In Silico Peptide and DNA Construct Design for a Cutaneous Piezoelectric Biointerface
Figure 1
Figure 1. Cabrera, A. (2026f). Screenshot of later-stage construct visualization for piezoelectric biointerface design [Project documentation image]. GitHub.
Figure 2
Figure 2. Screenshot documenting a later project page or workflow visualization related to the synthetic biology and biointerface design process. Source: Cabrera (2026i).
Figure 3
Figure 3. Screenshot documenting the final or advanced project page visualization for the proposed piezoelectric biointerface workflow. Source: Cabrera (2026j).
1. Project Summary
A Synthetic Biology Approach to Piezoelectric Biomaterials for Soft Robotic Muscle Rehabilitation
I. Problem Statement
Muscle rehabilitation after stroke, spinal cord injury, or neuromuscular disease is complex.
Current electrostimulation tools are often coarse, relying on large impulses with limited adaptability.
Spasticity remains a major unresolved challenge: muscles are overactive and resist controlled movement.
Need: a gentle, adaptive, tissue-compatible interface that can modulate muscle tone and support recovery.
II. The Proposed Solution — Concept Overview
The proposed solution is a soft robotic wearable with a piezoelectric biomaterial interface.
The material sits between the device and the skin or muscle.
It can:
Sense mechanical deformation
Deliver targeted micro-electrical impulses
Potentially sense temperature
Provide vibrotactile feedback
Goal at this stage: muscle tone modulation, not full movement restoration.
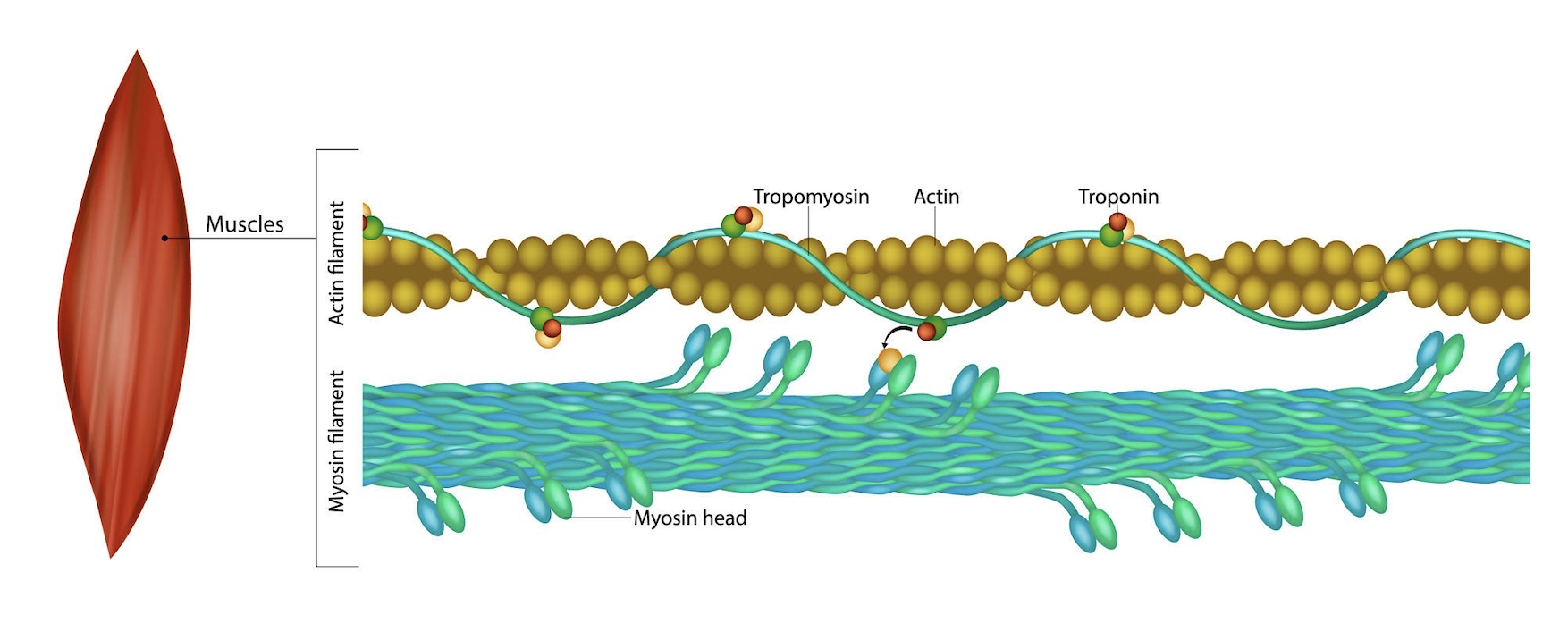
III. The Material — Piezoelectric Biomaterials
What is piezoelectricity? Piezoelectricity is the conversion between mechanical energy and electrical energy.
Biological piezoelectricity exists naturally in materials such as:
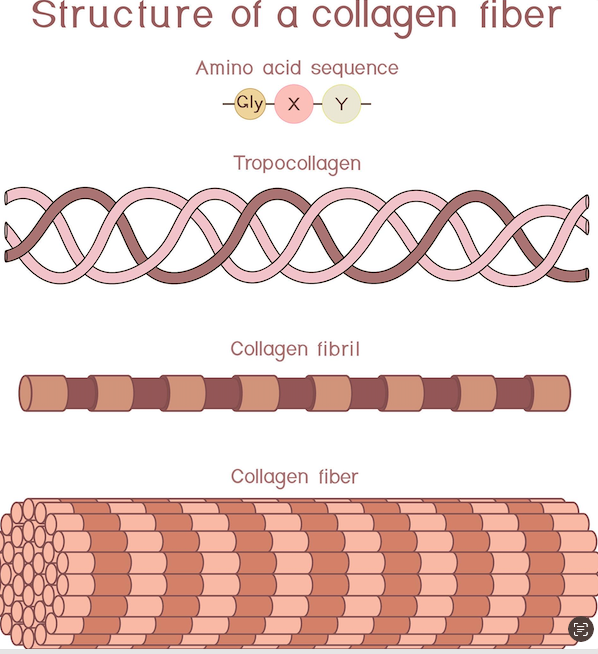
Collagen
Chitosan
Bone
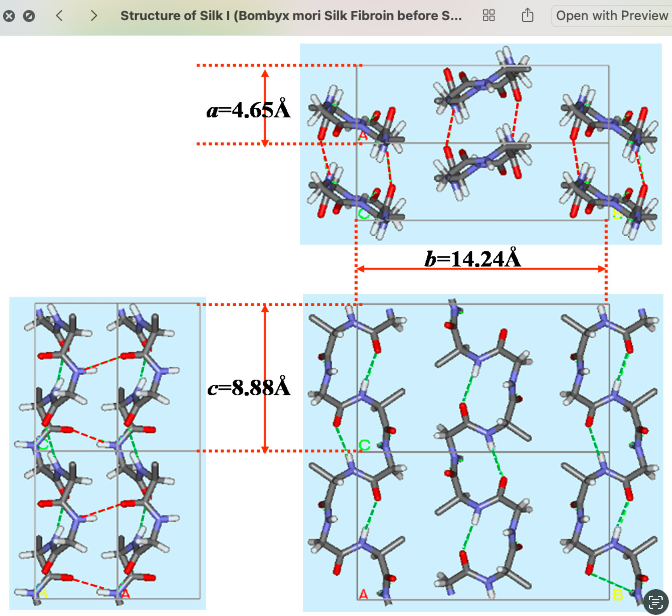
Silk
This project proposes designing a synthetic piezoelectric protein by combining amino acid sequences from collagen and chitosan.
The material can be 3D printed, enabling layered device architectures, such as:
A piezoelectric layer in contact with the skin
Supporting or functional materials layered above
IV. The Biological Engineering — Plasmid Design
The biological engineering approach is to express the piezoelectric protein in E. coli using a synthetic construct.
Plasmid components
Promoter, either constitutive or inducible
Ribosome binding site, such as the Shine-Dalgarno sequence
Target gene: piezoelectric protein sequence derived from collagen and chitosan
Linker sequences
Terminator
Tool used: Consensus / Benchling-style platform for sequence design.
Challenge: there is no existing validated plasmid for this specific piezoelectric construct. This makes the construct a novel contribution of the project.
V. Device Integration — Soft Robotics Interface
The soft robot provides the mechanical actuation, which may be:
Pneumatic
Hydraulic
Cable-driven
The piezoelectric layer functions as the smart interface, converting robot movement into electrical signals delivered to the muscle.
Advantages over conventional electrostimulation
Gentle, distributed impulses
Conformability to body geometry
Potential integration of sensing and actuation in one layer
Possible sensing of pressure and temperature
Alternative delivery formats
Patch
Micro-injection
Injectable hydrogel
Implant
VI. Clinical Targets
Potential clinical targets include:
Spasticity
Modulating overactive muscle tone in conditions such as:
Post-stroke spasticity
Cerebral palsy
Spinal cord injury
Muscle fatigue reduction
Supporting recovery during rehabilitation by reducing fatigue.
Muscle regeneration support
The material may act as a tissue scaffold while also providing stimulation.
Pelvic floor and fine motor applications
Highly customizable stimulation patterns could support applications requiring precise, localized modulation.
VII. Validation Plan
The validation plan includes:
Confirm protein expression: SDS-PAGE gel electrophoresis
Confirm protein identity: fluorescent protein tag, such as GFP, and western blot
Quantify yield: protein quantification assay
Characterize piezoelectric properties: measure electrical output under mechanical loading
Future translation: progress from in vitro testing to in vivo evaluation
VIII. Challenges and Next Steps
Key challenges and next steps include:
Completing the plasmid cassette, including promoter and terminator selection
Selecting an appropriate E. coli strain for quaternary protein structure formation
Translating the protocol from in vitro to in situ
Customizing stimulation patterns according to patient needs and anatomy
IX. Conclusion
This project proposes a first step toward a biologically derived, 3D-printable piezoelectric interface for soft robotic rehabilitation devices.
The novelty lies in combining:
Synthetic biology, through custom protein design
Soft robotics, through wearable and compliant actuation
The near-term goal is to:
Prove the construct
Characterize the material
Demonstrate muscle tone modulation
PiezoTone BioPatch is an in silico synthetic biology and biomaterials project that explores the design of a modular peptide and DNA construct for a future cutaneous piezoelectric hydrogel patch. The long-term motivation is to support research on pathological muscle-tone modulation, especially in conditions where abnormal tone, stiffness, or spasticity limits upper-limb movement, comfort, range of motion, and participation in daily life.
The project does not claim to directly treat spasticity or to prove therapeutic efficacy at this stage. Instead, it focuses on designing a molecular interface that could later be incorporated into a soft hydrogel patch placed on the skin. This cutaneous approach is safer and more feasible than an implantable scaffold because it avoids direct subcutaneous or intramuscular placement.
The project combines a chitosan–fibrin hydrogel concept, a collagen-like piezoelectric-inspired peptide motif, and cell-interface motifs such as RGD and IKVAV. The designed peptide was translated into a DNA coding sequence, annotated in Benchling, codon-optimized for E. coli, and developed toward a simulated expression cassette for future recombinant production.
2. Problematic and Research Gap
Pathological muscle tone is a complex rehabilitation challenge. In neurological conditions such as stroke, spinal cord injury, cerebral palsy, or other neuromotor disorders, abnormal tone may affect comfort, mobility, joint range of motion, and functional independence.
Across current studies, the main gap is not simply that the elbow or limb cannot be “de-spastic.” The larger gap is that elbow tone modulation is often:
not measured objectively;
not individualized to specific muscles;
not adapted to specific ranges of motion;
not clearly linked to sustained functional improvement;
not embedded in multimodal rehabilitation strategies;
not clearly connected to patient comfort, participation, and daily life.
Therefore, the project starts from the following idea:
The major gap is not only the lack of techniques to change limb muscle tone, but the lack of integrated, mechanism-informed, and patient-centered strategies that distinguish different sources of tone, target both central and peripheral contributors, and connect local tone changes to meaningful functional outcomes.
Muscle rehabilitation after stroke, spinal cord injury, or neuromuscular disease is complex.
Current electrostimulation tools are often coarse, relying on large impulses with limited adaptability.
Spasticity remains a major unresolved challenge: muscles are overactive and resist controlled movement.
Need: a gentle, adaptive, tissue-compatible interface that can modulate muscle tone and support recovery.
3. Opportunity
Piezoelectric biomaterials are interesting for rehabilitation and tissue-interface research because they can convert mechanical deformation into electrical signals. Available studies show that piezoelectric biomaterials can support nerve regeneration, reduce muscle atrophy, and improve motor recovery in animal models through self-powered or ultrasound-triggered electrical stimulation.
However, there is still no direct evidence that these materials reduce human muscle spasticity or pathological tone in neurological disorders. Therefore, this remains an important research gap.
This project uses that gap as an opportunity to ask:
Can a biofunctional cutaneous piezoelectric interface be designed in silico as a future platform for studying localized neuromuscular stimulation and pathological muscle-tone modulation?
4. Project Question
The main research question is:
How can an in silico synthetic biology design be used to create a modular peptide and DNA construct for a cutaneous piezoelectric hydrogel patch with future relevance for pathological muscle-tone modulation?
This question connects:
synthetic biology;
peptide design;
DNA construct design;
biomaterials;
hydrogel patch design;
piezoelectric interfaces;
neuromuscular rehabilitation.
5. Broad Objective
The broad objective of this project is to design, in silico, a DNA-encoded modular peptide that could functionalize a future chitosan–fibrin piezoelectric hydrogel patch for skin-contact neuromuscular stimulation research.
The project focuses on the molecular design stage, not on human testing.
6. Hypothesis
I hypothesize that a modular DNA-encoded peptide can be designed in silico to functionalize a chitosan–fibrin piezoelectric hydrogel patch, creating a skin-contact biointerface suitable for future studies of transcutaneous electroactive stimulation and pathological muscle-tone modulation.
7. Why a Cutaneous Patch?
The original concept considered a scaffold that could potentially interact with muscle or connective tissue. However, for implementation, a cutaneous patch is more feasible and safer as a first step.
A cutaneous patch would be placed on the skin rather than implanted under it.
Conceptually:
text
Skin surface
↓
Soft adhesive or hydrogel contact layer
↓
Peptide-functionalized chitosan–fibrin hydrogel
↓
Piezoelectric or collagen-inspired material layer
↓
Flexible protective backing
References:
Kamel, N. A. (2022). Bio-piezoelectricity: fundamentals and applications in tissue engineering and regenerative medicine. Biophysical Reviews, 14(3), 717–733. https://doi.org/10.1007/s12551-022-00969-z
Yogeswaran, N., Dang, W., Navaraj, W. T., Shakthivel, D., Khan, S., Polat, E. O., Gupta, S., Heidari, H., Kaboli, M., Lorenzelli, L., Cheng, G., & Dahiya, R. (2015). New materials and advances in making electronic skin for interactive robots. Advanced Robotics, 29(21), 1359–1373. https://doi.org/10.1080/01691864.2015.1095653
A related study demonstrated that biodegradable 3D piezoelectric scaffolds can deliver ultrasound-driven, wirelessly powered electrical stimulation and promote spinal cord injury repair in a rat model, supporting the relevance of piezoelectric biomaterials for regenerative neurorehabilitation (Chen et al., 2022).
Project Documentation Figures
The following screenshots document the development process of the synthetic sequence design, codon optimization, and plasmid-design workflow for the proposed piezoelectric biointerface construct.
Figure 4
Figure 4. Initial codon optimization and synthetic sequence design workflow for the proposed piezoelectric biointerface construct. Source: Cabrera (2026a).
Figure 5
Figure 5. Later stage of the sequence design workflow, including additional construct visualization or annotation. Source: Cabrera (2026b).
Figure 6
Figure 6. Refinement of the synthetic construct design and related sequence information. Source: Cabrera (2026c).
Figure 7
Figure 7. Continued development of the plasmid-design or sequence-design workflow. Source: Cabrera (2026d).
Figure 8
Figure 8. Updated design stage of the synthetic biology workflow for the biointerface construct. Source: Cabrera (2026e).
Figure 9
Figure 9. Final or later-stage visualization of the proposed construct design workflow. Source: Cabrera (2026f).
Figure 10
Figure 10. Additional stage of the project page or workflow for the proposed piezoelectric biointerface construct. Source: Cabrera (2026g).
Figure 11
Figure 11. Continued stage of the project page or workflow development. Source: Cabrera (2026h).
Project Title
Genetic Design of a Silk-Inspired Protein Module for Future Rehabilitation Biomaterials
SECTION 1: ABSTRACT
This project addresses a key challenge in wearable rehabilitation and soft robotics: many assistive devices still rely on rigid or non-biological materials that can limit comfort, adaptability, and integration with the body. Soft robotic systems offer a promising alternative, but there is still a gap in how biological material principles can be translated into programmable and manufacturable biomaterials for future wearable actuation. The overall objective of this project is to design and assemble a DNA construct encoding a protein-inspired material building block based on motifs from Bombyx mori silk fibroin and elastic protein domains, as a first step toward engineered biomaterials for soft rehabilitation devices.
The central hypothesis is that a genetically encoded silk-inspired or silk-elastin-like protein sequence can serve as a rational platform for future bio-derived films, fibers, or coatings with useful mechanical properties such as flexibility, resilience, and hierarchical assembly. To test this idea, the project will complete sequence design, codon optimization, plasmid planning, overlap design, Gibson Assembly, bacterial transformation, and clone validation. Methods include Benchling-based DNA design, PCR amplification, Gibson Assembly, E. coli transformation, colony screening, and sequence verification.
The expected outcome is a validated recombinant DNA construct that demonstrates the feasibility of integrating synthetic biology and protein design into a material-centered design workflow for future soft robotic textile applications. This project therefore functions as an enabling step between biomolecular design and the long-term development of adaptive rehabilitation wearables.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of my final project is to design, assemble, and validate a recombinant DNA construct encoding a silk-inspired or silk-elastin-like protein module by utilizing Benchling for sequence design, codon optimization, PCR, Gibson Assembly, bacterial transformation, and colony validation workflows. This aim focuses on creating a feasible genetic starting point for future biomaterial development and demonstrates how DNA design can be incorporated into a design research process for rehabilitation-oriented material systems.
Aim 2: Development Aim
The second aim of the project is to express and characterize the engineered protein material after successful plasmid validation, including small-scale protein production, purification, and exploratory material formation into films, coatings, or fibers. A successful Aim 1 would enable the next stage of testing whether the designed sequence shows desirable material behaviors such as film formation, flexibility, and compatibility with textile substrates.
Aim 3: Visionary Aim
The third aim of the project is to contribute to a long-term vision in which genetically designed protein materials become programmable components of wearable soft robotic systems for rehabilitation. If fully realized, this concept could support a new class of biomaterial-based soft actuators or structural interfaces that are lighter, more adaptive, and more biologically integrated than many current rehabilitation devices.
SECTION 3: BACKGROUND
Background and Literature Context
Soft robotics for rehabilitation is a rapidly growing field because soft devices can better conform to the body and reduce joint misalignment compared with rigid systems. Textile-based and soft actuator approaches are especially promising because they offer comfort, safety, and better integration into everyday life. However, important material challenges remain, including durability, controllability, biocompatibility, and the ability to closely adapt to the body while maintaining function.
At the same time, silk fibroin is highly relevant for biomaterial design because it combines strength, flexibility, hierarchical organization, and biocompatibility. Silk-inspired materials have strong potential for biomedical engineering and wearable systems. Synthetic biology methods such as Gibson Assembly also provide a practical way to construct recombinant sequences that encode designed protein materials. Despite progress in soft robotics and silk-based biomaterials, the integration of DNA-level protein design into rehabilitation-oriented material design is still underexplored. This project addresses that gap by positioning genetic design as the starting point of a future material system for wearable rehabilitation.
Two Peer-Reviewed Research Citations Relevant to the Project
Citation 1: Sanchez, V., Walsh, C. J., and Wood, R. J. Textile Technology for Soft Robotic and Autonomous Garments (2021).
This paper reviews how textile structures can function as robotic substrates rather than passive coverings. It shows that knitting, weaving, multilayer structures, and fiber orientation can contribute directly to sensing, actuation, and body-conforming performance in soft robotic garments. For this project, the paper is important because it supports the idea that future rehabilitation systems can benefit from material architectures inspired by biological systems, especially when movement and compliance are designed into the textile itself. It also helps justify why a biomaterial building-block approach could eventually feed into wearable actuator design rather than remaining purely molecular.
Citation 2: Recent review literature on silk fibroin-derived biomaterials for biomedical applications.
This body of research explains that silk fibroin-derived materials are highly versatile for regenerative and biomedical uses because they offer favorable mechanical properties, processability, and biocompatibility. The literature also highlights future directions involving intelligent biomaterials, sensors, and wearable health applications. For my project, this supports the use of Bombyx mori silk fibroin as a model for designing recombinant or inspired protein materials that may later be translated into films, fibers, or interfaces for soft systems. It therefore provides a bridge between molecular material design and rehabilitation-oriented device thinking.
Novelty and Innovation
This project is innovative because it does not begin with the actuator as the primary design object. Instead, it begins with the genetic design of a material building block that could later support soft robotic and textile applications. Rather than only using existing elastomers or fabrics, it explores whether protein-inspired sequence design can become part of a material-centered workflow for rehabilitation technology. The work is also novel because it connects synthetic biology tools such as Gibson Assembly with a design research question rooted in biomaterials, soft robotics, and future wearable rehabilitation systems.
Why This Project Matters and What Impact It Could Have
This project matters because rehabilitation devices are often limited by discomfort, poor adaptability, and a mismatch between rigid engineered materials and the soft, dynamic nature of the human body. Soft robotics has improved this situation, but there is still a major barrier in developing materials that combine compliance, structure, biocompatibility, and designability in one platform. By exploring recombinant protein design inspired by silk fibroin and elastic domains, this project proposes an upstream strategy for creating future materials that are programmable at the sequence level.
If successful, this work could contribute to new scientific and technical capabilities in the design of biologically inspired fibers, coatings, or composite interfaces for rehabilitation systems. Beyond the immediate project, the approach could help expand synthetic biology into design-led biomaterial development, opening possibilities in wearable health, biomedical manufacturing, and adaptive assistive technologies. At a field level, achieving these aims could shift part of soft robotics research from selecting existing materials toward encoding material function directly into designed biological sequences.
Ethical Implications
This project raises ethical questions related to responsibility, beneficence, and care. Because it uses genetic design and cloning methods, even at a small and non-pathogenic laboratory scale, it must be conducted with care regarding biosafety, containment, and responsible communication. Another ethical issue is translational overclaim: it would be inappropriate to suggest that an early-stage recombinant material construct is already a safe rehabilitation technology for patients. There are also broader concerns about access and fairness. If protein-designed biomaterials eventually enable advanced rehabilitation devices, those technologies should not become available only to well-funded laboratories or privileged health systems.
To ensure the project is ethical, the experimental scope should be limited to standard non-pathogenic laboratory strains, non-harmful recombinant sequences, and institutionally approved cloning practices. The project should clearly state that this work is a foundational material-design study, not a clinical intervention. Potential unintended consequences include failed assumptions about expression, folding, or material behavior, as well as overestimating the translational relevance of silk-inspired sequences. Alternatives include using non-recombinant silk fibroin, commercially available biomaterials, or simulation-based design approaches if biological uncertainty becomes too high. Ethical conduct in this project therefore requires transparent reporting of uncertainties, proportional claims, and attention to long-term access, sustainability, and safety in future applications.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Experimental Hypothesis
A recombinant DNA construct encoding a silk-inspired or silk-elastin-like protein module can be rationally designed and assembled using Gibson Assembly, creating a validated genetic platform for future biomaterial development relevant to rehabilitation-oriented soft systems.
Detailed Experimental Plan
Define the design target and functional logic In the first half day, I will define the biological rationale for the construct: a short recombinant protein containing a silk fibroin-inspired repetitive domain and, optionally, an elastic motif to introduce flexibility. Expected result: a clear design brief linking sequence motifs to desired material behavior.
Select protein motif sources from literature Over half a day to one day, I will review silk fibroin sequence features from Bombyx mori and identify a simplified motif suitable for classroom-scale DNA design. If appropriate, I will compare this with elastin-like motifs such as VPGXG repeats as a complementary domain. Expected result: a shortlist of feasible amino acid motifs for construct design.
Draft the protein architecture Over half a day, I will choose a modular protein layout such as His-tag + linker + silk-inspired repeat block + optional elastin-like block + stop codon. Expected result: a protein design schematic with module order and approximate length.
Codon-optimize the DNA sequence for E. coli In half a day, I will use Benchling or a similar design platform to codon-optimize the sequence for bacterial expression while minimizing problematic repeats or secondary structures when possible. Expected result: a codon-optimized DNA sequence ready for synthesis or PCR-based assembly.
Choose an expression plasmid backbone In half a day, I will select an appropriate plasmid backbone already available in class or lab, ideally one with a bacterial promoter, antibiotic resistance marker, and affinity-tag compatibility. Expected result: a plasmid map and insertion strategy.
Design Gibson overlaps In half a day, I will design 20–40 bp overlapping homology regions between insert and vector so the construct can be assembled by Gibson Assembly. Expected result: finalized primer or fragment overlap plan.
Plan fragment generation strategy In half a day, I will decide whether the insert will be obtained by gene synthesis, ordered fragment, or PCR amplification from designed oligos or templates, depending on course resources. Expected result: a practical build strategy and reagent list.
Prepare DNA fragments by PCR Over one day, I will amplify the vector backbone and/or insert fragments using a high-fidelity polymerase such as Phusion in order to reduce sequence errors. Expected result: visible DNA bands of expected size after gel verification.
Purify amplified DNA fragments In half a day, PCR products will be cleaned using spin-column purification or gel extraction if nonspecific bands are present. Expected result: purified DNA fragments suitable for assembly.
Perform Gibson Assembly reaction In half a day, purified overlapping fragments will be combined in the Gibson Assembly reaction according to the recommended molar ratios. Expected result: assembled plasmid molecules containing the designed insert.
Transform assembled plasmid into competent E. coli In half a day plus overnight incubation, I will transform the assembly product into competent E. coli and plate the cells on selective agar. Expected result: antibiotic-resistant colonies indicating successful uptake of plasmid DNA.
Screen colonies by colony PCR Over half a day to one day, several colonies will be screened using primers flanking the insertion site to identify clones with the expected insert size. Expected result: one or more positive colonies with the correct amplicon length.
Miniprep positive clones In half a day, promising colonies will be grown in liquid culture and plasmid DNA will be isolated using a miniprep protocol. Expected result: purified plasmid DNA from candidate correct clones.
Sequence-verify the construct Over two to four days depending on turnaround time, I will submit the plasmid for Sanger sequencing to verify insert identity and reading-frame integrity. Expected result: confirmed plasmid sequence matching the designed construct.
Analyze construct quality and feasibility In half a day, I will compare the sequencing result against the original design and note any mutations, assembly issues, or repeat instability. Expected result: a validated final plasmid map and a build assessment.
Optional expression test If time allows, over one to two days I will run a small exploratory expression test in E. coli and evaluate crude lysate or SDS-PAGE evidence of a protein band at the expected size. Expected result: preliminary indication of whether the construct is compatible with bacterial expression.
Interpretation for biomaterial relevance In half a day, I will relate the verified construct back to the larger design question: how sequence-defined biological materials could eventually support films, fibers, coatings, or reinforcement elements for wearable soft systems. Expected result: a design-oriented conclusion rather than only a cloning result.
Document the workflow visually In half a day, I will prepare a figure showing sequence design, plasmid assembly, clone validation, and future material translation. Expected result: a clear workflow figure for the report and presentation.
Approximate Timeline
Design and literature selection: 5 days
Sequence planning, codon optimization, and overlap design: 5 days
PCR, cleanup, and Gibson Assembly: 10 days
Transformation and colony growth: 1 day theory
Colony PCR and miniprep: 1 day theory
Sanger confirmation: 2–4 days theory
Optional expression test and interpretation: 1–2 days theory
Specific Methods, Tools, Technologies, and Concepts
Benchling for DNA and plasmid design
Codon optimization for bacterial expression
High-fidelity PCR
Gibson Assembly
E. coli transformation
Colony PCR
Miniprep and Sanger sequencing
Protein-inspired biomaterial design
Silk fibroin motif abstraction from Bombyx mori
Optional modular design using silk-elastin-like protein logic
Expected Overall Results
The most realistic expected result for this course is a bioispired outputverified recombinant plasmid encoding a protein-inspired biomaterial module. A strong outcome would be a sequence-confirmed construct with correct assembly and a clear rationale for future expression and material testing. If expression screening is possible, an additional expected result would be preliminary evidence that the construct is compatible with bacterial production, although this would be considered a stretch goal rather than a requirement.
Subsections of Individual Final Project
Imaging and measurement Final Project
Final Project: Measurement Plan
Recombinant Piezoelectric Biomaterial Interface for Soft Robotic Muscle Tone Modulation
Construct:His₆-FLAG-TEV-NQEQVSPL-(GGGGS)₃-GRGDS-IKVAV-(GPP)₁₀ in pcDNA3.1(+) / HEK293T expression system
Question 1: What aspects of the project will be measured?
This project proposes the design, synthesis, and initial characterisation of a recombinant piezoelectric fusion protein intended as the bioactive interface layer of a soft robotic rehabilitation device. The protein is engineered to: (1) be correctly expressed in human cells, (2) fold into a structurally active conformation — particularly the collagen-like (GPP)₁₀ domain — and (3) generate measurable electrical output under mechanical compression. Together, these three properties constitute the scientific claim of the project.
To evaluate whether each of these claims is substantiated, measurements are organised across three categories:
Category 1 — DNA-level measurements: Confirm that the plasmid construct is assembled correctly, the insert sequence matches the design, and the DNA is of sufficient purity for downstream experiments.
Category 2 — Protein-level measurements: Confirm that the recombinant protein is expressed in HEK293T cells, is present at detectable yield, adopts the correct secondary structure (triple helix in the collagen domain), and that the bioactive motifs (RGD and IKVAV) are functionally active.
Category 3 — Functional measurements: Confirm that the cast protein film generates voltage under mechanical deformation (the piezoelectric effect), is mechanically compatible with soft tissue, and is non-cytotoxic to muscle cells.
In total, 12 distinct measurements are performed, using 7 core analytical technologies.
Question 2: Description of all elements to be measured and how measurements will be performed
Category 1: DNA-level measurements
Measurement 1 — Plasmid insert sequence identity
What is measured:
The exact nucleotide sequence of the full 282 bp expression cassette inserted into pcDNA3.1(+), including the Kozak sequence, His₆ tag, FLAG tag, TEV protease site, piezoelectric domain (NQEQVSPL), three GGGGS flexible linkers, RGD adhesion motif (GRGDS), IKVAV neural attachment motif, (GPP)₁₀ collagen-like domain, and stop codon. Every base must match the codon-optimised design.
How it is performed:
To verify the correct insertion and sequence identity of the piezoelectric fusion protein cassette within the pcDNA3.1(+) expression vector, Sanger dye-terminator sequencing was performed on the final assembled plasmid. Following maxiprep purification (Qiagen HiSpeed Maxi Kit) of plasmid DNA propagated in E. coli DH5α, the concentration and purity of the DNA were confirmed by Nanodrop spectrophotometry, with acceptable samples yielding an A₂₆₀/A₂₈₀ ratio between 1.8 and 2.0 and a concentration of at least 100 ng/µL. Two sequencing reactions were submitted per sample: a forward read using the standard T7 promoter primer (5′-TAATACGACTCACTATA-3′), which anneals upstream of the multiple cloning site and reads into the insert from the CMV promoter end, and a reverse read using the BGH reverse primer (5′-TAGAAGGCACAGTCGAGG-3′), which anneals downstream of the insert and reads back through the 3′ end of the coding sequence. Together, these two reads provide overlapping coverage of the full 282 bp insert. Sequencing was performed by an external provider using capillary electrophoresis of fluorescently labelled chain-termination fragments. The resulting chromatogram trace files (.ab1 format) were uploaded to Benchling and aligned to the expected construct sequence using the built-in pairwise alignment tool.
Pass criterion: 100% base-call agreement with the expected sequence across the full 282 bp insert. Zero frameshifts, zero unexpected stop codons, all nine annotated domains present in the correct order and reading frame.
What is measured:
The size of DNA fragments produced after cutting the assembled plasmid with HindIII and XhoI restriction enzymes. This confirms that the insert was ligated into the backbone at the correct sites and is the expected size.
How it is performed:
Following sequence verification, 500 ng of maxiprep plasmid was digested in a 20 µL reaction containing 1 µL each of HindIII-HF and XhoI (New England Biolabs), 2 µL CutSmart buffer, and nuclease-free water. The reaction was incubated at 37°C for 60 minutes and heat-inactivated at 65°C for 20 minutes. The digested products were resolved on a 2% agarose gel prepared in 1× TAE buffer containing GelRed nucleic acid stain (1:10,000 dilution). A 1 kb Plus DNA ladder (NEB) was loaded alongside the samples. Electrophoresis was performed at 100V for 45 minutes. The gel was imaged under UV illumination using a gel documentation system. Band positions were compared to the expected fragment sizes calculated from the plasmid sequence.
Technologies used: Restriction enzyme digestion, agarose gel electrophoresis, UV gel imaging
Pass criterion: Two bands visible — approximately 282 bp (insert fragment) and 5,428 bp (linearised backbone). Absence of the smaller band indicates failed cloning or incorrect insertion orientation.
Measurement 3 — DNA purity and concentration (pre-transfection quality control)
What is measured:
Concentration of purified plasmid DNA in ng/µL and purity ratios A₂₆₀/A₂₈₀ and A₂₆₀/A₂₃₀, which reflect contamination by protein, phenol, or chaotropic salts respectively. This confirms the DNA preparation is suitable for HEK293T transfection.
How it is performed:
After maxiprep purification, the Nanodrop ND-1000 spectrophotometer (Thermo Fisher) was blanked with the elution buffer used in the final step of the purification protocol. A 1 µL aliquot of the plasmid preparation was loaded on the measurement pedestal. Absorbance readings at 230, 260, and 280 nm were recorded. Concentration was calculated from A₂₆₀ using the conversion factor for double-stranded DNA (50 ng·cm/µL). Samples were diluted to a working concentration of 1 µg/µL in TE buffer for transfection.
Technologies used: Nanodrop UV spectrophotometry
Pass criterion: A₂₆₀/A₂₈₀ ratio 1.8–2.0 (pure DNA). A₂₆₀/A₂₃₀ ≥ 1.8. Concentration ≥ 500 ng/µL. Samples outside these ranges were re-purified before use in cell transfection experiments.
Category 2: Protein-level measurements
Measurement 4 — Protein expression detection (western blot)
What is measured:
Presence of the recombinant piezoelectric fusion protein in HEK293T cell lysate, detected by antibody recognition of the His₆ or FLAG epitope tag. The expected molecular weight of the full fusion protein including tags is approximately 13.6 kDa.
How it is performed:
HEK293T cells were seeded at 2 × 10⁶ cells per well in 6-well plates and grown to 70–80% confluency in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were transiently transfected with 2 µg of the sequence-verified pcDNA3.1(+)-Piezo plasmid using Lipofectamine 3000 reagent (Thermo Fisher) according to the manufacturer’s protocol. An untransfected well was maintained as a negative control. At 48 hours post-transfection, cells were washed with ice-cold PBS and lysed in RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0) supplemented with protease inhibitor cocktail. Protein concentration in the clarified lysate was determined by BCA assay (Pierce). Twenty micrograms of total protein per lane were resolved on a 15% SDS-polyacrylamide gel under denaturing conditions and transferred to a 0.2 µm PVDF membrane by wet transfer at 100V for 60 minutes. The membrane was blocked with 5% non-fat dried milk in TBST for 1 hour at room temperature, then incubated overnight at 4°C with either mouse anti-His (1:2000, Abcam ab18184) or mouse anti-FLAG (1:1000, Sigma F1804) primary antibody. After washing (3 × 10 min in TBST), the membrane was incubated with HRP-conjugated anti-mouse IgG secondary antibody (1:5000) for 1 hour at room temperature. Signal was detected by enhanced chemiluminescence (ECL) using a ChemiDoc imaging system.
Pass criterion: A single band at approximately 13.6 kDa in the transfected lane, absent in the untransfected control. Presence of additional bands at higher molecular weight may indicate incomplete denaturation of the collagen triple helix domain (which is SDS-resistant in some contexts) and should be noted.
Measurement 5 — Protein yield quantification after affinity purification
What is measured:
Total mass (mg) of purified recombinant piezoelectric protein recovered per litre of HEK293T culture, following Ni-NTA affinity chromatography via the His₆ tag.
How it is performed:
HEK293T cells transfected as described in Measurement 4 were scaled to T-175 flasks. At 48–72 hours post-transfection, cells were harvested, lysed in native lysis buffer (50 mM NaH₂PO₄, 300 mM NaCl, 10 mM imidazole, pH 8.0) and the clarified lysate applied to a pre-equilibrated Ni-NTA agarose column (Qiagen). The column was washed with 20 column volumes of wash buffer (50 mM NaH₂PO₄, 300 mM NaCl, 20 mM imidazole, pH 8.0) and the protein eluted in elution buffer (50 mM NaH₂PO₄, 300 mM NaCl, 250 mM imidazole, pH 8.0). Eluted fractions were dialysed into PBS to remove imidazole. Protein concentration was determined by BCA assay using a BSA standard curve (0–2000 µg/mL). Absorbance at 562 nm was read on a plate reader (Tecan Infinite M200) after 30 minutes incubation at 37°C with BCA reagent.
Technologies used: Ni-NTA affinity chromatography, BCA protein assay, plate reader spectrophotometry at 562 nm
Pass criterion: ≥ 0.5 mg/L culture (transient HEK293T). Lower yields are acceptable at proof-of-concept stage but would necessitate stable cell line generation or bioreactor scale-up for material production.
Measurement 6 — Exact protein molecular mass and post-translational modifications (mass spectrometry)
What is measured:
The precise molecular mass of the purified protein to confirm identity and detect post-translational modifications — in particular hydroxyproline (+16 Da per modified residue) as evidence that the endogenous HEK293T prolyl-4-hydroxylase (P4H) has processed the (GPP)₁₀ collagen-like domain.
How it is performed:
Purified protein (≥ 5 µg in low-salt PBS, ≤ 150 mM NaCl) was submitted to the institutional mass spectrometry core facility for analysis by matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry. For intact protein analysis, the sample was co-crystallised with α-cyano-4-hydroxycinnamic acid (CHCA) matrix. For peptide-level confirmation of domain composition and modification sites, a parallel aliquot was digested with sequencing-grade trypsin (1:50 enzyme:protein ratio, 37°C overnight) and analysed by liquid chromatography tandem mass spectrometry (LC-MS/MS) on a Q Exactive Orbitrap instrument. Raw spectra were processed using MaxQuant and searched against the expected protein sequence. Hydroxyproline was included as a variable modification (+15.9949 Da on Pro residues).
Pass criterion: Intact mass within ±0.5% of the theoretical mass (6,900 Da for the tag-cleaved insert; 13,600 Da with His₆-FLAG-TEV tags). Detection of +16 Da shifts on proline residues in the (GPP)₁₀ domain confirms hydroxyproline formation and successful P4H activity, validating the use of HEK293T as expression host.
What is measured:
The secondary structure of the purified fusion protein — specifically whether the (GPP)₁₀ domain adopts a polyproline type-II (PPII) triple helix, which is both the structural and functional prerequisite for piezoelectric activity in collagen-derived materials. The melting temperature (Tm) is also measured as an indicator of thermostability at physiological temperature.
How it is performed:
Purified protein was dialysed into CD-compatible buffer (10 mM sodium phosphate, pH 7.4, no chloride ions which absorb in the far-UV) and diluted to 0.3–0.5 mg/mL. Circular dichroism spectra were recorded at 4°C on a Jasco J-815 spectropolarimeter using a 1 mm path-length quartz cuvette (Hellma). Wavelength scans were performed from 190 to 260 nm at 1 nm intervals, 1 nm bandwidth, 1 second response time, and three accumulations averaged per spectrum. The buffer baseline was subtracted. Data were expressed as mean residue ellipticity (MRE, deg·cm²·dmol⁻¹). A collagen triple helix produces a characteristic signature: a positive peak near 225 nm and a negative peak near 200 nm. To determine thermal stability, a thermal denaturation scan was performed by monitoring ellipticity at 225 nm while ramping temperature from 4°C to 70°C at 1°C/min. Tm was calculated as the inflection point of the sigmoidal melting curve using a Boltzmann fit.
Pass criterion: A positive CD signal at approximately 225 nm confirms PPII triple helix formation. Tm ≥ 30°C confirms the domain is stable approaching physiological temperature. A Tm ≥ 37°C would be required for any future in vivo application. Absence of the 225 nm signal indicates the collagen domain is unfolded — likely due to insufficient GPP repeat length or absent hydroxyproline — and the construct would require redesign.
Measurement 8 — Functional activity of RGD and IKVAV bioactive motifs (cell adhesion assay)
What is measured:
The ability of the purified fusion protein — specifically the GRGDS and IKVAV domains — to promote adhesion of C2C12 skeletal muscle myoblasts to a coated surface relative to an uncoated control and a fibronectin positive control.
How it is performed:
Flat-bottomed 96-well plates were coated overnight at 4°C with 100 µL per well of the purified piezoelectric protein at 10 µg/mL in PBS. Uncoated wells (PBS only) served as negative controls; fibronectin-coated wells (10 µg/mL) served as positive controls. Wells were blocked with 1% BSA in PBS for 1 hour at room temperature to prevent non-specific adhesion, then washed three times with PBS. C2C12 myoblasts (ATCC CRL-1772) were detached with trypsin-EDTA, resuspended in serum-free DMEM, and 10,000 cells per well seeded in 100 µL. Plates were incubated at 37°C with 5% CO₂ for 2 hours. Non-adherent cells were removed by three gentle washes with PBS. Adherent cells were fixed with 4% paraformaldehyde for 10 minutes, stained with 0.1% crystal violet in 25% methanol for 20 minutes, washed five times with distilled water, and air-dried. Crystal violet was solubilised by addition of 10% acetic acid (100 µL/well) with shaking for 10 minutes. Absorbance at 590 nm was measured on a plate reader. All conditions were performed in triplicate (n = 3 independent experiments).
Pass criterion: ≥ 2× A₅₉₀ signal on protein-coated wells relative to uncoated control (p < 0.05 by one-way ANOVA with Tukey post-hoc). Adhesion comparable to fibronectin positive control would constitute a strong result. Failure to exceed uncoated control suggests the RGD/IKVAV domains are sterically occluded in the folded protein conformation.
Category 3: Functional measurements
Measurement 9 — Piezoelectric voltage output under mechanical compression
What is measured:
The voltage (mV) and piezoelectric coefficient (d₃₃, expressed in pC/N) generated by the cast protein film when subjected to controlled, reproducible compressive mechanical stress. This is the primary functional readout of the entire project — the measurement that directly tests whether the designed material does what it is intended to do.
How it is performed:
Purified protein was cast as a film on gold-coated ITO glass substrates at a concentration of 20 µg/cm² by slow evaporation at room temperature in a humidity-controlled environment. Films were air-dried for 24 hours and their thickness measured by profilometry (expected: 50–200 nm). For macroscale electrical characterisation, a top gold electrode was deposited by sputter coating. Films were connected to a high-impedance electrometer (Keithley 6517B, input impedance > 200 TΩ) to prevent charge dissipation. A sinusoidal compressive force (1 Hz, amplitude 1–10 N) was applied perpendicular to the film surface using a dynamic mechanical analyser (TA Instruments DMA Q800) fitted with a flat compression clamp. The voltage output waveform was recorded simultaneously on a digital oscilloscope (Tektronix TBS1052B) at a sampling rate of 10 kHz. The piezoelectric coefficient d₃₃ was calculated as d₃₃ = Q/F, where Q is the generated charge (integrated current) and F is the applied force. For nanoscale domain mapping, piezoresponse force microscopy (PFM) was performed on a Bruker Dimension Icon AFM in contact mode using a conductive Pt/Ir-coated cantilever, applying an AC bias of 2V at 20 kHz to map the piezoelectric response across the film surface.
Technologies used: Dynamic mechanical analyser, high-impedance electrometer, digital oscilloscope, piezoresponse force microscopy (PFM), gold sputter coating, film profilometry
Pass criterion: A measurable, reproducible voltage output in phase with the applied mechanical force, with d₃₃ ≥ 1 pC/N. This threshold is benchmarked against natural collagen (0.7–2 pC/N) and represents the minimum signal relevant for neuromuscular stimulation. Even a sub-threshold signal at this stage constitutes a positive scientific result, as no prior study has measured piezoelectric output from a sequence-designed recombinant protein of this composition.
Measurement 10 — Mechanical stiffness of the protein film (Young’s modulus)
What is measured:
The elastic modulus (Young’s modulus, kPa) of the hydrated protein film, which determines whether the material is mechanically compatible with direct skin and muscle tissue contact in a wearable device. A material that is too stiff creates compliance mismatch; a material that is too soft lacks structural integrity.
How it is performed:
Protein films were cast on glass substrates as described above and hydrated in PBS at 37°C for 1 hour prior to measurement to replicate physiological conditions. Nanoindentation was performed using atomic force microscopy (AFM, Bruker BioScope Catalyst) in force-volume mode. A pyramidal silicon nitride cantilever (spring constant k = 0.03 N/m, tip radius approximately 20 nm, Bruker MLCT) was calibrated against a glass slide. Force-indentation curves were collected at 25 locations distributed across the film surface at an indentation depth of 200–500 nm. Young’s modulus was calculated from each curve by fitting to the Hertz contact model for a pyramidal indenter using NanoScope Analysis software. For bulk mechanical characterisation, dog-bone shaped film specimens were punched with a custom die (gauge length 5 mm, width 2 mm) and tested on a micro-tensile tester (Instron 5943) at a crosshead displacement rate of 1 mm/min. Stress-strain curves were recorded and Young’s modulus extracted from the linear elastic region (strain 0–5%).
Pass criterion: Young’s modulus 1–100 kPa (skeletal muscle stiffness: 8–17 kPa; soft tissue wearable contact range: 1–100 kPa). Films with modulus > 500 kPa would be considered mechanically mismatched for skin-contact applications and would require reformulation as a composite hydrogel.
Measurement 11 — Cytotoxicity of the protein film (Live/Dead fluorescence assay)
What is measured:
The percentage of live C2C12 skeletal muscle myoblasts after 72 hours of direct culture on the protein film surface, compared to a tissue-culture polystyrene control and a known toxic positive control (0.1% Triton X-100). This establishes that the material is not cytotoxic — a prerequisite for any future in vivo or clinical application.
How it is performed:
Protein films were cast directly into the wells of a 24-well tissue culture plate (10 µg/cm²) and allowed to dry. Films were sterilised by exposure to UV light (254 nm, 30 minutes) in a biosafety cabinet. Wells were rehydrated with PBS, blocked with 1% BSA, and then 50,000 C2C12 cells per well were seeded in complete DMEM. After 72 hours of culture at 37°C with 5% CO₂, media was aspirated and wells washed gently twice with PBS. The Live/Dead staining solution was prepared by diluting Calcein AM to 2 µM and ethidium homodimer-1 (EthD-1) to 4 µM in PBS. One hundred microlitres of staining solution was added per well and incubated at room temperature for 20 minutes protected from light. Cells were imaged immediately on an inverted fluorescence microscope (Zeiss Axio Observer) using FITC filter (Calcein AM, live cells: green fluorescence, excitation 495 nm, emission 515 nm) and TRITC filter (EthD-1, dead cells: red fluorescence, excitation 528 nm, emission 617 nm). Five randomly selected fields per well were imaged at 10× magnification. Live and dead cells were counted using the Cell Counter plugin in Fiji/ImageJ. Percentage viability was calculated as (live cells / total cells) × 100.
Pass criterion: ≥ 80% cell viability on the protein film compared to uncoated tissue-culture plastic control (baseline viability expected ≥ 95%). ISO 10993-5 standard for in vitro cytotoxicity: less than 30% reduction in viability relative to control = non-cytotoxic classification. A result below 70% viability would require investigation of residual imidazole contamination from purification or osmolarity effects from film preparation.
Measurement 12 — Protein thermal stability (differential scanning fluorimetry)
What is measured:
The melting temperature (Tm, °C) of the full fusion protein — the temperature at which 50% of the protein population has unfolded. This confirms the protein is globally stable at and above physiological temperature (37°C), and is particularly important given the marginal thermal stability expected from the (GPP)₁₀ domain in the absence of hydroxylation.
How it is performed:
Differential scanning fluorimetry (DSF, also known as ThermoFluor or protein thermal shift assay) was performed in a 96-well PCR plate format. Each well contained 18 µL of purified protein at 0.1–0.5 mg/mL in PBS and 2 µL of 100× SYPRO Orange dye (Thermo Fisher S6651; working concentration 5×). SYPRO Orange is environmentally sensitive and fluoresces strongly when bound to the hydrophobic core of unfolded proteins but is quenched in aqueous environments. Plates were sealed, briefly centrifuged, and loaded into a Bio-Rad CFX96 real-time PCR system. A temperature gradient from 25°C to 95°C was applied at a ramp rate of 0.5°C per 30 seconds, with fluorescence measured at each step using the FRET channel (excitation 490 nm, emission 575 nm). Raw fluorescence data were exported and processed in the DSFworld online tool or in GraphPad Prism. The melting temperature Tm was determined as the minimum of the first derivative (−dF/dT) of the melting curve. Experiments were performed in triplicate.
Pass criterion: Tm ≥ 37°C, confirming the protein does not unfold at body temperature. Given that the (GPP)₁₀ domain has a predicted Tm of approximately 30–35°C without hydroxyproline and ≥ 40°C with hydroxyproline (Hyp), the DSF result directly reports whether HEK293T P4H activity was sufficient. A Tm below 37°C would constitute a failure of the collagen folding aim and would require either extended (GPP) repeat length or confirmed P4H co-expression.
Question 3: Technologies used — detailed descriptions
1. Sanger sequencing
Sanger (chain-terminator) sequencing is based on the selective incorporation of dideoxynucleotides (ddNTPs) during in vitro DNA synthesis. In the modern fluorescent variant, each of the four ddNTPs carries a different fluorophore. During PCR, the polymerase randomly incorporates a ddNTP instead of a dNTP, terminating chain elongation at that position. This produces a population of fragments of every possible length, each terminated by a fluorescently labelled base. These fragments are separated by capillary electrophoresis — passing through a polymer-filled capillary under an electric field — and detected by a laser as they elute. The output is a chromatogram where each peak position represents a nucleotide position and the colour of the peak identifies the base (A, T, G, or C). Read lengths of 700–900 bp are routinely achieved with high accuracy (>99.9% per base). In this project, Sanger sequencing confirms the identity of the codon-optimised insert at single-base resolution.
2. Agarose gel electrophoresis
Agarose gel electrophoresis separates nucleic acid molecules by size. Agarose — a polysaccharide derived from seaweed — forms a porous matrix when cast in buffer. When an electric field is applied, negatively charged DNA (due to its phosphate backbone) migrates toward the positive electrode. Smaller fragments migrate faster through the matrix than larger ones, creating separation by size over time. A fluorescent intercalating dye (GelRed or ethidium bromide) is incorporated into the gel or loading buffer and intercalates between base pairs; these dye-DNA complexes fluoresce brightly under UV light, allowing bands to be visualised. The size of each band is estimated by comparison to a DNA ladder — a mixture of DNA fragments of known sizes run in an adjacent lane. In this project, a 2% agarose gel resolves the ~282 bp insert released by HindIII/XhoI digest.
3. SDS-PAGE and western blotting
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) separates proteins by molecular weight under denaturing conditions. SDS — a negatively charged detergent — binds to proteins in proportion to their mass, giving all proteins a uniform negative charge-to-mass ratio. Proteins are then driven through a polyacrylamide matrix by an electric field, with smaller proteins migrating faster. After electrophoresis, proteins are transferred from the gel to a PVDF membrane by applying an electric current perpendicular to the gel (wet or semi-dry transfer). The membrane is then probed with a primary antibody specific to an epitope on the target protein (in this case, the His₆ or FLAG tag), followed by a horseradish peroxidase (HRP)-conjugated secondary antibody that binds the primary. Addition of an ECL substrate causes HRP to catalyse a chemiluminescent reaction, and the emitted light is captured on film or a digital imager to reveal the position of the target protein as a band.
4. MALDI-TOF mass spectrometry
Matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry determines the mass of intact molecules with high precision. The purified protein is co-crystallised with a light-absorbing organic matrix (typically CHCA or sinapinic acid) on a metal target plate. A short pulse from a UV laser ablates and ionises the matrix-protein co-crystals, transferring the energy to the protein without fragmenting it. The ionised proteins are accelerated through a vacuum tube by a high-voltage electric field. Because all ions receive the same kinetic energy, lighter ions travel faster and arrive at the detector first — their arrival time (time of flight) is directly proportional to their mass-to-charge ratio (m/z). For protein identification at the peptide level, tryptic digestion followed by LC-MS/MS provides sequence coverage and identifies sites of post-translational modification such as hydroxyproline (+15.9949 Da).
5. Circular dichroism spectroscopy
Circular dichroism (CD) spectroscopy measures the differential absorption of left- and right-circularly polarised light by optically chiral molecules. Because the peptide bonds of proteins are chiral, and because different secondary structures (α-helix, β-sheet, random coil, polyproline II helix) position these bonds in distinct spatial arrangements, each secondary structure produces a characteristic CD spectrum in the far-UV region (190–260 nm). The polyproline type-II (PPII) helix characteristic of collagen triple helices produces a positive peak near 225 nm and a negative peak near 200 nm — a signature distinct from α-helices (which show two negative peaks at 208 and 222 nm) or disordered coils. The collagen triple helix is the specific target structure for the (GPP)₁₀ domain, as this conformation is required for piezoelectric dipole alignment. Thermal denaturation monitored by CD at 225 nm directly reports the melting temperature of this domain.
6. Fluorescence microscopy and Live/Dead assay
Fluorescence microscopy detects the emission of specific fluorescent molecules (fluorophores) after excitation with light of a defined wavelength. In the Live/Dead assay, two fluorophores with distinct spectral properties are used simultaneously. Calcein AM is a membrane-permeant dye that is cleaved by intracellular esterases in live cells to produce green-fluorescent calcein, which is retained in the cytoplasm — only metabolically active (live) cells produce a signal. Ethidium homodimer-1 (EthD-1) is a membrane-impermeant dye that can only enter cells with compromised plasma membranes (dead cells), where it intercalates into DNA and produces bright red fluorescence. Imaging cells stained with both dyes on a fluorescence microscope with appropriate filter sets simultaneously reveals the proportion of live (green) and dead (red) cells, enabling quantitative viability assessment.
7. Atomic force microscopy (AFM) nanoindentation and piezoresponse force microscopy (PFM)
AFM uses a sharp nanoscale tip (radius 2–50 nm) mounted on a flexible cantilever to interact with a sample surface. In nanoindentation mode, the tip is pressed into the sample surface with a controlled force, and the deflection of the cantilever (measured by a reflected laser beam) is recorded as a function of tip position. By fitting the resulting force-indentation curve to the Hertz contact mechanics model, the local Young’s modulus of the material is extracted. In piezoresponse force microscopy (PFM) mode, an alternating voltage is applied between the conductive tip and the grounded sample. If the material is piezoelectric, it locally deforms in phase with the applied AC voltage (converse piezoelectric effect), causing a detectable cantilever oscillation. The amplitude and phase of this oscillation, measured by lock-in detection, provide a nanoscale map of the piezoelectric response across the film surface — directly confirming the piezoelectric nature of the material at sub-micron resolution.
Summary table
#
Measurement
Category
Technology
Pass criterion
1
Insert sequence identity
DNA
Sanger sequencing
100% match, no frameshifts
2
Plasmid size and insert presence
DNA
Restriction digest + gel
~282 bp + ~5,428 bp bands
3
DNA purity and concentration
DNA
Nanodrop spectrophotometry
A₂₆₀/A₂₈₀ 1.8–2.0
4
Protein expression
Protein
SDS-PAGE + western blot
Band at ~13.6 kDa
5
Protein yield
Protein
BCA assay
≥ 0.5 mg/L culture
6
Exact mass + modifications
Protein
MALDI-TOF / LC-MS/MS
Mass ±0.5%; +16 Da on Pro
7
Triple helix formation
Protein
Circular dichroism
Positive peak at 225 nm; Tm ≥ 30°C
8
RGD/IKVAV functional activity
Protein
Cell adhesion assay
≥ 2× adhesion vs control
9
Piezoelectric voltage output
Functional
PFM + electrometer
d₃₃ ≥ 1 pC/N
10
Film mechanical stiffness
Functional
AFM nanoindentation
1–100 kPa
11
Cytotoxicity
Functional
Live/Dead fluorescence
≥ 80% viability
12
Protein thermal stability
Functional
DSF / ThermoFluor
Tm ≥ 37°C
Document prepared for HTGAA Final Project submission.Sequence: NQEQVSPL-(GGGGS)₃-GRGDS-IKVAV-(GPP)₁₀ | Vector: pcDNA3.1(+) | Host: HEK293T
Individual Final Project
Project Title
Genetic Design of a Silk-Inspired Protein Module for Future Rehabilitation Biomaterials
Project inspiration:
Inspiration 1 —
This screenshot documents the protein peptide/DNA sequence could be organized as a modular synthetic biology design.
Inspiration 2
This image supports the design logic of the project by showing how the biological sequence can be represented, checked, and connected to functional motifs such as scaffold anchoring, flexible linkers, cell-interface motifs, and collagen-like domains.
Inspiration 3 —
the transition from conceptual peptide design toward a possible creation.
SECTION 1: ABSTRACT
This project addresses a key challenge in wearable rehabilitation and soft robotics: many assistive devices still rely on rigid or non-biological materials that can limit comfort, adaptability, and integration with the body. Soft robotic systems offer a promising alternative, but there is still a gap in how biological material principles can be translated into programmable and manufacturable biomaterials for future wearable actuation. The overall objective of this project is to design and assemble a DNA construct encoding a protein-inspired material building block based on motifs from Bombyx mori silk fibroin and elastic protein domains, as a first step toward engineered biomaterials for soft rehabilitation devices.
The central hypothesis is that a genetically encoded silk-inspired or silk-elastin-like protein sequence can serve as a rational platform for future bio-derived films, fibers, or coatings with useful mechanical properties such as flexibility, resilience, and hierarchical assembly. To test this idea, the project will complete sequence design, codon optimization, plasmid planning, overlap design, Gibson Assembly, bacterial transformation, and clone validation. Methods include Benchling-based DNA design, PCR amplification, Gibson Assembly, E. coli transformation, colony screening, and sequence verification.
The expected outcome is a validated recombinant DNA construct that demonstrates the feasibility of integrating synthetic biology and protein design into a material-centered design workflow for future soft robotic textile applications. This project therefore functions as an enabling step between biomolecular design and the long-term development of adaptive rehabilitation wearables.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of my final project is to design, assemble, and validate a recombinant DNA construct encoding a silk-inspired or silk-elastin-like protein module by utilizing Benchling for sequence design, codon optimization, PCR, Gibson Assembly, bacterial transformation, and colony validation workflows. This aim focuses on creating a feasible genetic starting point for future biomaterial development and demonstrates how DNA design can be incorporated into a design research process for rehabilitation-oriented material systems.
Aim 2: Development Aim
The second aim of the project is to express and characterize the engineered protein material after successful plasmid validation, including small-scale protein production, purification, and exploratory material formation into films, coatings, or fibers. A successful Aim 1 would enable the next stage of testing whether the designed sequence shows desirable material behaviors such as film formation, flexibility, and compatibility with textile substrates.
Aim 3: Visionary Aim
The third aim of the project is to contribute to a long-term vision in which genetically designed protein materials become programmable components of wearable soft robotic systems for rehabilitation. If fully realized, this concept could support a new class of biomaterial-based soft actuators or structural interfaces that are lighter, more adaptive, and more biologically integrated than many current rehabilitation devices.
SECTION 3: BACKGROUND
Background and Literature Context
Soft robotics for rehabilitation is a rapidly growing field because soft devices can better conform to the body and reduce joint misalignment compared with rigid systems. Textile-based and soft actuator approaches are especially promising because they offer comfort, safety, and better integration into everyday life. However, important material challenges remain, including durability, controllability, biocompatibility, and the ability to closely adapt to the body while maintaining function.
At the same time, silk fibroin is highly relevant for biomaterial design because it combines strength, flexibility, hierarchical organization, and biocompatibility. Silk-inspired materials have strong potential for biomedical engineering and wearable systems. Synthetic biology methods such as Gibson Assembly also provide a practical way to construct recombinant sequences that encode designed protein materials. Despite progress in soft robotics and silk-based biomaterials, the integration of DNA-level protein design into rehabilitation-oriented material design is still underexplored. This project addresses that gap by positioning genetic design as the starting point of a future material system for wearable rehabilitation.
Two Peer-Reviewed Research Citations Relevant to the Project
Citation 1: Sanchez, V., Walsh, C. J., and Wood, R. J. Textile Technology for Soft Robotic and Autonomous Garments (2021).
This paper reviews how textile structures can function as robotic substrates rather than passive coverings. It shows that knitting, weaving, multilayer structures, and fiber orientation can contribute directly to sensing, actuation, and body-conforming performance in soft robotic garments. For this project, the paper is important because it supports the idea that future rehabilitation systems can benefit from material architectures inspired by biological systems, especially when movement and compliance are designed into the textile itself. It also helps justify why a biomaterial building-block approach could eventually feed into wearable actuator design rather than remaining purely molecular.
Citation 2: Recent review literature on silk fibroin-derived biomaterials for biomedical applications.
This body of research explains that silk fibroin-derived materials are highly versatile for regenerative and biomedical uses because they offer favorable mechanical properties, processability, and biocompatibility. The literature also highlights future directions involving intelligent biomaterials, sensors, and wearable health applications. For my project, this supports the use of Bombyx mori silk fibroin as a model for designing recombinant or inspired protein materials that may later be translated into films, fibers, or interfaces for soft systems. It therefore provides a bridge between molecular material design and rehabilitation-oriented device thinking.
Novelty and Innovation
This project is innovative because it does not begin with the actuator as the primary design object. Instead, it begins with the genetic design of a material building block that could later support soft robotic and textile applications. Rather than only using existing elastomers or fabrics, it explores whether protein-inspired sequence design can become part of a material-centered workflow for rehabilitation technology. The work is also novel because it connects synthetic biology tools such as Gibson Assembly with a design research question rooted in biomaterials, soft robotics, and future wearable rehabilitation systems.
Why This Project Matters and What Impact It Could Have
This project matters because rehabilitation devices are often limited by discomfort, poor adaptability, and a mismatch between rigid engineered materials and the soft, dynamic nature of the human body. Soft robotics has improved this situation, but there is still a major barrier in developing materials that combine compliance, structure, biocompatibility, and designability in one platform. By exploring recombinant protein design inspired by silk fibroin and elastic domains, this project proposes an upstream strategy for creating future materials that are programmable at the sequence level.
If successful, this work could contribute to new scientific and technical capabilities in the design of biologically inspired fibers, coatings, or composite interfaces for rehabilitation systems. Beyond the immediate project, the approach could help expand synthetic biology into design-led biomaterial development, opening possibilities in wearable health, biomedical manufacturing, and adaptive assistive technologies. At a field level, achieving these aims could shift part of soft robotics research from selecting existing materials toward encoding material function directly into designed biological sequences.
Ethical Implications
This project raises ethical questions related to responsibility, beneficence, and care. Because it uses genetic design and cloning methods, even at a small and non-pathogenic laboratory scale, it must be conducted with care regarding biosafety, containment, and responsible communication. Another ethical issue is translational overclaim: it would be inappropriate to suggest that an early-stage recombinant material construct is already a safe rehabilitation technology for patients. There are also broader concerns about access and fairness. If protein-designed biomaterials eventually enable advanced rehabilitation devices, those technologies should not become available only to well-funded laboratories or privileged health systems.
To ensure the project is ethical, the experimental scope should be limited to standard non-pathogenic laboratory strains, non-harmful recombinant sequences, and institutionally approved cloning practices. The project should clearly state that this work is a foundational material-design study, not a clinical intervention. Potential unintended consequences include failed assumptions about expression, folding, or material behavior, as well as overestimating the translational relevance of silk-inspired sequences. Alternatives include using non-recombinant silk fibroin, commercially available biomaterials, or simulation-based design approaches if biological uncertainty becomes too high. Ethical conduct in this project therefore requires transparent reporting of uncertainties, proportional claims, and attention to long-term access, sustainability, and safety in future applications.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Experimental Hypothesis
A recombinant DNA construct encoding a silk-inspired or silk-elastin-like protein module can be rationally designed and assembled using Gibson Assembly, creating a validated genetic platform for future biomaterial development relevant to rehabilitation-oriented soft systems.
Detailed Experimental Plan
Define the design target and functional logic In the first half day, I will define the biological rationale for the construct: a short recombinant protein containing a silk fibroin-inspired repetitive domain and, optionally, an elastic motif to introduce flexibility. Expected result: a clear design brief linking sequence motifs to desired material behavior.
Select protein motif sources from literature Over half a day to one day, I will review silk fibroin sequence features from Bombyx mori and identify a simplified motif suitable for classroom-scale DNA design. If appropriate, I will compare this with elastin-like motifs such as VPGXG repeats as a complementary domain. Expected result: a shortlist of feasible amino acid motifs for construct design.
Draft the protein architecture Over half a day, I will choose a modular protein layout such as His-tag + linker + silk-inspired repeat block + optional elastin-like block + stop codon. Expected result: a protein design schematic with module order and approximate length.
Codon-optimize the DNA sequence for E. coli In half a day, I will use Benchling or a similar design platform to codon-optimize the sequence for bacterial expression while minimizing problematic repeats or secondary structures when possible. Expected result: a codon-optimized DNA sequence ready for synthesis or PCR-based assembly.
Choose an expression plasmid backbone In half a day, I will select an appropriate plasmid backbone already available in class or lab, ideally one with a bacterial promoter, antibiotic resistance marker, and affinity-tag compatibility. Expected result: a plasmid map and insertion strategy.
Design Gibson overlaps In half a day, I will design 20–40 bp overlapping homology regions between insert and vector so the construct can be assembled by Gibson Assembly. Expected result: finalized primer or fragment overlap plan.
Plan fragment generation strategy In half a day, I will decide whether the insert will be obtained by gene synthesis, ordered fragment, or PCR amplification from designed oligos or templates, depending on course resources. Expected result: a practical build strategy and reagent list.
Prepare DNA fragments by PCR Over one day, I will amplify the vector backbone and/or insert fragments using a high-fidelity polymerase such as Phusion in order to reduce sequence errors. Expected result: visible DNA bands of expected size after gel verification.
Purify amplified DNA fragments In half a day, PCR products will be cleaned using spin-column purification or gel extraction if nonspecific bands are present. Expected result: purified DNA fragments suitable for assembly.
Perform Gibson Assembly reaction In half a day, purified overlapping fragments will be combined in the Gibson Assembly reaction according to the recommended molar ratios. Expected result: assembled plasmid molecules containing the designed insert.
Transform assembled plasmid into competent E. coli In half a day plus overnight incubation, I will transform the assembly product into competent E. coli and plate the cells on selective agar. Expected result: antibiotic-resistant colonies indicating successful uptake of plasmid DNA.
Screen colonies by colony PCR Over half a day to one day, several colonies will be screened using primers flanking the insertion site to identify clones with the expected insert size. Expected result: one or more positive colonies with the correct amplicon length.
Miniprep positive clones In half a day, promising colonies will be grown in liquid culture and plasmid DNA will be isolated using a miniprep protocol. Expected result: purified plasmid DNA from candidate correct clones.
Sequence-verify the construct Over two to four days depending on turnaround time, I will submit the plasmid for Sanger sequencing to verify insert identity and reading-frame integrity. Expected result: confirmed plasmid sequence matching the designed construct.
Analyze construct quality and feasibility In half a day, I will compare the sequencing result against the original design and note any mutations, assembly issues, or repeat instability. Expected result: a validated final plasmid map and a build assessment.
Optional expression test If time allows, over one to two days I will run a small exploratory expression test in E. coli and evaluate crude lysate or SDS-PAGE evidence of a protein band at the expected size. Expected result: preliminary indication of whether the construct is compatible with bacterial expression.
Interpretation for biomaterial relevance In half a day, I will relate the verified construct back to the larger design question: how sequence-defined biological materials could eventually support films, fibers, coatings, or reinforcement elements for wearable soft systems. Expected result: a design-oriented conclusion rather than only a cloning result.
Document the workflow visually In half a day, I will prepare a figure showing sequence design, plasmid assembly, clone validation, and future material translation. Expected result: a clear workflow figure for the report and presentation.
Approximate Timeline
Design and literature selection: 5 days
Sequence planning, codon optimization, and overlap design: 5 days
PCR, cleanup, and Gibson Assembly: 10 days
Transformation and colony growth: 1 day theory
Colony PCR and miniprep: 1 day theory
Sanger confirmation: 2–4 days theory
Optional expression test and interpretation: 1–2 days theory
Specific Methods, Tools, Technologies, and Concepts
Benchling for DNA and plasmid design
Codon optimization for bacterial expression
High-fidelity PCR
Gibson Assembly
E. coli transformation
Colony PCR
Miniprep and Sanger sequencing
Protein-inspired biomaterial design
Silk fibroin motif abstraction from Bombyx mori
Optional modular design using silk-elastin-like protein logic
Expected Overall Results
The most realistic expected result for this course is a bioispired outputverified recombinant plasmid encoding a protein-inspired biomaterial module. A strong outcome would be a sequence-confirmed construct with correct assembly and a clear rationale for future expression and material testing. If expression screening is possible, an additional expected result would be preliminary evidence that the construct is compatible with bacterial production, although this would be considered a stretch goal rather than a requirement.