cells.
Apply the protein lysate to a Ni-NTA column.
Allow the His-tagged PiezoTone protein to bind to the nickel resin.
Wash away non-specific proteins.
Elute the His-tagged protein using imidazole.
Analyze the eluted fractions by SDS-PAGE.
Expected result:
The purified fraction should show a stronger and cleaner band at the expected molecular weight.
Western Blot
I would use Western blotting with an anti-His antibody to confirm that the detected protein contains the His-tag.
Procedure:
Run the expressed protein on SDS-PAGE.
Transfer the proteins to a membrane.
Incubate the membrane with an anti-His antibody.
Detect the signal.
Confirm whether the band appears at the expected size.
Expected result:
A positive band at the expected molecular weight would confirm the presence of the His-tagged PiezoTone protein.
Protein Concentration Assay
I would measure protein concentration using a Bradford assay, BCA assay, or Nanodrop-based protein measurement.
Procedure:
Prepare a standard curve using known protein concentrations.
Add the protein sample to the assay reagent.
Measure absorbance using a spectrophotometer or plate reader.
Calculate the concentration of the purified protein.
Expected result:
A quantitative value in mg/mL or µg/mL, showing how much PiezoTone protein was produced.
Mass Spectrometry
For stronger confirmation, I would use mass spectrometry to verify the molecular mass and identity of the protein. Protein purification facilities often use mass spectrometry to confirm protein identity after purification. oai_citation:2‡embl.org
Procedure:
Excise the protein band from an SDS-PAGE gel or prepare the purified protein in solution.
Digest the protein into peptides, commonly using trypsin.
Analyze the peptides by mass spectrometry.
Compare the detected peptide masses with the expected PiezoTone sequence.
Expected result:
The detected peptide fragments should match the designed PiezoTone sequence.
4. Material Integration into a Soft Robotic Scaffold
What I would measure
Once the PiezoTone protein or peptide is produced, I would measure whether it can be integrated into a material system, such as:
Hydrogel
Biofilm
Textile coating
Flexible polymer scaffold
Soft robotic actuator layer
The measurable elements are:
Protein distribution in the material
Protein retention after washing or deformation
Film or scaffold thickness
Surface morphology
Mechanical stability
Adhesion to textile or soft substrate
Technologies and methods
Microscopy
I would use optical microscopy or fluorescence microscopy if the protein is labeled.
Purpose:
Observe whether the material coating is homogeneous.
Check whether the protein or peptide is distributed across the scaffold.
Detect cracks, aggregation, or irregular deposition.
SEM: Scanning Electron Microscopy
If available, I would use SEM to observe the microstructure of the scaffold.
Purpose:
Analyze surface morphology.
Observe fibers, pores, or crystalline structures.
Compare untreated and PiezoTone-coated samples.
FTIR Spectroscopy
I would use FTIR spectroscopy to detect chemical bonds and confirm whether the protein or peptide is present in the material.
Purpose:
Identify characteristic amide peaks from proteins.
Compare the base material with the PiezoTone-integrated material.
Verify chemical interaction between protein and scaffold.
Contact Angle Measurement
If the material is intended to interact with skin or biological fluids, I would measure the contact angle.
Purpose:
Determine whether the surface is hydrophilic or hydrophobic.
Understand how the material might behave when placed on skin.
Compare before and after protein coating.
5. Piezoelectric or Electromechanical Response
What I would measure
Because the project is related to piezoelectric tone modulation, I would measure whether the material generates an electrical signal when mechanically deformed.
The measurable elements are:
Voltage output under pressure or bending
Current output
Signal stability over repeated cycles
Sensitivity to deformation
Response time
Durability after repeated mechanical loading
Why this is important
The key functional hypothesis is that the PiezoTone-based material or hybrid scaffold could participate in mechanical-electrical interaction. If the material is compressed, stretched, or bent, it should ideally generate a measurable electrical response or modify the mechanical/electrical behavior of the wearable system.
Technologies and methods
Oscilloscope or Digital Multimeter
I would use an oscilloscope or sensitive digital multimeter to measure voltage output.
Procedure:
Place electrodes on the PiezoTone-integrated material.
Apply controlled pressure, bending, or stretching.
Record the voltage response.
Repeat the test under different forces and frequencies.
Compare the response with a control sample without PiezoTone.
Expected result:
The PiezoTone-integrated material should show a measurable electrical response under mechanical deformation.
Force Sensor + Voltage Measurement
To quantify the relationship between force and voltage, I would combine:
Force sensor
Mechanical testing setup
Oscilloscope or data acquisition board
Procedure:
Apply known forces to the sample.
Measure the generated voltage.
Plot voltage output against applied force.
Calculate sensitivity.
Possible output data:
Voltage-force curve
Peak voltage
Signal repeatability
Response under cyclic loading
Cyclic Mechanical Testing
I would test the material under repeated bending or compression cycles.
Purpose:
Evaluate durability.
Measure whether the signal decreases over time.
Understand whether the material is suitable for wearable use.
6. Muscle Tone / Wearable Performance Measurements
What I would measure
If the project advances into a wearable prototype, I would measure how the system interacts with muscle activity or muscle tone.
The measurable elements could include:
Muscle activation
Muscle contraction
Muscle stiffness or tone
Movement range
User comfort
Pressure applied by the wearable
Response of the actuator to body movement
Technologies and methods
EMG: Electromyography
I would use surface electromyography to measure electrical activity of muscles.
Purpose:
Detect muscle activation.
Compare muscle activity before, during, and after using the wearable.
Understand whether the system supports or modulates muscle effort.
Procedure:
Place surface EMG electrodes on the target muscle.
Record baseline muscle activity.
Activate or apply the wearable system.
Record muscle activity during movement or assisted movement.
Compare EMG amplitude and frequency changes.
Expected result:
If the wearable supports movement, the EMG signal may show reduced effort for the same movement task, or a change in activation pattern.
Mechanomyography / Piezoresistive Sensing
Mechanomyography measures mechanical vibrations or movements produced by muscle contraction. Wearable force-sensitive or piezoresistive sensors have been explored as alternatives or complements to EMG for measuring muscle contraction. oai_citation:3‡PMC
Purpose:
Measure the mechanical behavior of the muscle.
Detect contraction intensity.
Compare muscle mechanical response with and without the wearable.
Procedure:
Place a piezoresistive or vibration sensor over the target muscle.
Ask the participant or test system to perform controlled movements.
Record the mechanical signal.
Compare the signal to EMG and actuator output.
Ultrasound or Wearable Ultrasonic Sensing
For advanced validation, I could use ultrasound to measure muscle thickness or contraction parameters. Wearable ultrasonic sensors based on PVDF piezoelectric films have been used to measure skeletal muscle contractile parameters. oai_citation:4‡MDPI
Purpose:
Measure changes in muscle thickness during contraction.
Observe deeper muscle movement.
Validate whether the wearable affects muscle contraction.
Procedure:
Place the ultrasound sensor over the target muscle.
Record muscle thickness during rest and contraction.
Compare data before and after wearable assistance.
Analyze contraction timing and amplitude.
7. Biocompatibility and Skin Interaction
What I would measure
If the PiezoTone material is intended to be used close to the body or skin, I would measure basic biocompatibility and comfort-related properties.
The measurable elements are:
Skin irritation potential
Cytotoxicity
Surface pH
Breathability
Flexibility
Comfort
Moisture interaction
Technologies and methods
Cell Viability Assay
For early biocompatibility testing, I would use a cell viability assay such as MTT or Live/Dead staining.
Purpose:
Test whether the material is toxic to cells.
Compare cells exposed to the material with control cells.
Expected result:
Cells exposed to the material should maintain high viability compared with controls.
Wearability Observation
For a non-clinical prototype, I would evaluate:
Comfort
Flexibility
Skin contact
Stability during movement
Ease of wearing and removing the device
This would be done first with non-invasive user feedback and mechanical testing, not clinical claims.
Summary Table of Measurements
PiezoTone Project — Validation & Characterisation Plan
Project Element
What I Will Measure
Technology / Method
Expected Result
DNA construct
Correct sequence and size
Agarose gel electrophoresis; Sanger sequencing
Correct PiezoTone insert in plasmid
Protein expression
Presence of PiezoTone protein
SDS-PAGE
Band at expected molecular weight
Protein identity
His-tag and sequence confirmation
Western blot; mass spectrometry
Confirmed His-tagged PiezoTone protein
Protein quantity
Concentration and yield
Bradford assay; BCA assay; Nanodrop
Quantified protein concentration
Protein purification
Purity of eluted protein
Ni-NTA chromatography; SDS-PAGE
Clean purified protein band
Material integration
Distribution and morphology
Optical microscopy; fluorescence microscopy; SEM
Homogeneous coating or scaffold integration
Chemical composition
Protein–material interaction
FTIR spectroscopy
Protein-related chemical signatures
Piezoelectric response
Voltage generated by deformation
Oscilloscope; multimeter; force sensor
Measurable voltage under pressure or bending
Mechanical durability
Stability over repeated movement
Cyclic bending/compression testing
Stable response after repeated cycles
Muscle activity
Muscle electrical activation
Surface EMG
Change in muscle activation pattern
Muscle mechanical response
Contraction or vibration
Mechanomyography; piezoresistive sensors
Measurable muscle contraction signal
Muscle morphology
Muscle thickness or contraction
Ultrasound / wearable ultrasonic sensing
Change in muscle thickness during movement
Skin interaction
Comfort and biocompatibility
Cell viability assay; user comfort observation
Non-toxic and wearable material behaviour
Final Measurement Strategy
The most important measurements for my final project would be organized in three levels.
Level 1: Biological verification
First, I would confirm that the PiezoTone DNA construct is correct using agarose gel electrophoresis and Sanger sequencing. Then, I would express the protein in E. coli or in a cell-free system and verify expression using SDS-PAGE. If the protein has a His-tag, I would purify it using Ni-NTA chromatography and confirm its identity using Western blot and, ideally, mass spectrometry.
Level 2: Material and electromechanical characterization
Second, I would integrate the purified PiezoTone protein or peptide into a soft scaffold, hydrogel, coating, or textile-based material. I would measure its distribution using microscopy, its chemical presence using FTIR, and its morphology using SEM. Then, I would test whether the material produces an electrical response under mechanical deformation using an oscilloscope, force sensor, and cyclic bending or compression setup.
Level 3: Wearable and muscle-related validation
Finally, I would evaluate the wearable system as a soft robotic interface for muscle tone modulation. I would use EMG to measure muscle activation and mechanomyography or piezoresistive sensing to measure mechanical contraction. In a more advanced stage, ultrasound could be used to measure changes in muscle thickness and contraction dynamics. These measurements would help determine whether the PiezoTone-based soft wearable system can interact with muscle movement and support rehabilitation-oriented applications.
Overall, these measurements would allow me to evaluate the project from DNA design to protein production, from biomaterial integration to electromechanical response, and finally from wearable prototype to possible muscle tone modulation performance.
Correction about the expression system
Initially, I considered expressing the PiezoTone peptide/protein in E. coli. However, after reviewing the biological requirements of the target protein, I realized that E. coli may not be the most appropriate system if the protein requires a correct quaternary structure, complex folding, or post-translational modifications.
Because E. coli is a prokaryotic system, it is very useful for producing simple recombinant proteins, peptides, and bacterial proteins. However, it has limitations when expressing complex eukaryotic proteins, especially proteins that need:
Correct folding into multi-subunit or quaternary structures
Disulfide bond formation
Glycosylation or other post-translational modifications
Mammalian-like cellular processing
Membrane localization or complex protein assembly
For this reason, if the PiezoTone concept requires a protein that functions through a complex quaternary structure or needs mammalian post-translational modifications, a mammalian cell expression system may be more suitable than E. coli.
Possible mammalian expression systems include:
HEK293 cells
CHO cells
COS-7 cells
These systems would allow better protein folding, mammalian post-translational modifications, and more realistic functional behavior for proteins related to mechanosensing, ion channels, or cellular tone modulation.
Therefore, the expression strategy should be adjusted as follows:
Use E. coli only for early-stage plasmid amplification, cloning, and possibly simple peptide expression.
Use a mammalian expression system if the target protein requires complex folding, quaternary structure, or mammalian post-translational modifications.
Validate the expression using SDS-PAGE, Western blot, immunofluorescence, and possibly functional assays depending on the target protein.
Although I initially considered expressing the PiezoTone construct in E. coli, this may not be suitable if the protein requires a correct quaternary structure, complex folding, or mammalian post-translational modifications. In that case, a mammalian cell expression system such as HEK293 or CHO cells would be more appropriate.
The predicted molecular weight of the provided eGFP construct, including the LE linker and C-terminal 6×His purification tag, is approximately 28,006.6 Da for the unmodified polypeptide. However, mature eGFP undergoes chromophore formation, which involves dehydration and oxidation, resulting in a mass loss of approximately 20 Da. Therefore, the expected molecular weight of mature eGFP is approximately 27,986.6 Da.
In LC-MS analysis, the protein is expected to appear as a multiply charged ion series. Under denaturing LC-MS conditions, eGFP will unfold and typically show a broader distribution of higher charge states compared with native MS conditions, where the folded protein usually presents fewer and lower charge states.
Question 2: Adjacent Charge State Approach
Background
In electrospray ionization (ESI), a protein acquires multiple protons, producing a charge state envelope — a series of peaks at different m/z values corresponding to different numbers of charges z. The adjacent charge state method uses two neighboring peaks to simultaneously solve for z and the molecular weight M.
2.1 Determine z for each adjacent pair of peaks (n, n + 1)
Charge state determination (in short)
For each adjacent pair of peaks, corresponding to charge states (z_n) and (z_{n+1}), the charge state can be estimated using:
[
z = \frac{m/z_{n+1}}{(m/z_n) - (m/z_{n+1})}
]
where:
(m/z_n) is the mass-to-charge ratio of one peak
(m/z_{n+1}) is the mass-to-charge ratio of the adjacent peak at the next higher charge state
(z) is the charge state of the peak at (m/z_n)
z = (m/z of lower-mass adjacent peak) /
[(m/z of higher-mass peak) - (m/z of lower-mass adjacent peak)]
Then the neutral molecular weight can be estimated with: M = z × (m/z - proton mass)
where the proton mass is approximately: 1.0073 Da
For peak $n$ carrying charge $z$, and peak $n+1$ carrying charge $z + 1$ (at lower m/z), the observed m/z values are (ignoring the small proton mass as an approximation):
z is the charge state of peak n (the higher m/z peak of the pair). Peak n+1 has charge $z + 1$. Round the result to the nearest integer — charge states must be whole numbers.
2.2 Calculate M from z
Once z is known (rounded), recover the molecular weight using either peak:
From peak n:
$$M = z \cdot \frac{m}{z_n} - z \cdot 1.00728$$
Both should give the same M. Small differences reflect reading uncertainty from the spectrum.
2.3 Step-by-Step Procedure
For each adjacent pair selected from the LC-MS spectrum (Figure 1):
Read off $\left(\frac{m}{z}\right)n$ and $\left(\frac{m}{z}\right){n+1}$ from the spectrum
Plug into the formula to calculate z
Round z to the nearest integer
Calculate M using the rounded z
Repeat for a second adjacent pair
Average the M values → report as experimental MW
Compare to the theoretical value from Question 1
2.4 Worksheet Template
Peak pair
$(m/z)_n$
$(m/z)_{n+1}$
z (calc)
z (rounded)
M (Da)
Pair 1
Pair 2
Average M
2.5 Interpreting the Charge State Distribution
State
Conditions
Expected Charge States
Appearance in Spectrum
Native (folded)
Aqueous, near-physiological pH
Lower z (fewer charges)
Peaks at higherm/z, narrow envelope
Denatured (unfolded)
Acidic pH, organic solvent
Higher z (more charges)
Peaks at lowerm/z, broad envelope
Key concept: In the native state, the compact folded structure shields many basic residues from protonation. In the denatured state, the fully unfolded chain exposes all basic sites. Despite different charge envelopes, both states yield the same molecular weight M.
3. Calculate the measurement accuracy / relative error
Using the labelled adjacent charge-state peaks in the intact eGFP LC-MS spectrum, the charge states were assigned from approximately 33+ to 28+. The molecular weight was calculated using the relationship:
[
MW = z \times (m/z - H)
]
where (H = 1.0073) Da. Across the selected charge states, the calculated molecular weights were highly consistent, giving an average experimental molecular weight of approximately 27,983.2 Da.
The predicted molecular weight of mature eGFP containing the LE linker and C-terminal 6×His tag is approximately 27,986.6 Da. Therefore, the relative error of the measurement is:
This is excellent mass accuracy. The small residual error arises from reading peak positions off a printed figure; the Xevo G3 QTof achieves < 5 ppm under calibrated conditions with lockspray.
eGFP Intact MS — Questions 2.2, 3, and Charge State Observation
Q2.2 — Determine MW from Adjacent Charge State Pairs
Using labeled peaks from the denatured-state envelope in Figure 1 and the formula:
$$z = \frac{\dfrac{m}{z_{n+1}}}{\dfrac{m}{z_n} - \dfrac{m}{z_{n+1}}} \qquad M = z \cdot \frac{m}{z_n} - z \cdot 1.00728$$
This is excellent mass accuracy. The small residual error arises from reading peak positions off a printed figure; the Xevo G3 QTof achieves < 5 ppm under calibrated conditions with lockspray.
Q — Can You Observe the Charge State from the Zoomed-In Peak?
Yes — the charge state is z = 19.
Why the charge state is readable here
At 30,000 resolution, the instrument just resolves individual isotope peaks within the native-state charge state envelope. Since consecutive isotopes differ by exactly 1 Da in mass, their spacing in m/z is:
The Xevo G3 at 30,000 resolution just clears this threshold. On a lower-resolution instrument (e.g. a single quadrupole or low-res QTof), the isotope peaks would merge into a single unresolved hump and the charge state could not be read this way — you would only see a broad peak and would need to use the adjacent charge state formula instead.
Summary table
Property
Value
Charge state (z) from isotope spacing
19
Isotope spacing observed (Δ m/z)
0.054
Predicted m/z at z = 19
1473.8
Observed m/z (most abundant peak)
1473.79
Minimum resolution required
28,006
Instrument resolution
30,000 ✓
Q — Can You Observe the Charge State from the Zoomed-In Peak?
Yes — the charge state is z = 19.
Why the charge state is readable here
At 30,000 resolution, the instrument just resolves individual isotope peaks within the native-state charge state envelope. Since consecutive isotopes differ by exactly 1 Da in mass, their spacing in m/z is:
The Xevo G3 at 30,000 resolution just clears this threshold. On a lower-resolution instrument (e.g. a single quadrupole or low-res QTof), the isotope peaks would merge into a single unresolved hump and the charge state could not be read this way — you would only see a broad peak and would need to use the adjacent charge state formula instead.
Trypsin cleavage sites marked with |. K = Lysine (bold), R = Arginine (italic).
MVSK|GEELFTGVVPILVELDGDVNGHK|FSVSGEGEGDATYGK|LTLK|FICTTGK|
K K K K K
LPVPWPTLVTTLTYGVQCFSR|YPDHMK|QHDFFK|SAMPEGYVQER|TIFFK|
R K K R K
DDGNYK|TR|AEVK|FEGDTLVNR|IELK|GIDFK|EDGNILGHK|
K R K R K K K
LEYNYNSHNVYIMADK|QK|NGIK|VNFK|IR|
K K K K R
HNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSK|DPNEK|R|
K K R
DHMVLLEFVTAAGITLGMDELYK|LEHHHHHH
K
Note: There are no KP or RP motifs in this sequence, so trypsin cleaves at all 26 K and R residues without exception.
MW calculated as average isotope masses (Da), including water (+18.02 Da).
Notes on specific peptides
Peptide 23 (HNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSK, 41 residues, 4473.84 Da) is unusually long because R(169) and K(210) are separated by a stretch containing no K or R — these large peptides can be difficult to detect by LC-MS due to poor chromatographic retention.
Peptides 12, 19, 22, 25 (TR, QK, IR, R) are very small (1–2 residues) and will likely not be retained on a reversed-phase LC column — they are typically not observed in a standard bottom-up peptide mapping experiment.
Peptide 6 (LPVPWPTLVTTLTYGVQCFSR) contains multiple prolines and W, making it hydrophobic and challenging to detect; however, it contains a Cys residue (from C49 of the full protein), which is typically alkylated (+57 Da, carbamidomethylation) prior to digestion.
Molecular Weight Calculation Using ExPASy PeptideMass
The theoretical molecular weight of the eGFP construct was calculated using the ExPASy PeptideMass tool. The input sequence included the full eGFP sequence, the LE linker, and the C-terminal 6×His purification tag.
The result showed a theoretical pI of 5.90, an average molecular weight of 28,006.60 Da, and a monoisotopic molecular weight of 27,988.96 Da.
eGFP Peptide Map — Questions 4 & 5
Question 4 — Does the Chromatogram Peak Count Match the Prediction?
From the tryptic digest prediction (Question 2): 27 peptides predicted with 0 missed cleavages.
The chromatogram shows fewer peaks than predicted because:
Reason
Examples
Very small peptides not retained on reversed-phase column
$$\boxed{[M+H]^+ = M + 1.00728 = \mathbf{1050.527 \ \text{Da}}}$$
Verification
The main spectrum shows a singly charged peak ($z = 1$) at 1050.524 — matching the calculated value with a difference of only 0.003 Da (~2.8 ppm), confirming $z = 2$ is correct.
Value
Most abundant m/z
525.767
Charge state z
2
Calculated [M+H]⁺
1050.527 Da
Observed [M+H]⁺ (z = 1 peak)
1050.524 Da
Mass error
~2.8 ppm ✓
Peptide identity
FEGDTLVNR (residues 115–123)
Theoretical [M+H]⁺ (monoisotopic)
1050.522 Da
eGFP Peptide Map — Questions 6 & 7
Question 6 — Peptide Identification and Mass Accuracy
Peptide Identity
From Question 5, the peptide at 2.78 min has $[M+H]^+ = 1050.527$ Da. Comparing to the predicted tryptic peptide list, this matches:
This is excellent mass accuracy, consistent with the Waters BioAccord QTof performance specification of < 5 ppm.
Question 7 — Sequence Coverage Confirmed by Peptide Mapping
From Figure 6 (Amino Acid Coverage Map):
$$\boxed{\textbf{88% sequence coverage}}$$
Value
Total residues in eGFP (with His-tag)
247
Residues confirmed by peptide mapping
~217
Sequence coverage
88%
The blue highlighted regions in Figure 6 show the residues confirmed by detected and identified tryptic peptides. The ~12% not covered corresponds to residues in peptides that were either:
Too small to be retained on the reversed-phase column (e.g. TR, QK, R, IR)
Not detected above the signal threshold
Present as missed cleavage products outside the search window
As part of Week 11, I participated in the HTGAA 1536 Pixel Artwork Canvas, a collective bioart experiment where each participant could contribute at least one pixel to a shared global artwork. The artwork was connected to cell-free reaction compositions, where each pixel represented a small contribution to a larger collaborative biological and visual system.
For my contribution, I added pixels to the shared canvas as part of the collective image composition. I enjoyed the idea that many small individual actions could come together to create a larger community artwork. This made the project feel playful, experimental, and collaborative, while also connecting visual design with biological systems and cell-free expression.
What I liked most about the project was the combination of art, biology, and community participation. It was interesting to see how a simple pixel-based interface could represent a much larger experiment involving biological reagents, reaction design, and collective authorship.
Cell-Free Master Mix Composition: Component Roles
E. coli Lysate
BL21 (DE3) Star Lysate, including T7 RNA Polymerase The lysate provides the biological machinery needed for transcription and translation, including ribosomes, tRNAs, enzymes, and metabolic components from E. coli. Because it includes T7 RNA polymerase, it can efficiently transcribe DNA templates controlled by a T7 promoter into mRNA for protein expression.
Salts / Buffer
Potassium Glutamate Potassium glutamate helps recreate an intracellular-like ionic environment for the cell-free reaction. It supports proper ribosome function, protein folding, and overall enzyme activity during transcription and translation.
HEPES-KOH pH 7.5 HEPES-KOH acts as a buffer to maintain the reaction at a stable pH around 7.5. This is important because transcription, translation, and enzyme activity are sensitive to pH changes during incubation.
Magnesium Glutamate Magnesium ions are essential cofactors for ribosome function, nucleotide interactions, and many enzymatic reactions in cell-free protein synthesis. The glutamate counterion also helps maintain a biologically compatible salt environment.
Potassium Phosphate Monobasic Potassium phosphate monobasic contributes phosphate ions and helps support the buffering capacity of the reaction. It also participates in maintaining the correct phosphate balance needed for energy metabolism and nucleotide-related reactions.
Potassium Phosphate Dibasic Potassium phosphate dibasic works together with the monobasic form to create a phosphate buffer system. The balance between monobasic and dibasic phosphate helps stabilize pH and supports long-duration cell-free reactions.
Energy / Nucleotide System
Ribose Ribose provides a sugar precursor that can be used by enzymes in the lysate to regenerate nucleotide monophosphates and support energy metabolism. In the 20-hour system, it helps sustain long-term protein production more gradually than direct high-energy substrates.
Glucose Glucose serves as a metabolic energy source that can be processed by enzymes in the lysate to help regenerate ATP and other energy carriers. This supports longer incubation times by feeding the reaction’s internal energy regeneration pathways.
AMP AMP is a nucleotide monophosphate that can be converted into higher-energy nucleotide forms needed for RNA synthesis and energy cycling. It contributes to the nucleotide pool required for transcription and sustained reaction activity.
CMP CMP provides the cytidine nucleotide precursor needed for RNA synthesis. During the reaction, it can be converted into CTP, which is incorporated into mRNA during transcription.
GMP GMP provides the guanosine nucleotide precursor needed for RNA synthesis. It can be converted into GTP, which is used in transcription and also plays roles in translation-related energy processes.
UMP UMP provides the uridine nucleotide precursor needed for RNA synthesis. It can be converted into UTP, which is incorporated into mRNA during transcription.
Guanine Guanine acts as an additional nucleobase precursor that can support nucleotide regeneration pathways. It helps maintain the supply of guanine-containing nucleotides during longer cell-free reactions.
Translation Mix: Amino Acids
17 Amino Acid Mix The 17 amino acid mix supplies most of the amino acids required to build the expressed protein. These amino acids are used by ribosomes during translation to assemble the polypeptide chain.
Tyrosine Tyrosine is added separately because it can have solubility or stability limitations in amino acid mixtures. Providing it separately helps ensure enough tyrosine is available for protein synthesis.
Cysteine Cysteine is also added separately because it is chemically reactive and can be unstable in solution. It is important for proteins that require cysteine residues, including those that may form disulfide bonds or need specific structural features.
Additives
Nicotinamide Nicotinamide supports cofactor-related metabolism and may help maintain the activity of enzymes involved in energy regeneration. In long-duration cell-free systems, it can contribute to sustaining reaction performance over time.
Backfill
Nuclease-Free Water Nuclease-free water is used to bring the reaction to the correct final volume without introducing enzymes that could degrade DNA or RNA. It ensures that the concentrations of all components are adjusted accurately while protecting the genetic template and transcripts.
Main Differences Between the 1-Hour PEP-NTP Mix and the 20-Hour NMP-Ribose-Glucose Mix
The 1-hour optimized PEP-NTP master mix uses high-energy components such as PEP and NTPs directly, making it suitable for fast protein expression over a short incubation time. In contrast, the 20-hour NMP-Ribose-Glucose system uses nucleotide monophosphates, ribose, and glucose to regenerate energy and nucleotides more gradually through enzymatic pathways in the lysate.
This makes the 20-hour system more sustainable and cost-effective for longer fluorescent protein production, while the 1-hour system is more immediate but likely less suitable for extended incubation.
Fluorescent Protein Properties Relevant to Cell-Free Expression
sfGFP sfGFP, or superfolder GFP, is useful in cell-free systems because it folds efficiently and matures rapidly compared with many other GFP variants. This strong folding behavior can improve fluorescence readout even when protein expression conditions are not ideal. oai_citation:0‡FPbase
mRFP1 mRFP1 is a red fluorescent protein that is monomeric and relatively acid tolerant, but it is somewhat slow to mature and has lower brightness compared with newer red fluorescent proteins. In a cell-free system, this means fluorescence may appear later or be weaker even if protein translation is successful. oai_citation:1‡FPbase
mKO2 mKO2 is an orange fluorescent protein with good photostability and red-shifted emission, but it has moderate acid sensitivity and a maturation time of around 108 minutes. In long cell-free reactions, maintaining pH is important so that the fluorescent signal is not reduced by acidification of the reaction mixture. oai_citation:2‡PMC
mTurquoise2 mTurquoise2 is a cyan fluorescent protein reported to mature rapidly and have very low acid sensitivity. These properties make it suitable for cell-free expression because fluorescence can develop relatively quickly and remain more stable if the reaction pH changes slightly. oai_citation:3‡FPbase
mScarlet-I mScarlet-I is a bright red fluorescent protein that is reported to be rapidly maturing, but it still has moderate acid sensitivity. In a 36-hour incubation, buffering capacity may strongly affect the final red fluorescence intensity. oai_citation:4‡FPbase
Electra2 Electra2 is a blue fluorescent protein derived from Entacmaea quadricolor. As with other fluorescent proteins, its final fluorescence depends on correct folding and chromophore maturation; blue fluorescent proteins can be more challenging to read out clearly because they may have lower brightness or require careful excitation and detection settings. oai_citation:5‡FPbase
Hypothesis for Improving Fluorescence Over a 36-Hour Incubation
Hypothesis: For mScarlet-I, increasing the buffering capacity of the 36-hour artwork master mix by optimizing HEPES-KOH pH 7.5 and the potassium phosphate monobasic/dibasic buffer system will help maintain a stable pH during long incubation. Because mScarlet-I has moderate acid sensitivity, better pH stability should reduce fluorescence loss and improve the final red signal after 36 hours.
A possible adjustment would be to keep the reaction close to pH 7.5 by testing slightly higher HEPES-KOH and phosphate buffer concentrations while maintaining magnesium and potassium levels compatible with translation. The expected effect is stronger and more stable fluorescence because the protein can fold, mature, and remain fluorescent under less acidic reaction conditions.
Short Note for the Next Experimental Phase
In the next phase, the exact reagent concentrations for the assigned artwork wells should be chosen based on the fluorescent protein in each well. For proteins with acid sensitivity, such as mKO2 and mScarlet-I, buffer optimization should be prioritized; for slower-maturing proteins such as mRFP1, the reaction should support long-term energy regeneration and oxygen availability to allow chromophore maturation over the full 36-hour incubation.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
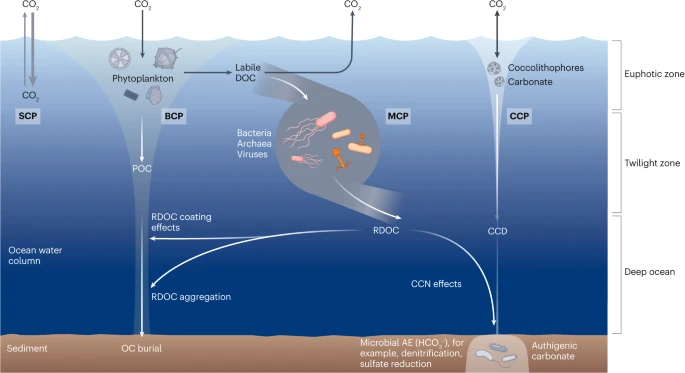
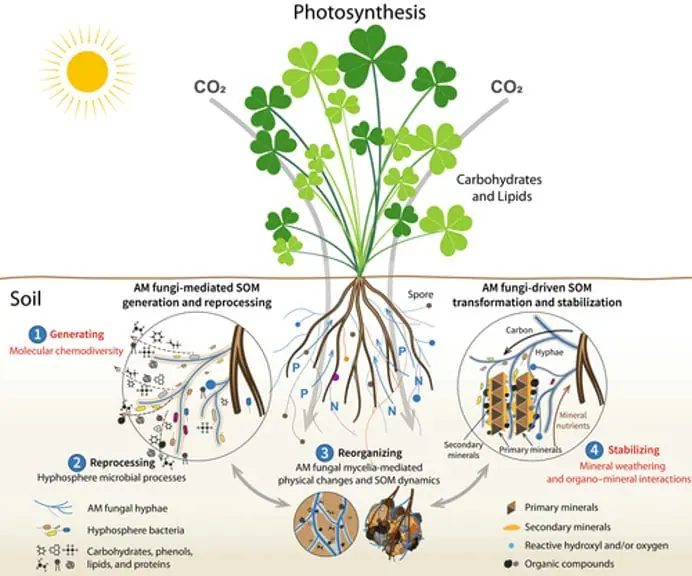
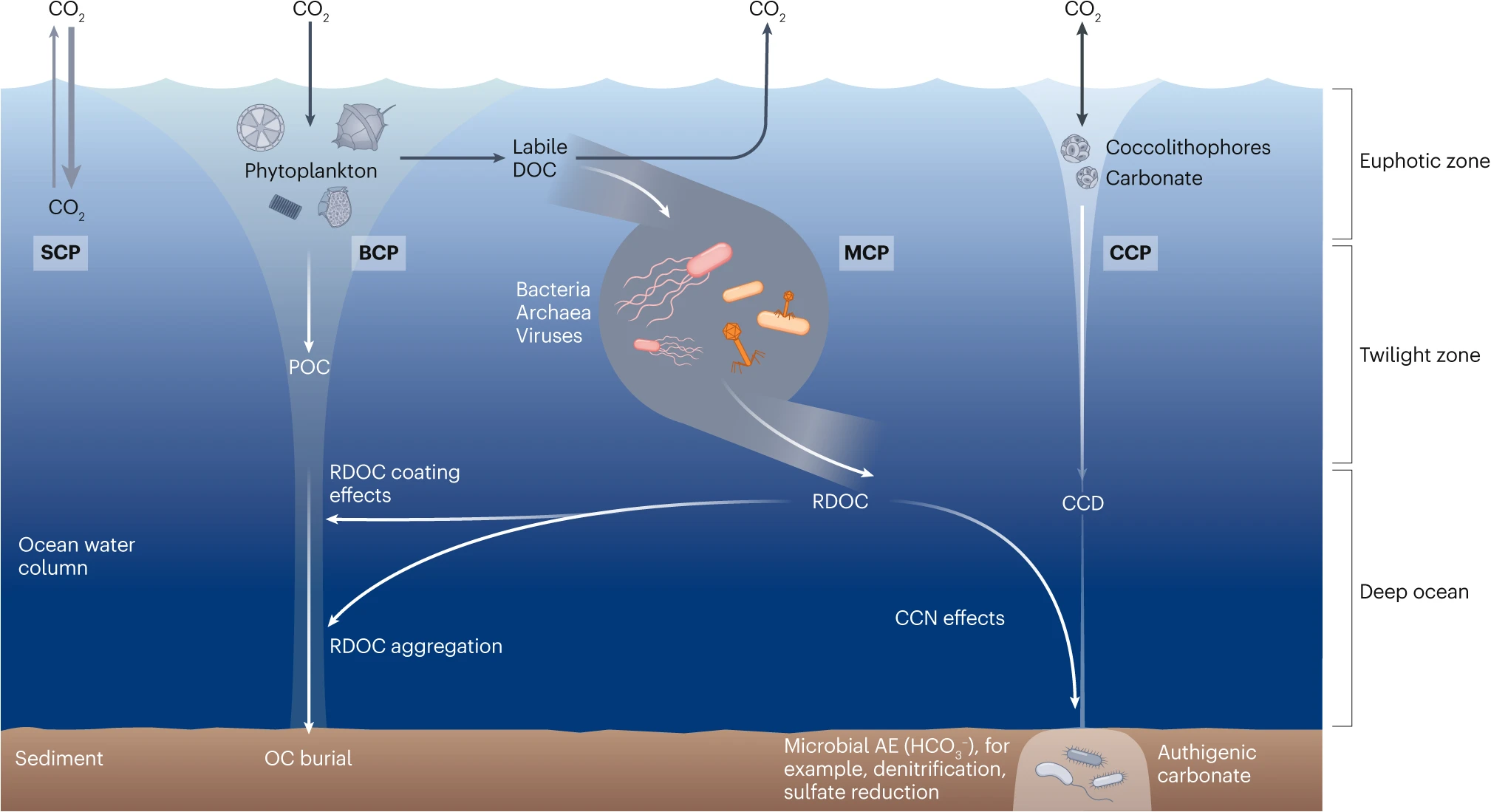
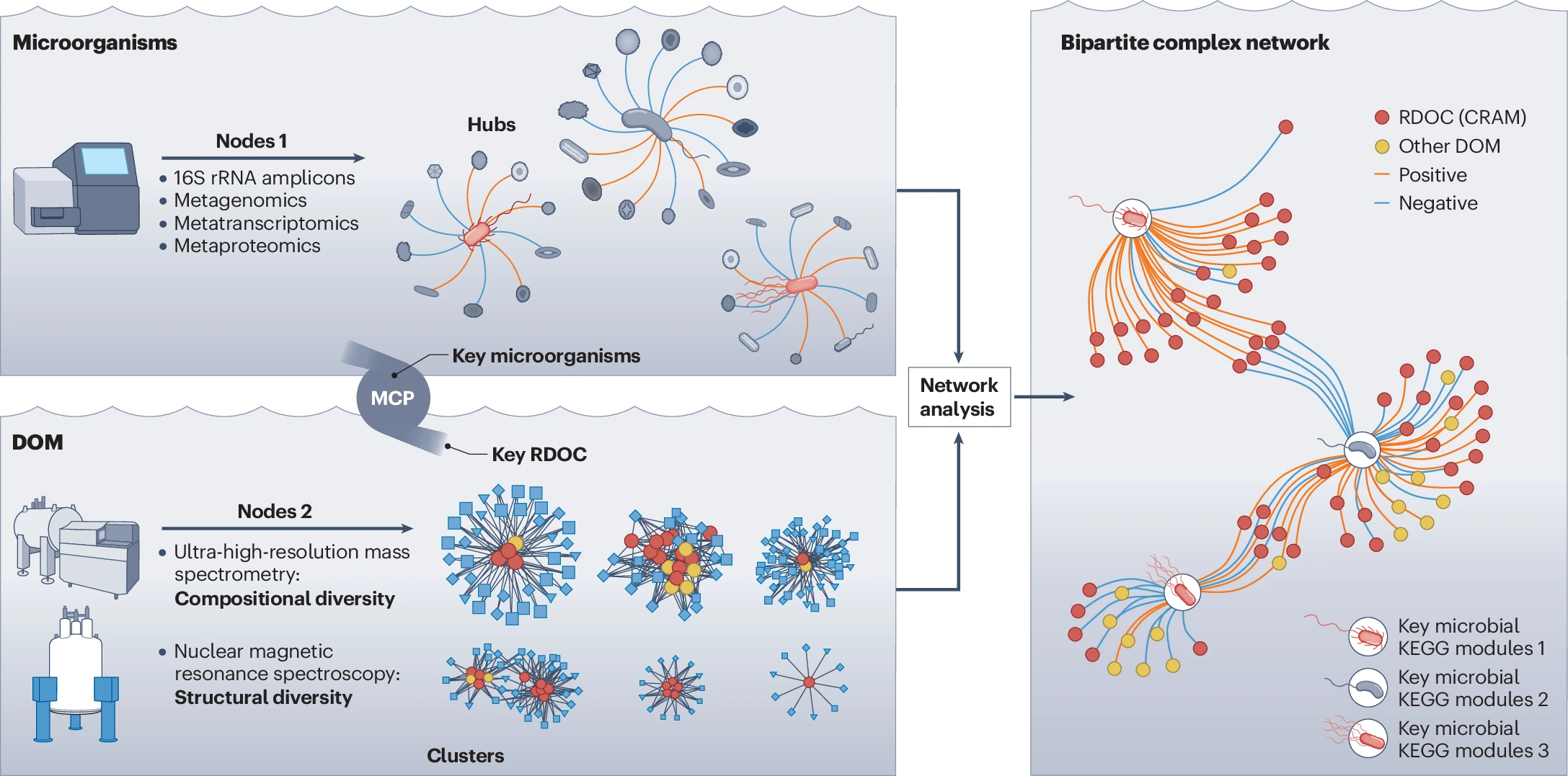
























{kind=link}
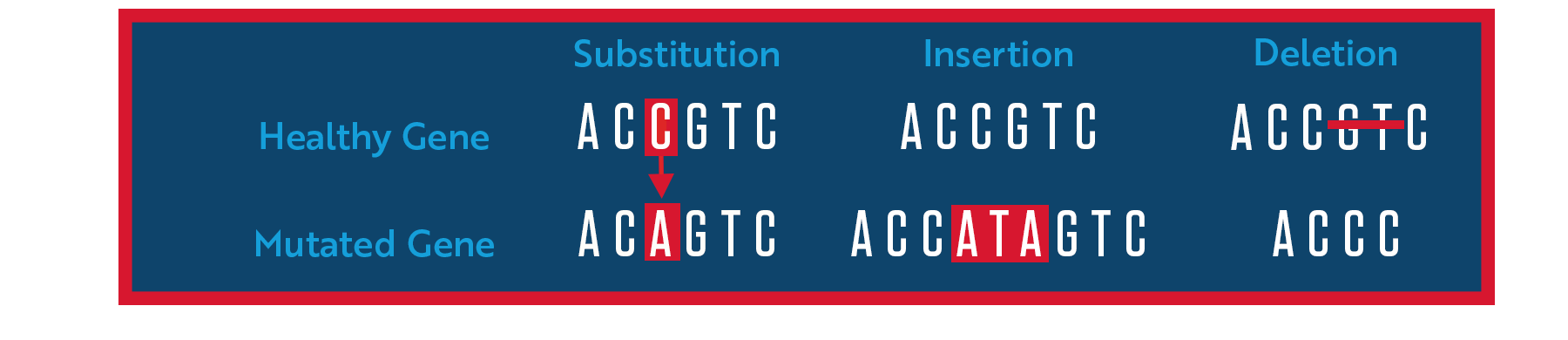{kind=link}