First, describe a biological engineering application or tool you want to develop and why. The engineering of extracted hematopoietic stem cells so their B-cell progeny produces bnAbs (broadly neutralizing antibodies), so after exposure to an immunogen (a highly mutable virus like HIV, Influenza, and so on) they can provide protection for a long time after autologous engraftment.
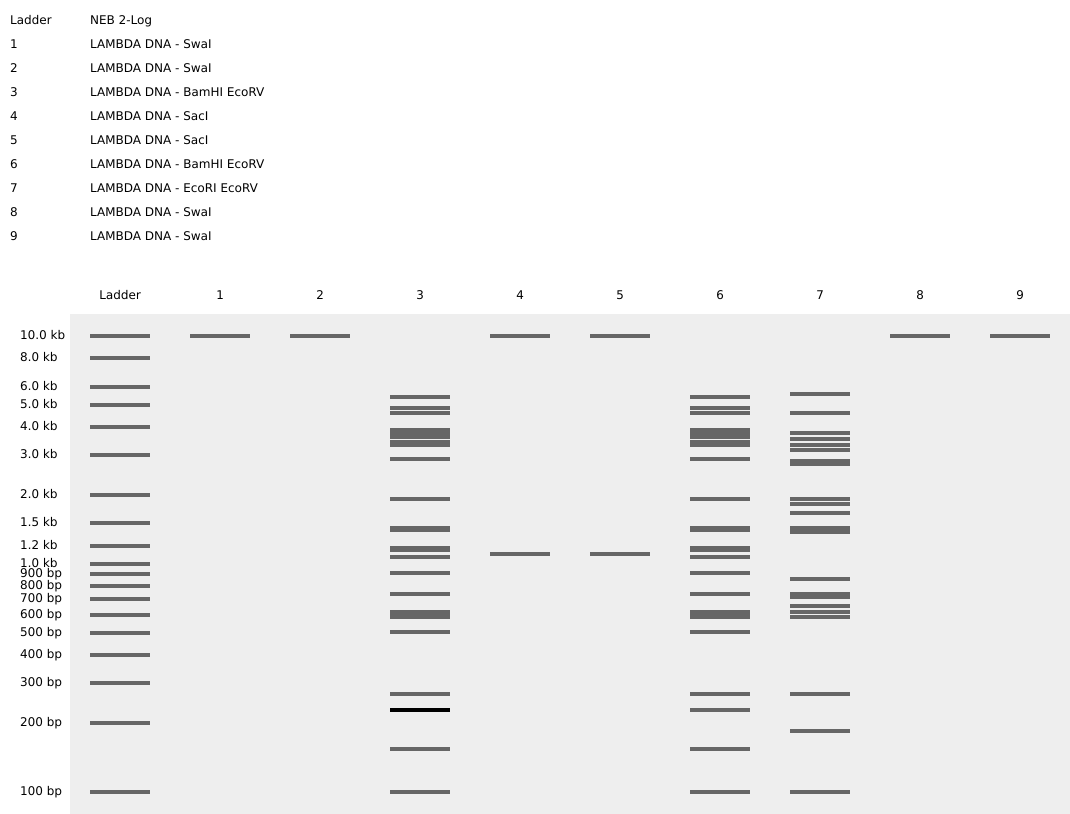
Part 0: Basics of Gel Electrophoresis Attend or watch all lecture and recitation videos. Optionally watch bootcamp. Done :checkmark:
Part 1: Benchling & In-silico Gel Art See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
Homework Assignment: Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME! Committed Listeners Required
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Week 4 — Protein Design Part I This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip) How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Meat contains at around 26g of protein per 100g of meat, so, in 500g of meat, this would be 26 g x 5 = 130g of protein
Homework DUE BY START OF MAR 10 LECTURE Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? As stated in the thermofisher website, “Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2” The Phusion DNA Polymerase will be the one replicating the DNA at a really high fidelity The nucleotides will be ‘inserted’ into the replicated DNA strands And the reaction buffer (Including MgCl2, which is a co-factor needed for the enzyme), which is optimized, meaning, it has the favorable conditions for the enzyme such as pH.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Scalability for one, and for second, way more outputs; comparing this to, for example, the lac operon, if I recall correctly it only has a represor, an operator, and the lactose gene. IANNs would have way more repressor, operators, and so way more outputs, and ways said outputs are regulated, by multiple inputs.
Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis not only is a robust and efficient system compared to in vivo methods, in which yields are comparatively lower, but if we go in terms of flexibility and experimental variables, there’s a lot of control to be had here given that we only use the components that are essential, this means that variables such as metabolism, possible toxicity, and laborous work such as transformation, transfection, just the whole genetically enginering part (and the assays that come with it, such as, verifying if my organism is indeed genetically modified!) is something to not worry about anymore. Now, because of all the previously mentioned, we do have more control too on elements needed for protein synthesis, such as, nucleotides, salts, DNA concentration, pH; said elements would typically be directed towards other cellular processes, but not here because there is no cell.
Homework Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Homework Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Assignees for this section MIT/Harvard students Required Committed Listeners Required
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. Make a note on your HTGAA webpages including: what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”) If I recall correctly, I was helping on the HTGAA spelling in the bottom left corner!
Subsections of Homework
Week 1 HW: Principles and Practices
1. First, describe a biological engineering application or tool you want to develop and why.
The engineering of extracted hematopoietic stem cells so their B-cell progeny produces bnAbs (broadly neutralizing antibodies), so after exposure to an immunogen (a highly mutable virus like HIV, Influenza, and so on) they can provide protection for a long time after autologous engraftment.
The reason why I want to develop this is because it is near within our reach to be able to create a method of viral protection against a lot of viruses which we thought we’d never be able to get rid of.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Governance/policy goal: Trials for healthiness.
First and foremost: the evaluation if this should be something to be researched, weighing the benefits and the possible risks of bringing such tools into the world, the same way that chiral lifeforms was evaluated.
Secondly: biological safety for the patient. Rigorous and extensive research, as it goes for any kind of treatment! Specifically, see if this treatment leads to the development of an autoimmunity, or causes any harm in trials using an in-vivo model, then murine model, then human model.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
–First governance action: Authorization by prescription/indication
Purpose: To ensure this therapy doesn't drift abroad, it is required to make sure that only people go through the same approval process as gene therapies or CAR-T cell therapy.
Design: To make it work, we'd need FDA approval for risk groups in which conventional vaccines (if any were to be approved) or drugs like lenacapavir do not work. Once that's done, this immunotherapy could be standarized in capable hospitals. Hopefully can be funded by government, alternatively, donations, or both.
Assumptions: Assuming that non-authorized use is detectable/enforceable.
Risks of Failure & “Success”: Failure would be slowing down their the translational part of immunotherapy, success would be the exact opposite, but could lead to 'medical tourism'.
–Second governance action: Long term surveillance
Purpose: To ensure no harm happens off-target and there are no adverse effects, there is the need for long term surveillance.
Design: Hospitals collect periodic data, which incude checkups for any symptoms of autoimmunity. Funded by the patient's health insurance.
Assumptions: Data is secured and used ethically.
Risks of Failure & “Success”: Underfunded registries may lead to loss of the follow ups, which would lead to (if present) any effect going undetected. Success would lead to privacy concerns.
–Third governance action: Contain by design
Purpose: Standarize the protocol (as much as it can be, considering this is a personalized immunotherapy) to ensure it doesn't go off-target,
Design: by establishing the gene(s) to be inserted, promoter(s), and the locus/loci to target. Lastly, a kill switch in case the immunotherapy goes wrong, such as an over-expressed protein to target with antibodies.
Assumptions: Assumes that the protocol and go wrong can also be 'reversed' by the addition of a kill switch.
Risks of Failure & “Success”: By design there's the assumption that the safeguards will generate confidence (that could be false confidence if the design does not work). Success can bring costs higher by adding complexity (particularly the kill switch).
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
🥉
🥈
🥇
• By preventing incidents
✅
✅
✅✅
• By helping respond
✅
✅✅
✅✅✅
Foster Lab Safety
🥉
🥈
🥇
• By preventing incident
✅
✅✅
✅✅✅
• By helping respond
✅
✅✅
✅✅✅
Protect the environment
• By preventing incidents
N/A
N/A
N/A
• By helping respond
N/A
N/A
N/A
Other considerations
🥉
🥈
🥇
• Minimizing costs and burdens to stakeholders
✅
✅
✅✅✅
• Feasibility?
✅✅
✅✅
✅✅
• Not impede research
✅
✅
✅✅
• Promote constructive applications
✅
✅
✅✅
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Definitely the third option, contain by design. Not only because of the scoring, but because I believe the first 2 options already are a must, given how common the those logistics are in medical proceedures alike. I believe the trade-offs mostly come from the risks (of failure and the ones that come from success in every option) rather than from the operation itself. I believe this would be the most relevant to academic medical centers.
–Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
The possibility of the immunotherapy causing autoimmunity, after a long time. I wasn’t accounting on the duration of the immune cells in the body. The governance actions appropriate for those issues would be option 3 and/or 2.
Assignment (Week 2 Lecture Prep) — DUE BY START OF FEB 10 LECTURE
Homework Questions from Professor Jacobson
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate is 1:10 to the power of 6!
This doesn’t compare much to the length of the human genome, which is 3:10 to the power of 9.
Biology deals with this through proofreading activity; through 3’ to 5’ exonuclease activity.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
So the average human protein has 1036 base pairs, and the average aminoacid has 4 ways of synthesis, and because this is permutations (how many combinations there are), it would be 4 to the power of 1036.
There can be many reasons such as, unoptimal within cellular context (such as, pH is not optimal for the final product when using a certain combination of aminoacids, and so the protein’s faulty, but it would be for a different combination of aminoacids in which the pH is actually suitable for the protein, so the second one would be conserved), another one reason could be rare tRNA’s that would slow down the synthesis, paradoxically, it could also be a combination of only optimal codons, which would make ribosome speed go beyond its capability, as in, make it go too fast to the point where the folding does not occur correctly (this last one I read in a paper that I’d like to be able to find again, it was something about mechanistic properties of synthesis from the ribosome).
Homework Questions from Dr. LeProust
1. What’s the most commonly used method for oligo synthesis currently?
Phosphodiester method, I believe that’s what the majority has access to.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because each nucleotide addition requires all steps such as coupling, capping, oxidation, deblocking, at around 200 cycles is where it begins to truncate
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
Because truncation seems to happen after 200 cycles. There are methods that can go up to 500nt, but that’s pushing it!
Homework Question from George Church
1. Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
No AI.
(Kansas State University, n. d). The 10 aminoacids are lysine, methionine, tryptophan, threonine, valine, isoleucine, leucine, arginine, histidine and phenylalanine.
Peculiar, that does nothing… and even if they were to do that, couldn’t the dinosaurs just get their lysine from regular animals or plants? I think this contingency plan would work if it was instead a deletion or anything that were to stop the translation of the enzyme(s) that synthesize an aminoacid or metabolite that is produced by dinosaurs (and is rarely ever found in diet).
Attend or watch all lecture and recitation videos. Optionally watch bootcamp.
Done :checkmark:
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs.
I title it “A greeting..”
Part 2: Benchling & In-silico Gel Art
Unable to do, lack the lab access.
Part 3: DNA Design Challenge
3.1. Choose your protein.
Which protein have you chosen and why?
I have chosen Lactose. I wanted to do Lactase instead, but it seemed to big.
In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Codon optimization is needed in case a sequence contains too many rare tRNAs (which, may be common from the original organism), which could mess up with protein translation; it also may impact the protein’s stability depending on the cellular context, specifically, the pH, so having compatible codon’s for once the protein’s done is also something to keep in mind.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
First, it needs a promoter, and a terminator; once that’s edited into the sequence, it can be inserted into a yeast (I chose yeast and not a bacteria because there are some post-translational-modifications that bacteria is unable to do!)
Oh, also, it would be a plasmid. In order to insert this plasmid, a method that can be used is Heat Shock of Yarrowia lipolytica (forgive me if I forget to name the organism in italics). Would just need to follow the protocol, but also design a selection method, such as the introduction of an antibiotic-resistant gene, or the usage of a strain that lacks the capability of producing a metabolite that is essential for metabolism, transform, and then pass it onto a medium that also does not have said metabolite.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I think something interesting would be the DNA of all, if not, multiple flowers that produce the color blue. Blue is quite a rare color in nature, and it would be wonderful if it could be figured out what genes are responsible for the blue of the flower… the reason why is because, with genetic engineering, surely blue cotton could be possible!
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Illumina’s sequencing-by-synthesis. It has a great output and, because plant genomes are immense, I think a technology that can do a whole-genome sequencing would be the best fit for the DNA I want to sequence as answered above.
Is your method first-, second- or third-generation or other? How so?
Second generation! Because it’s a high output and massive-in-parallel sequencing method
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
Purified plant DNA, quantify DNA (with nanodrop), fragmentation, NGS protocol (usage of adapters and then the fragments are amplified with PCR. Then this library is ready for the sequencer).
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Using the sequencer, the DNA fragments are absorbed by the flow cell, new strands of DNA (one base at a time) are synthesized with fluorescent labeled nucleotides. Given that each nucleotide will have a different color, each new base added will be captured.
What is the output of your chosen sequencing technology?
A digital file with reads, which are these sequences that are meant to be put together to reconstruct the whole genome.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would want to synthesize a mRNA vaccine that uses the principle of those superadjuvant HIV vaccines, but the immunogen would be a cancer molecule. It would be interesting to see the effects of bnAbs against cancer.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
A DNA plasmid into RNA with the use of phage RNA polymerase. The reason why is because it is an mRNA vaccine, so that polymerase is very essential.
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
Design the plasmid template with compatible elements like the promoter for the phage RNA polymerase. Then, do in vitro transcription.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
This is a method that is relatively simple and does not really require much speed given that it is a small synthesis (in terms of nucleotides), accurate-wise it can be ~70-80% because capping sometimes can be incomplete. Since it is an enzymatic in vitro process, it is suitable to be scaled up pretty quickly.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I would want to edit the p53 gene. I believe that elephants do not have risky rates of cancer despite their size/number of cells, and that’s because (If I am not misremembering) they have a bunch of p53 copies. Reason I would do this is for the better health of a lot of people.
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR. Because I think this would be the safest option given how many gene editing therapies that use CRISPR are being approved lately.
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
CRISPR-Cas9 uses a guide RNA that matches a specific DNA sequence, and it has the enzyme Cas9 which is directed by said guide to the target. This enzyme makes a double-strand break, and then the cell tries to repair this break, while it is repairing it, there can be the insertion of a corrected sequence if a repair template is provided.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Target DNA is identified in order to design the guide RNA. The input includes Cas9, guide RNA and donor DNA template.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I’m not aware of any as CRISPR seems to be described as a very efficient and precise gene editing tool in literature.
Week 3 HW: Lab Automation
Homework
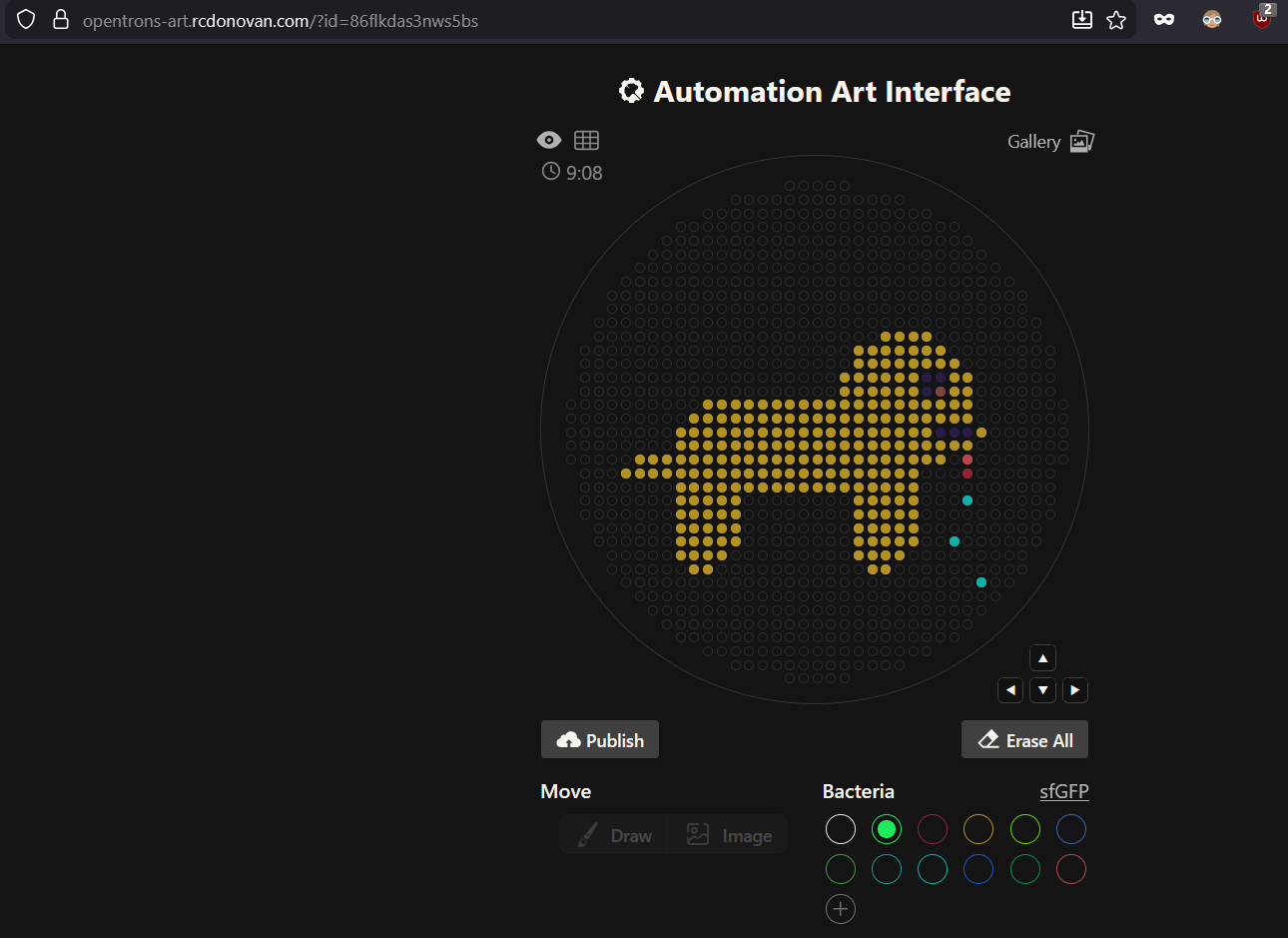
Assignment: Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME!
Committed Listeners Required
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
Ask for help early!
If you are having any trouble with scripting, contact your TAs as soon as possible for help.
Do not wait until your scheduled robot time slot or you may not be able to complete this assignment!
If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that:
Use the download icon pointed to by the red arrow in this diagram.
Use the download icon pointed to by the red arrow in this diagram.
If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
Sign up for a robot time slot if you are at MIT/Harvard/Wellesley or at a Node offering Opentrons automation. The Python script you created will be run on the robot to produce your work of art!
At MIT/Harvard? Lab times are on Thursday Feb.19 between 10AM and 6PM.
At other Nodes? Please coordinate with your Node.
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Bryant Jr, J. A., Kellinger, M., Longmire, C., Miller, R., & Wright, R. C. (2023). AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots. Synthetic Biology, 8(1), ysac032.
They published an open-source Python software package called “AssemblyTron” and it automates DNA assembly worfklows using the Opentrons OT-2 liquid handling opentrons.. it can automate PCR setupts, gradient optimization, Golden Gate assembly and homology based in vivo assembly.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
Example 1: You are creating a custom fabric, and want to deposit art onto specific parts that need to be intertwined in odd ways. You can design a 3D printed holder to attach this fabric to it, and be able to deposit bio art on top. Check out the Opentrons 3D Printing Directory.
Example 2: You are using the cloud laboratory to screen an array of biosensor constructs that you design, synthesize, and express using cell-free protein synthesis.
Echo transfer biosensor constructs and any required cofactors into specified wells.
Bravo stamp in CPFS reagent master mix into all wells of a 96-well / 384-well plate.
Multiflo dispense the CFPS lysate to all wells to start protein expression.
PlateLoc seal the plate.
Inheco incubate the plate at 37°C while the biosensor proteins are synthesized.
XPeel remove the seal.
PHERAstar measure fluorescence to compare biosensor responses.
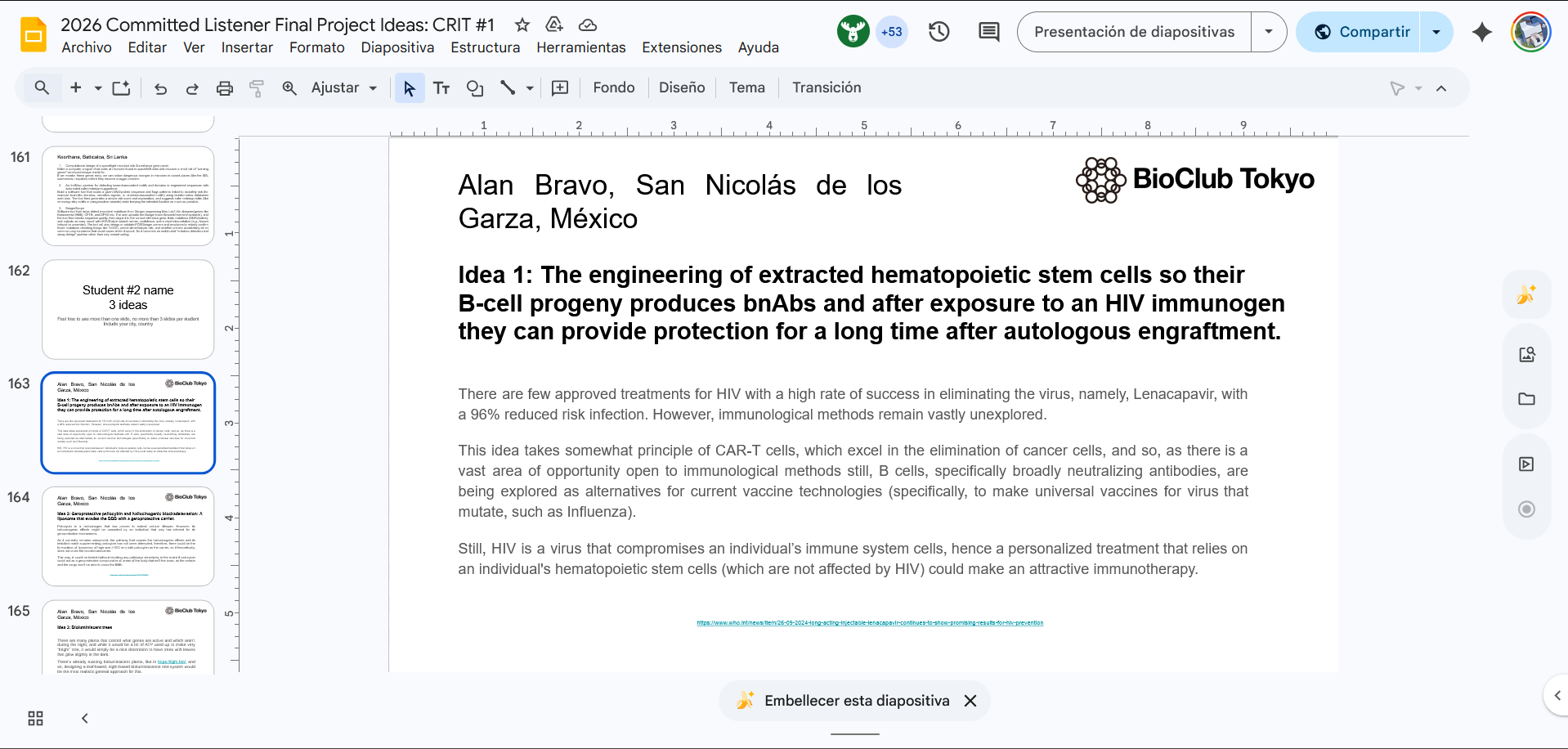
Idea 1, The engineering of extracted hematopoietic stem cells so their B-cell progeny produces bnAbs and after exposure to an HIV immunogen they can provide protection for a long time after autologous engraftment: AUTOMATION
So, with python I could generate safety kill-switch modules. I could Echo transfer plasmids into 96-well plates, Inhecho incubate the cells, automate ELISA plate reader for measuring antibody secretion.
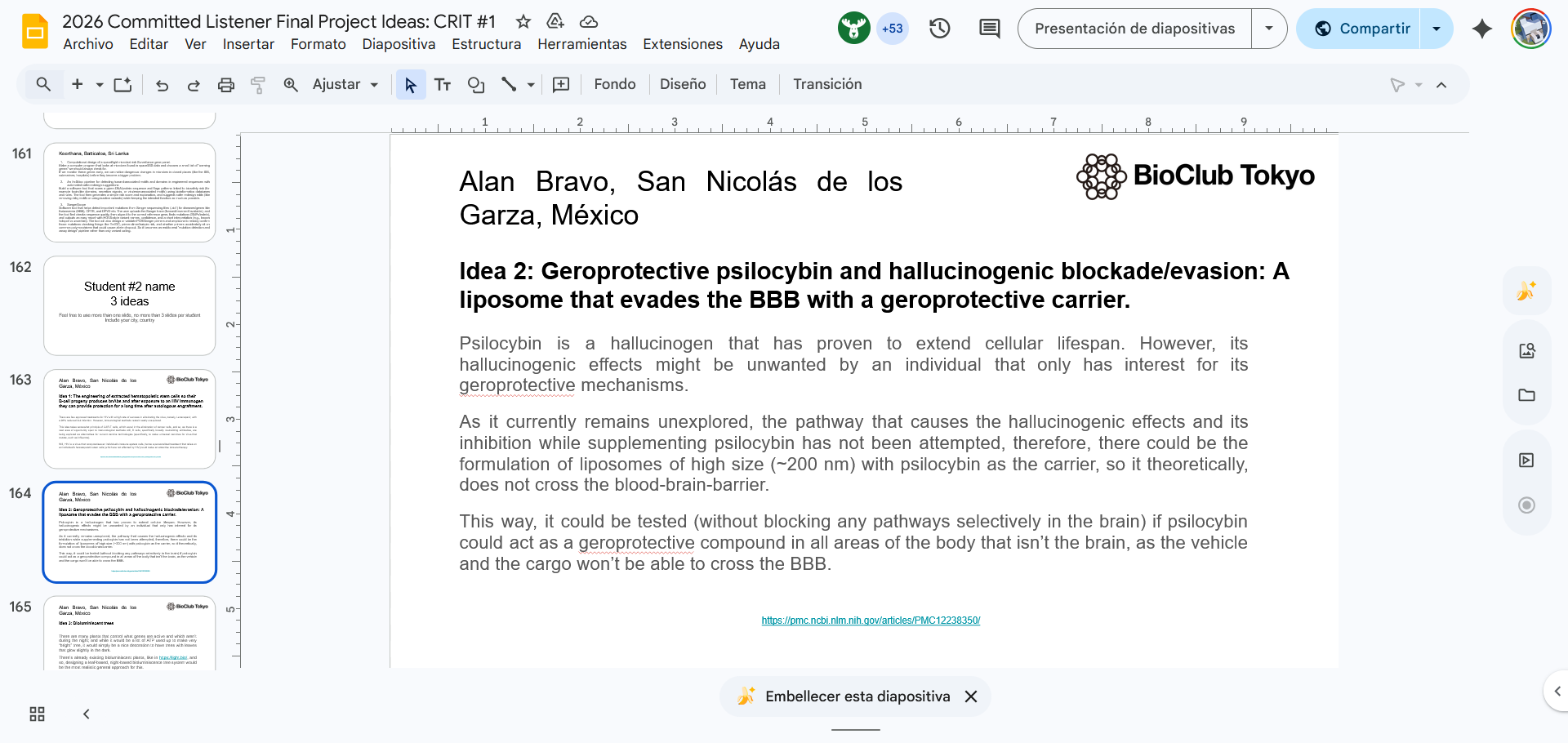
Idea 2, Geroprotective psilocybin and hallucinogenic blockade/evasion: A liposome that evades the BBB with a geroprotective carrier: AUTOMATION
Automated drug loading quantification via plate reader,
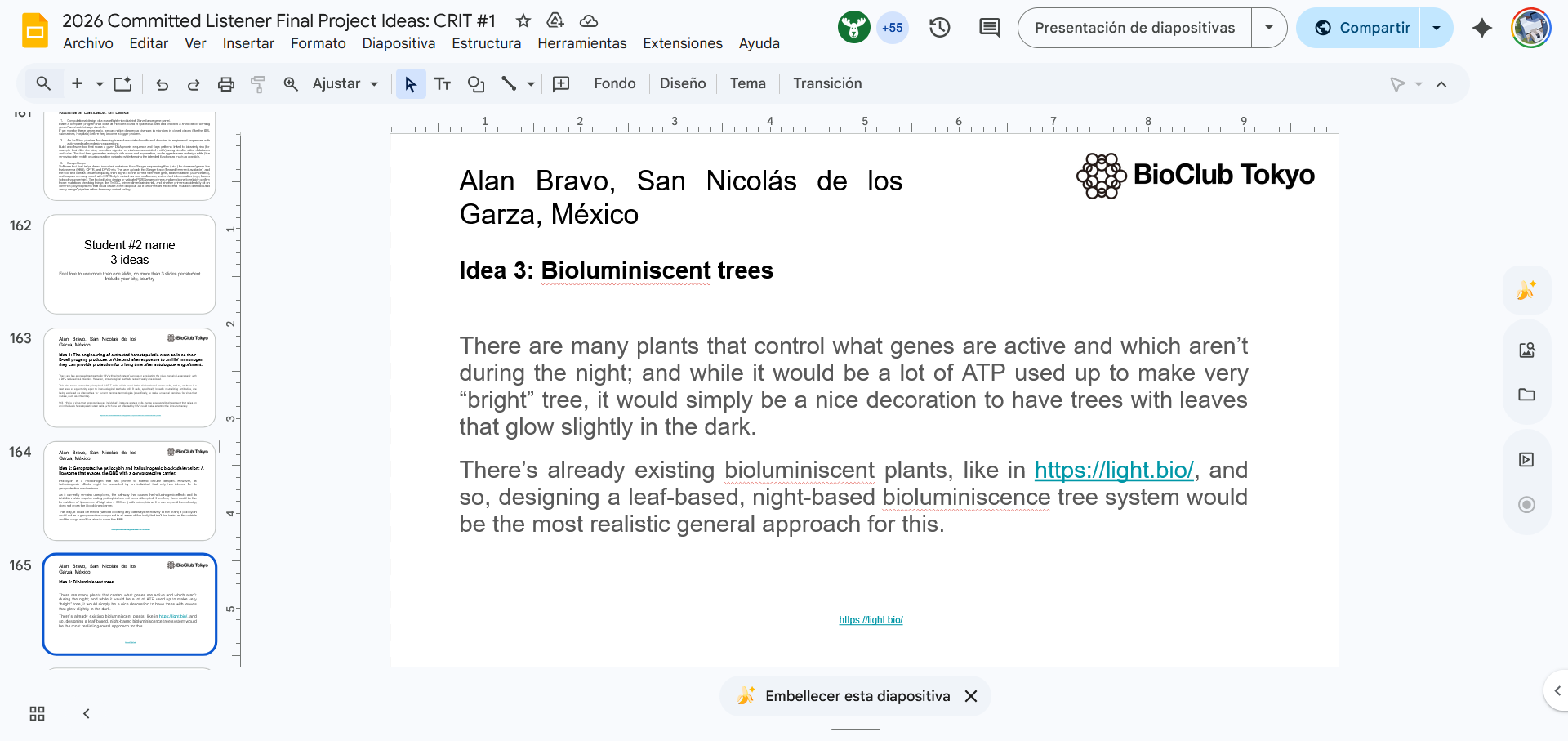
Idea 3, Bioluminiscent trees: AUTOMATION
Echo transfer DNA fragments into 96-well plates, automate Agrobacterium transformation mixes, and potentially a Python image analysis that quantifies glow intensity.
There’s a lot that can be automated; it is not my strength just yet.
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
Committed Listeners Required
As explained in this week’s recitation, add 1-3 slides in your Node’s section of this slide deck with 3 ideas you have for an Individual Final Project. Be sure to put your name, city, and country on your slide!
Week 4 HW: Protein Design Part I
Week 4 — Protein Design Part I
This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat contains at around 26g of protein per 100g of meat, so, in 500g of meat, this would be
26 g x 5 = 130g of protein
Average aminoacid would be ~100 g per mol, so
130g / (100g/mol) = 1.3 mol of aminoacids
Mole = 6.02 x 1023, so
1.3 x Mole = 7.8x1023
So, 7.8x10^23
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because there is no horizontal gene transfer, or at least an effective one
Why are there only 20 natural amino acids?
I’d like to think that it is because that is as optimal life gets
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
The primordial soup!
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handedness
Can you discover additional helices in proteins?
Yes
Why are most molecular helices right-handed?
Almost all aminoacids are L-aminoacids, which means they’re gonna be right-handed helices as that is as optimal as it gets for energy.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Because of their hydrogen bonds, their structure sets them up for said hydrogen bombs. I believe this happens in Alzheimer’s?
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Proteins that aren’t folded properly tend be these β-sheets, and so aggregation happens, and amyloid-β peptides instead of doing their functino, they aggregate.
Design a β-sheet motif that forms a well-ordered structure.
Part B. Protein Analysis and Visualization
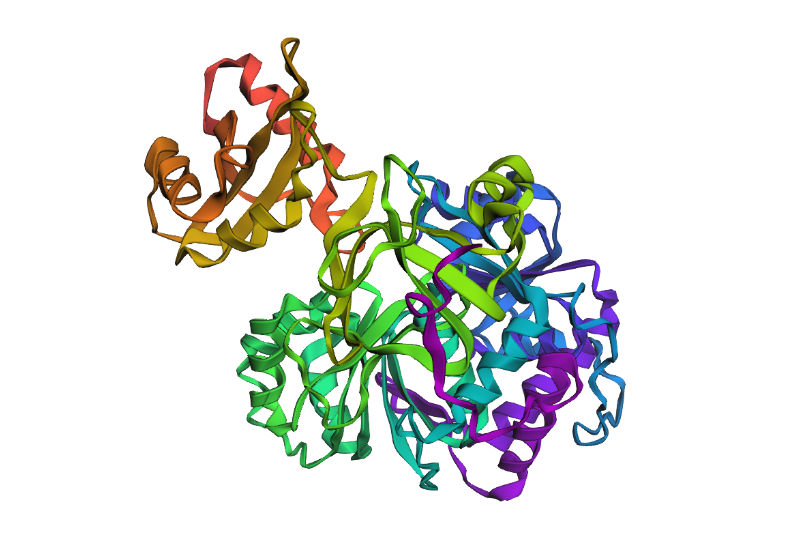
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
Luciferase. I think it’s just a neat protein with a neat name, has a neat function too, which is, emiting light!
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
550 aminoacids long.
Most frequent: L (52 times)
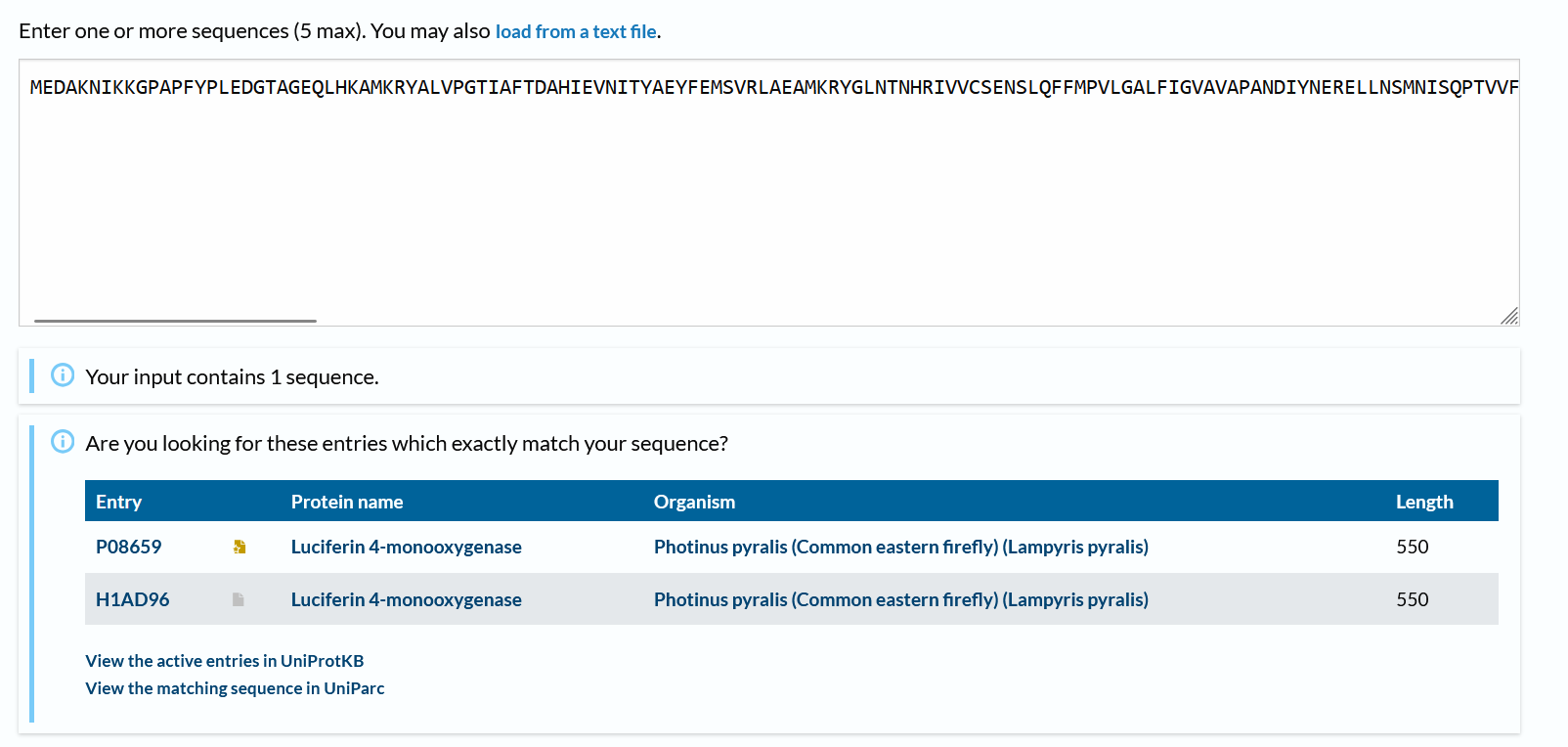
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
There are 2 more!
Does your protein belong to any protein family?
I believe the most correct family would be “oxidative enzymes that produce bioluminescence”.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Well, the resolution is 2.00 Å, so yes!
Are there any other molecules in the solved structure apart from protein?
“Unique protein chains: 1”, so no
Does your protein belong to any structure classification family?
I believe oxidoreductases
Open the structure of your protein in any 3D molecule visualization software:
The chosen software is Chimera
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
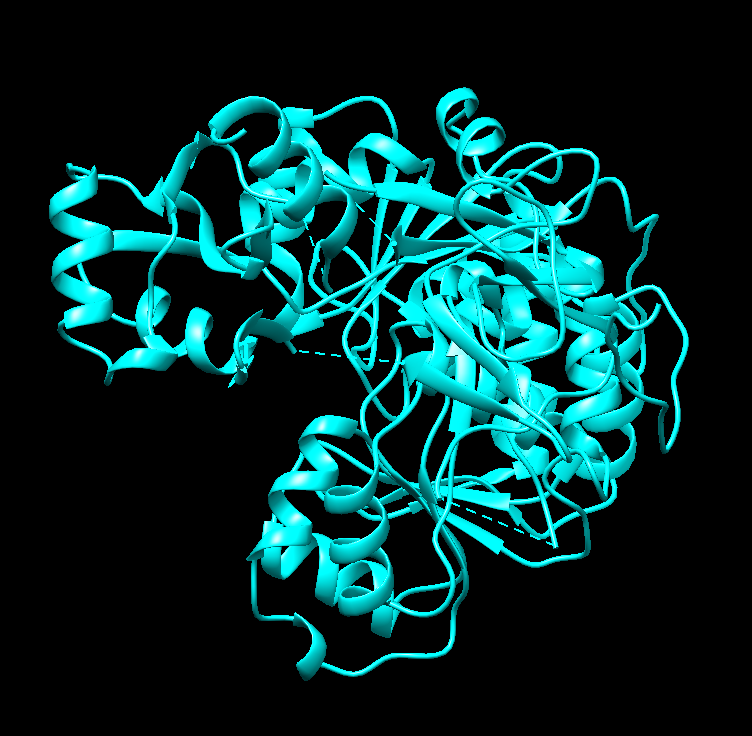
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Nothing seems to happen for “ball and stick” and seemingly there’s no option for cartoon. This happens when only “ribbon” is colored differently:
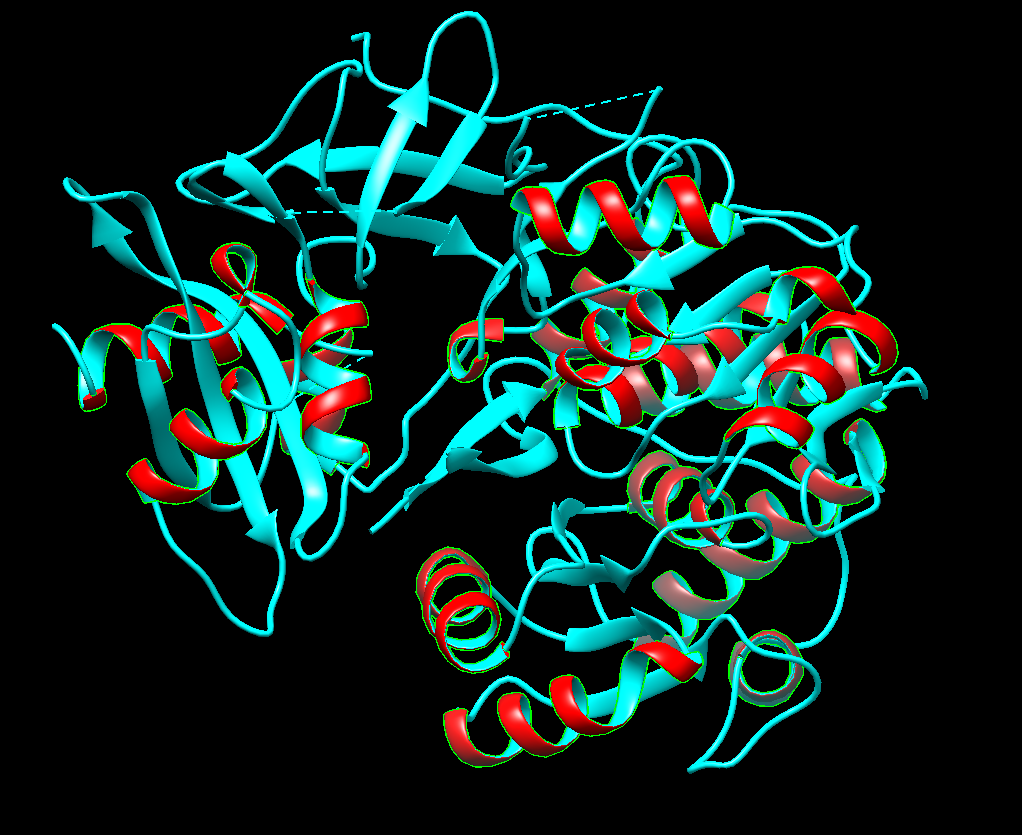
Color the protein by secondary structure. Does it have more helices or sheets?
Right now I’m selecting helices (the red ones). There seems to be more sheets
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Right now I’m selecting hydrophobic regions (the pink ones). There seems to be quite a tie
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
It does! I think that’s what I’m seeing.
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Deep Mutational Scans
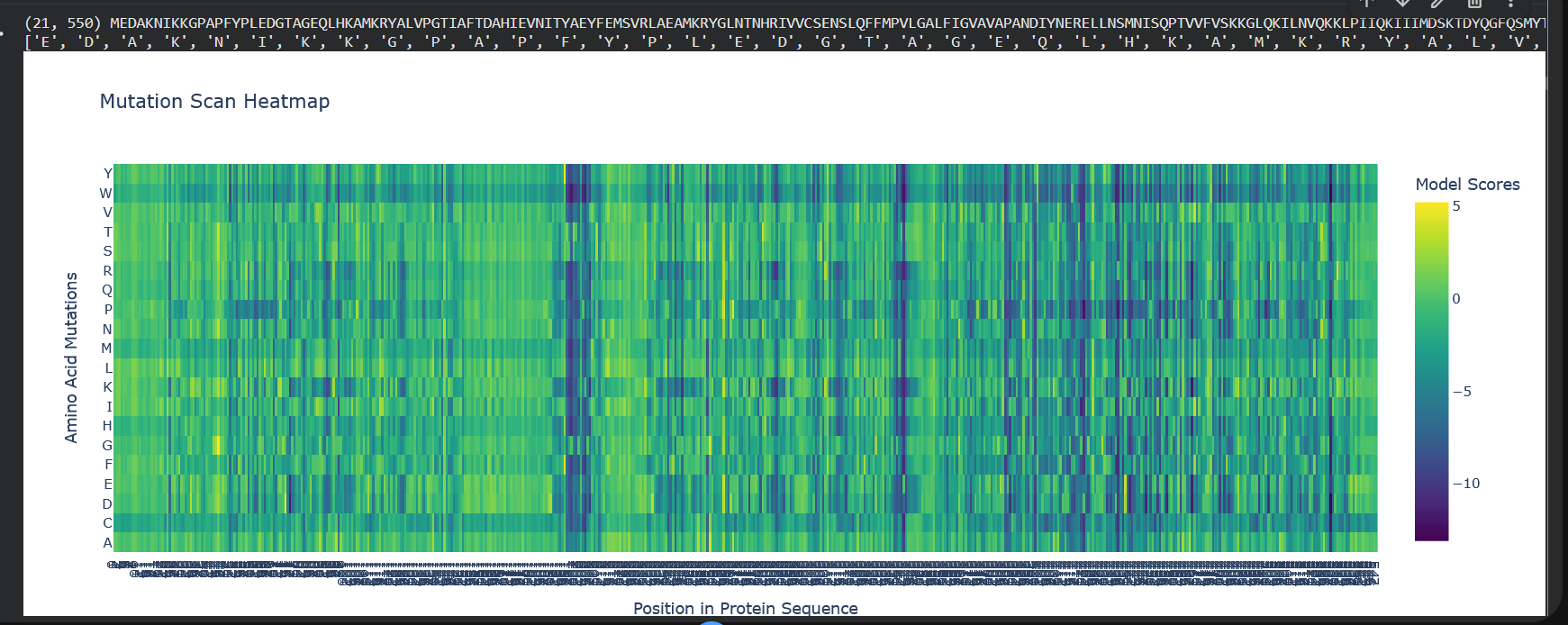
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
So W stands out a lot here, which is tryptophan, the color blue indicates that mutations are not well tolerated. And also notably, from position 197 to 207 there’s a lot of dark blue, so it means that this region’s quite conserved.
Latent Space Analysis
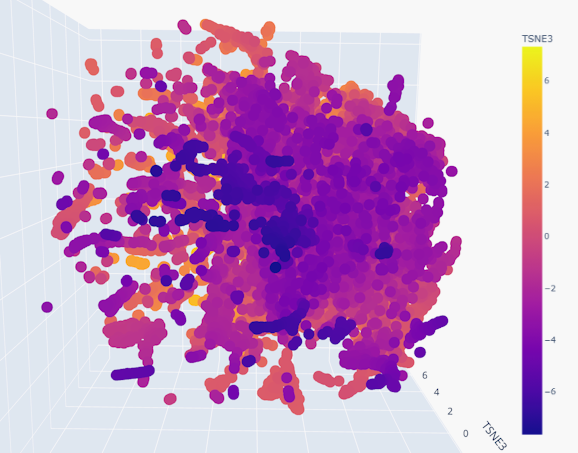
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
Indeed, the neighborhoods do appear to group similar proteins together, given the smoothness of the gradient
C2. Protein Folding
Folding a protein
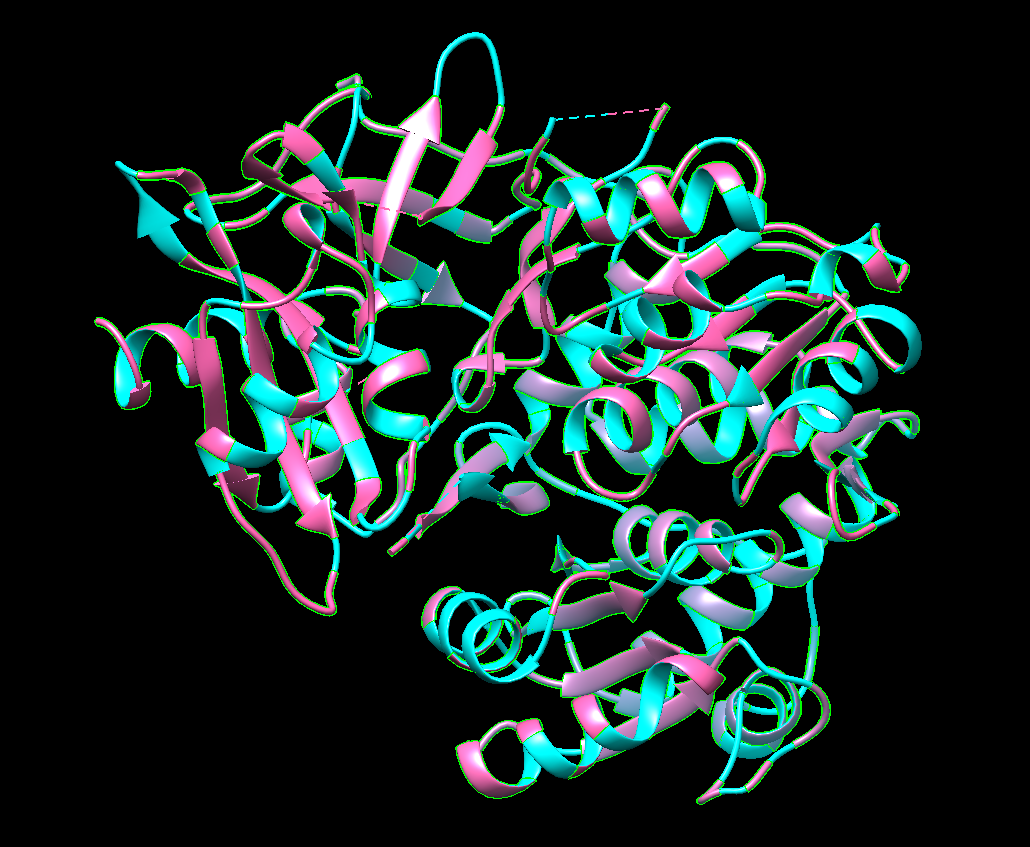
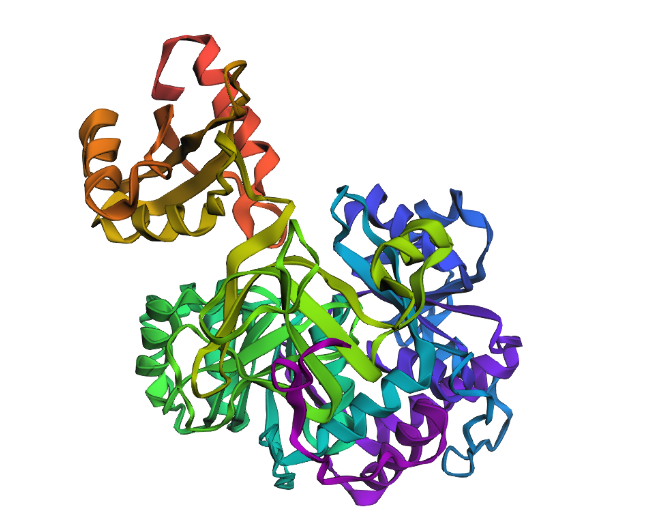
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes! It really does! This is the predicted ESMFold
FAnd this is the original one!
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
For the most part, it is resilient
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
Very very similar
Week 5 HW: Protein Design Part II
Homework
DUE BY START OF MAR 10 LECTURE
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
PeptiVerse: therapeutic property prediction
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
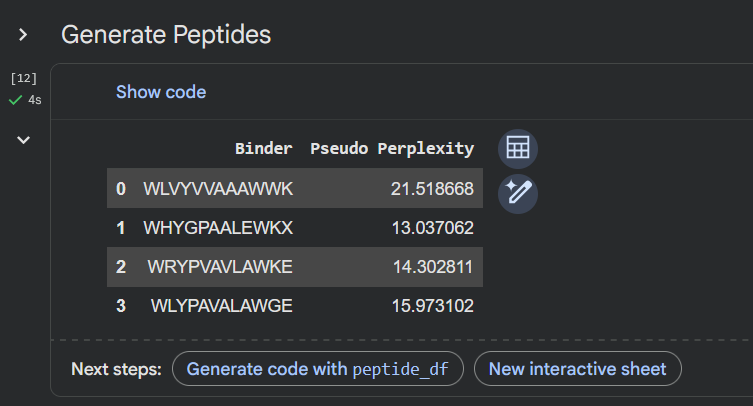
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Apologies, couldn’t figure out how to compare the perplexity score for the known pepdite
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
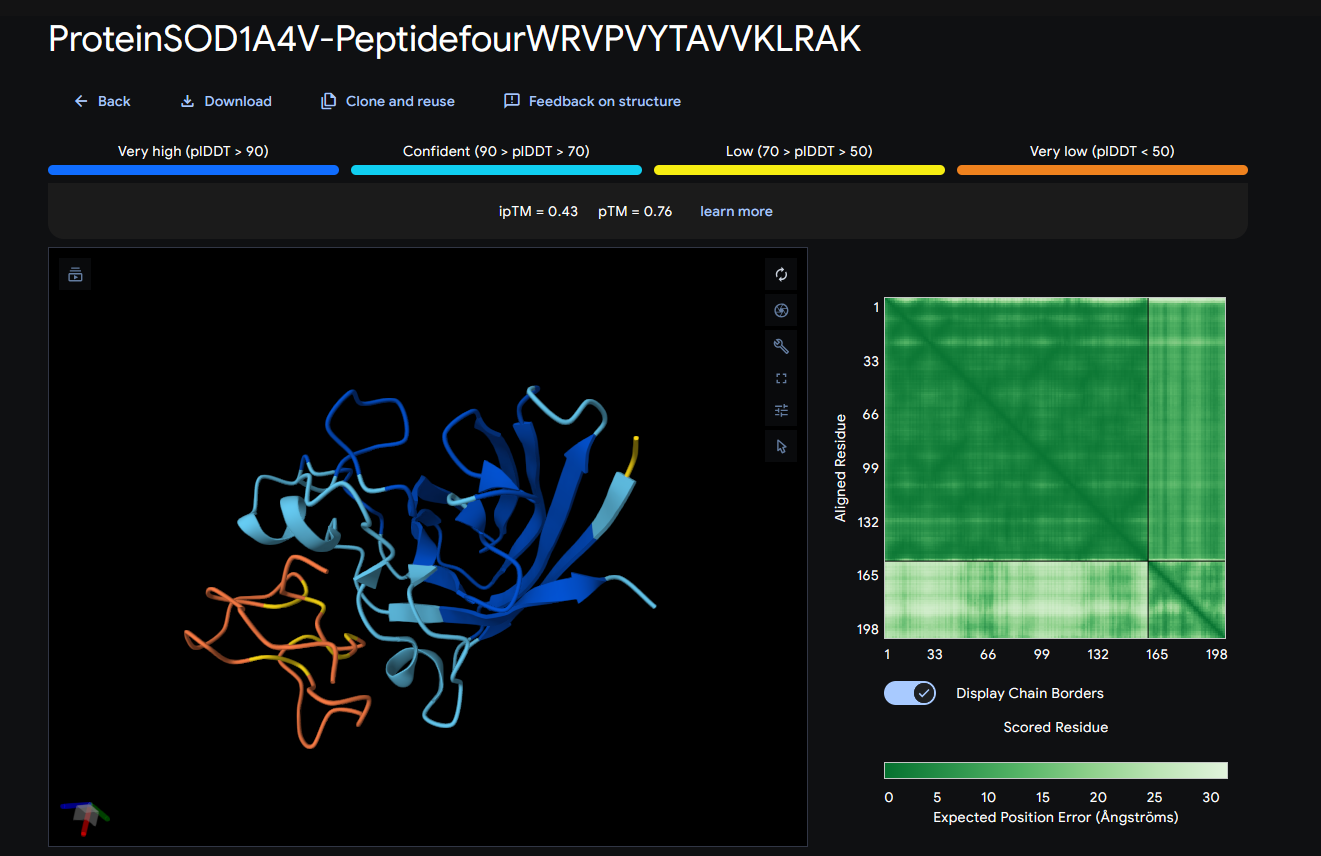
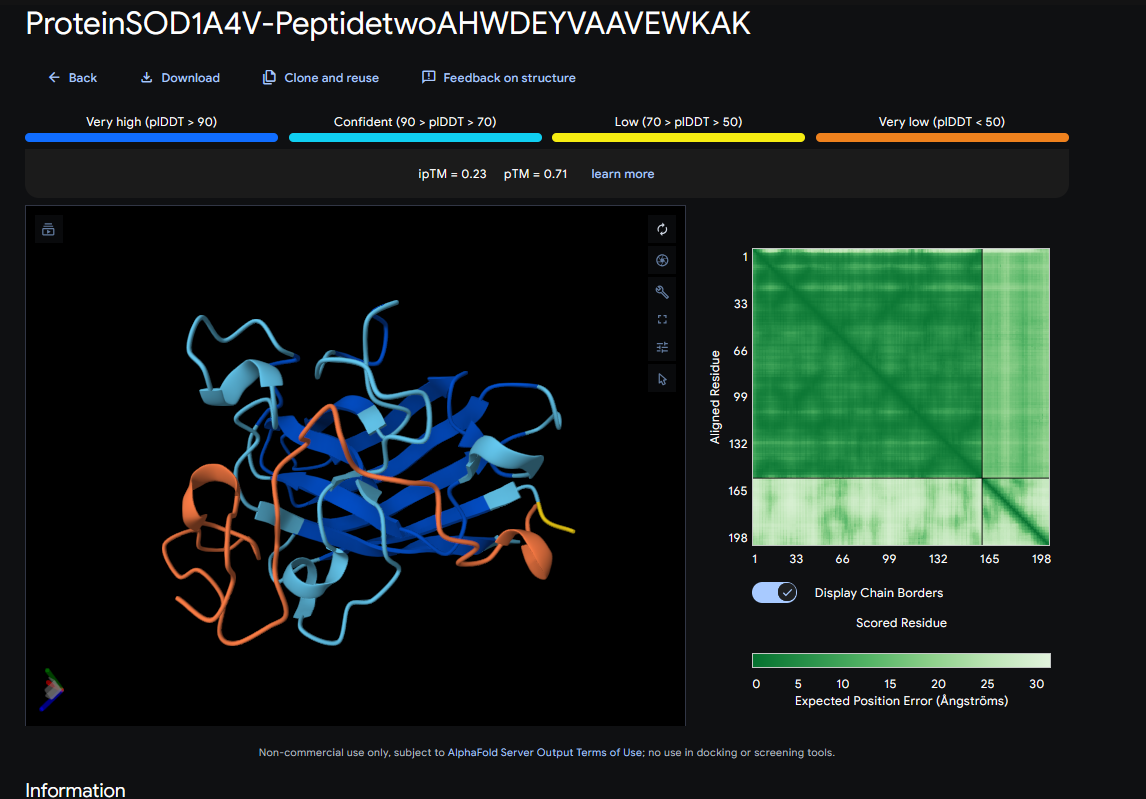
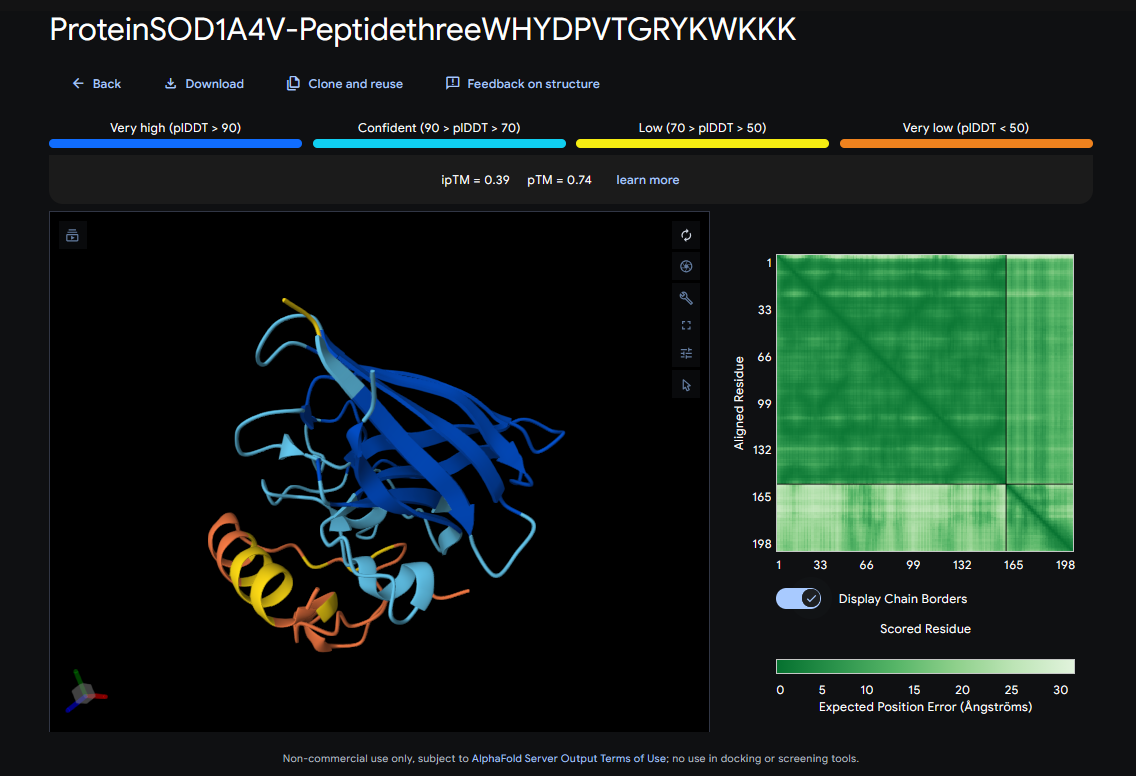
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
ipTM score = 0.36
It seems to bind at around aminoacids 60-64
It does not localize near A4V
It does engage with the β-barrel region, very much on the surface
It appears to be surface-bound
ipTM score = 0.23
It seems to bind just a little bit around A4V and 142-145
It barely does engage with the β-barrel region, near the A4V
It appears to be surface-bound
ipTM score = 0.39
It seems to bind to 60-66, and 143-146
It does not localize near A4V
It does not engage with the β-barrel region
It appears to be surface-bound
ipTM score = 0.43
It seems to bind to 59-64
It does not localize near A4V
It does not engage with the β-barrel region
It appears to be surface-bound
These ipTM scores indicate that the predicted structures are likely wrong, because they’re under 0.5
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Admittedly, I concede for just this one. I have a really tough time with these, sometimes the code just doesn’t load
Week 6 — Genetic Circuits Part I: Assembly Technologies
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
As stated in the thermofisher website, “Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2”
The Phusion DNA Polymerase will be the one replicating the DNA at a really high fidelity
The nucleotides will be ‘inserted’ into the replicated DNA strands
And the reaction buffer (Including MgCl2, which is a co-factor needed for the enzyme), which is optimized, meaning, it has the favorable conditions for the enzyme such as pH.
What are some factors that determine primer annealing temperature during PCR?
The GC content %, the amount of nucleotides, self-dimers, and homodimers
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR uses the thermomixer, which is a machine that controls the temperature of a minitube at 3 distinct temperatures: denaturation, primer annealing, and extension. This is going to be the biggest difference in protocols compared to restriction enzyme digestions. As for when PCR might be preferable: it is when we have a physical nucleotidic sequence to amplify, as in, we don’t have enough copies for whatever we may want to do with it.
As for restriciton enzyme digests, this would only really take place with one temperature; the optimal temperature for our restriction enzyme(s). No thermomixer needed. As for when we would prefer this method; it would be when we have a lot of a nucleotidic sequence, but it is still within our sample, maybe within a plasmid. So in order to isolate the nucleotides we really care about, we use digestion enzymes.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The ends of fragments have to have homologous overlaps, around 20-40 bp of matching sequence (this includes primers to add those overlap regions)
How does the plasmid DNA enter the E. coli cells during transformation?
The membrane is more permeable, and this can be through different methods like electroporation, heatshock, and chemical transformation.
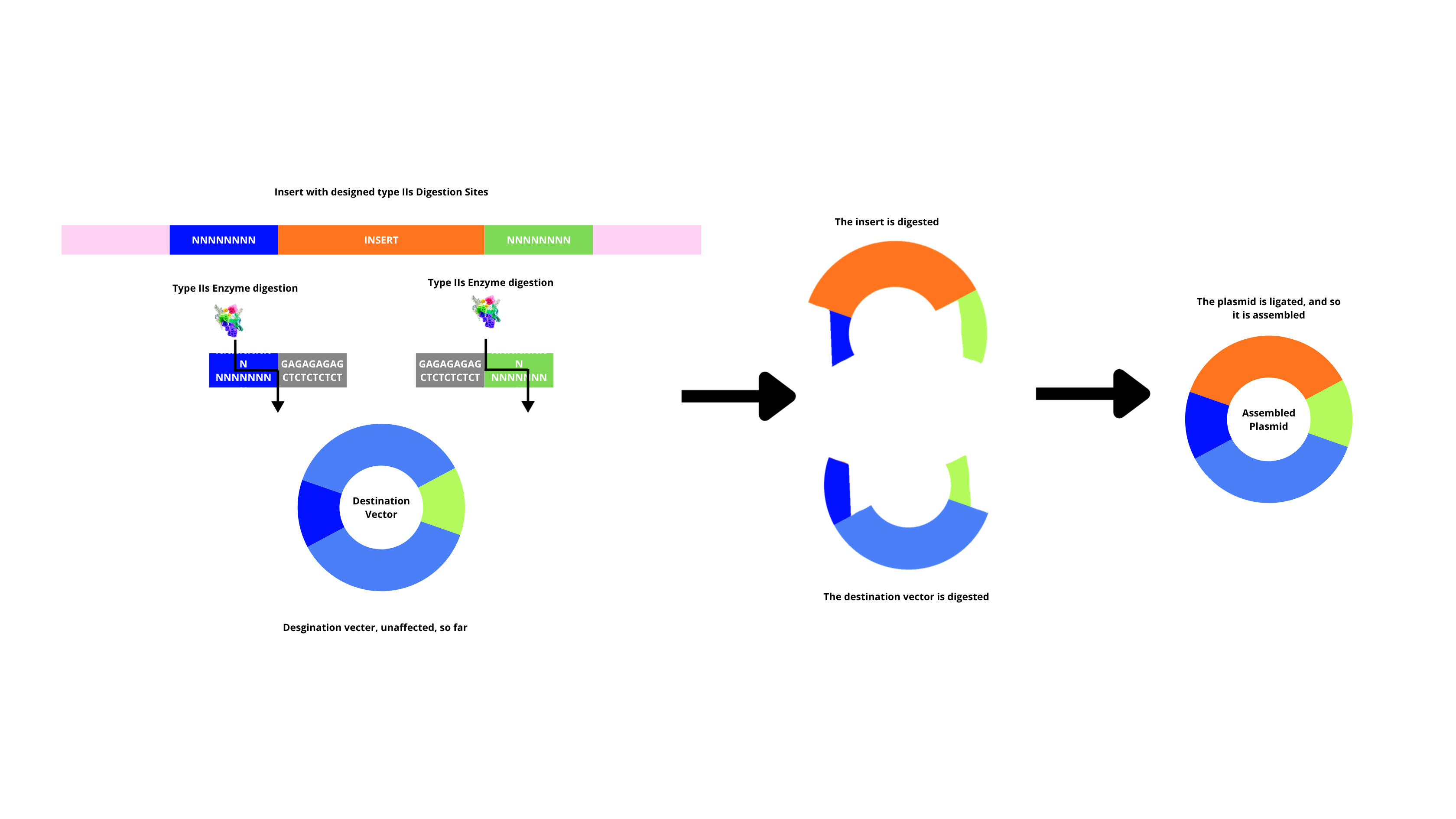
Describe another assembly method in detail (such as Golden Gate Assembly)
By using IIs restriction enzymes together with a ligase, multiple fragments are assembled in order and in a single reaction. This is done by having the enzyme cut fragments that generate overgangs, and complementary overhangs annel between adjacent fragments, the ligase seals them (after this, original restriction sites are removed, so they’re not digested again). This method too has its precautions, such as, fragments that have overhangs designed to be unique, so wrong annealing doesn’t happen.
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Part 2 of this week’s homework
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
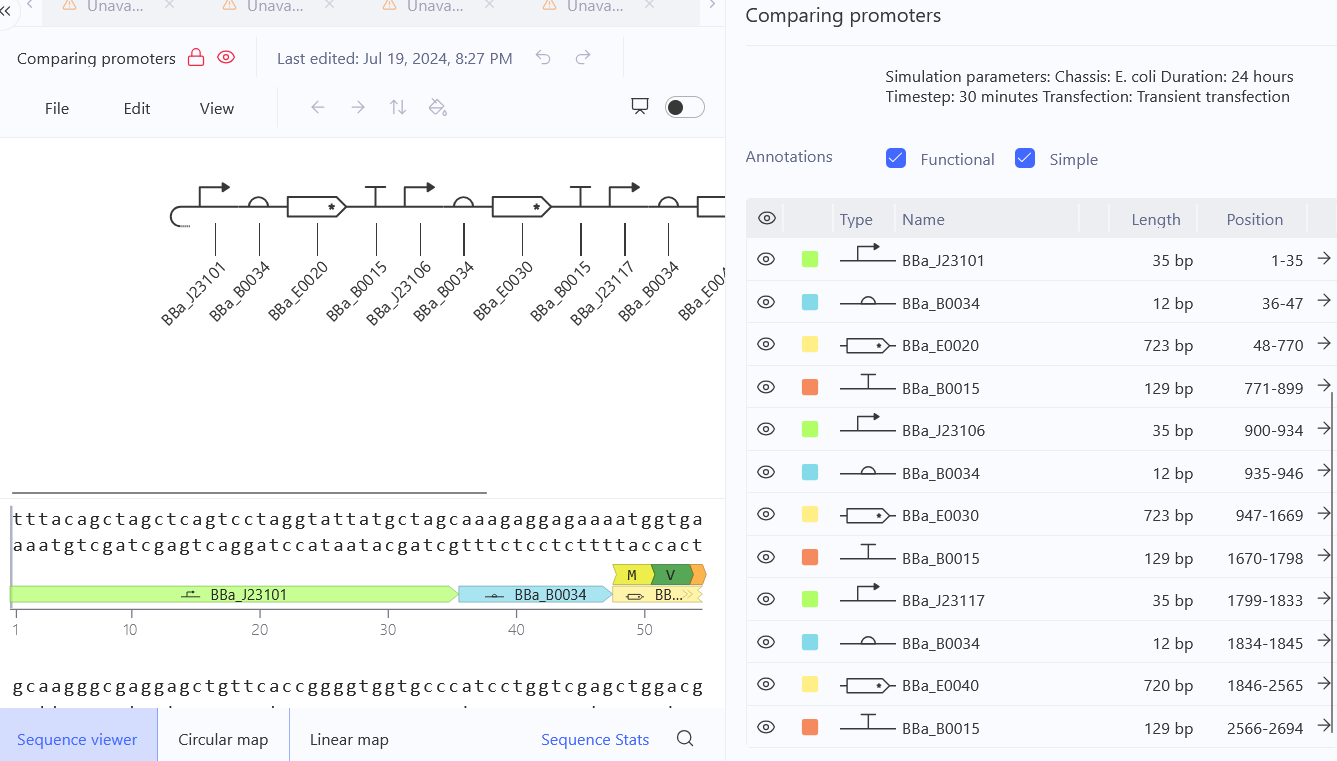
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
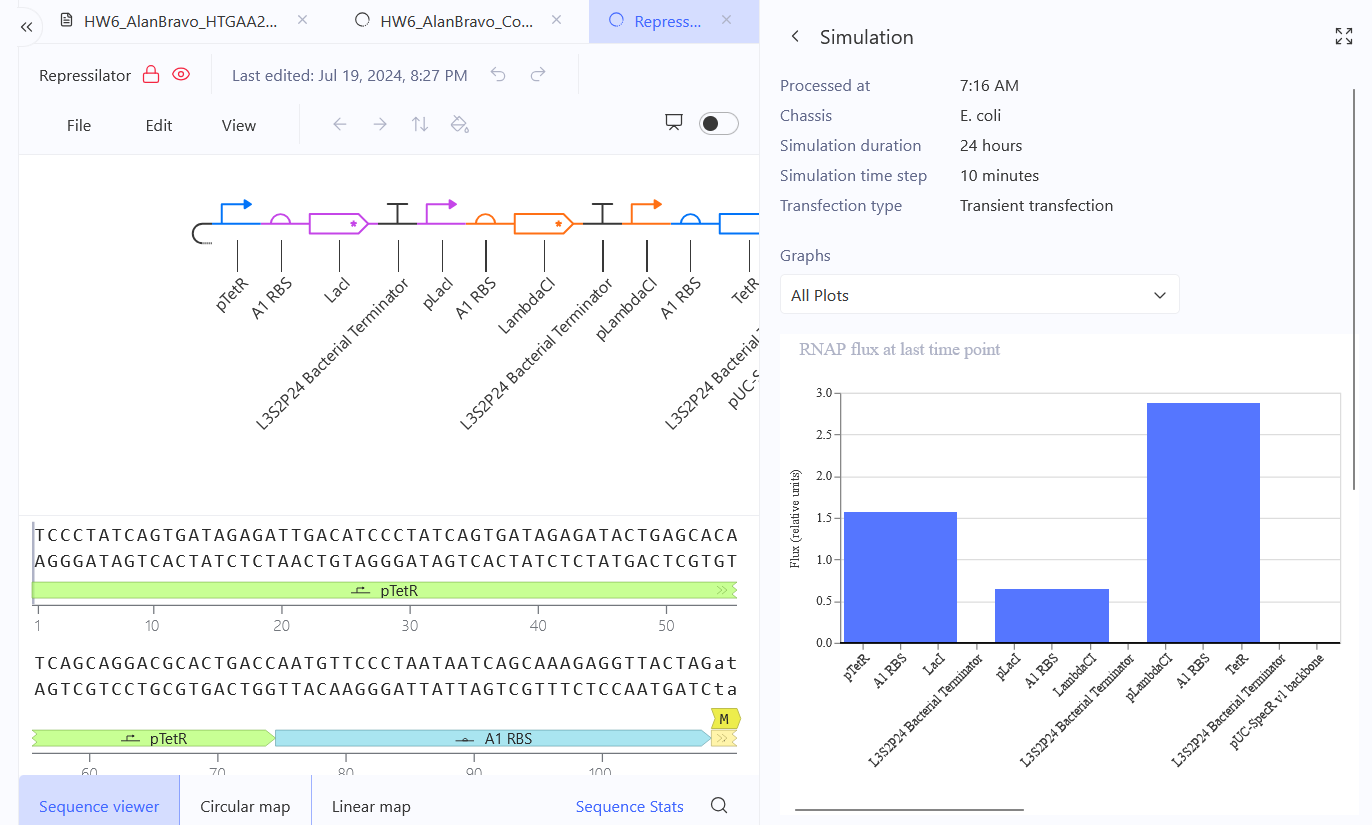
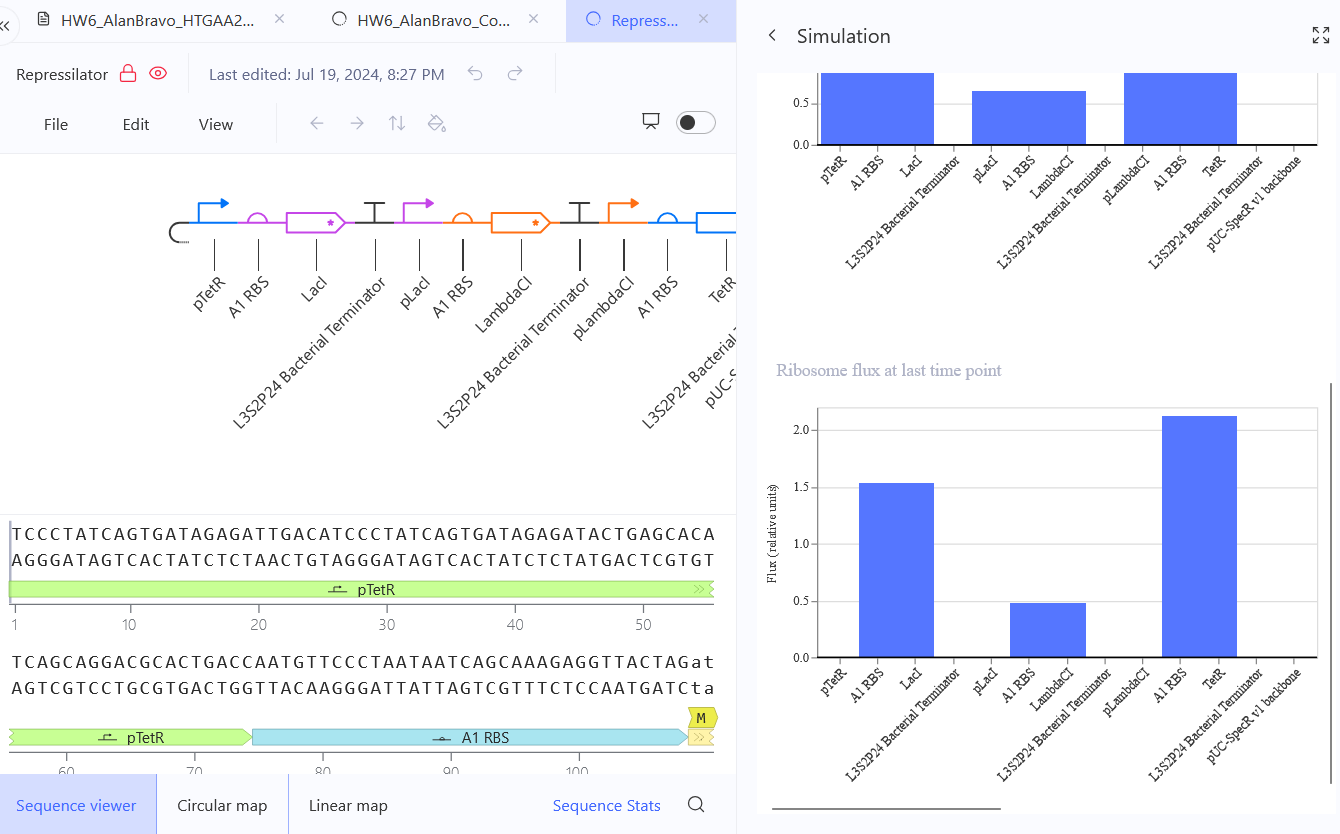
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
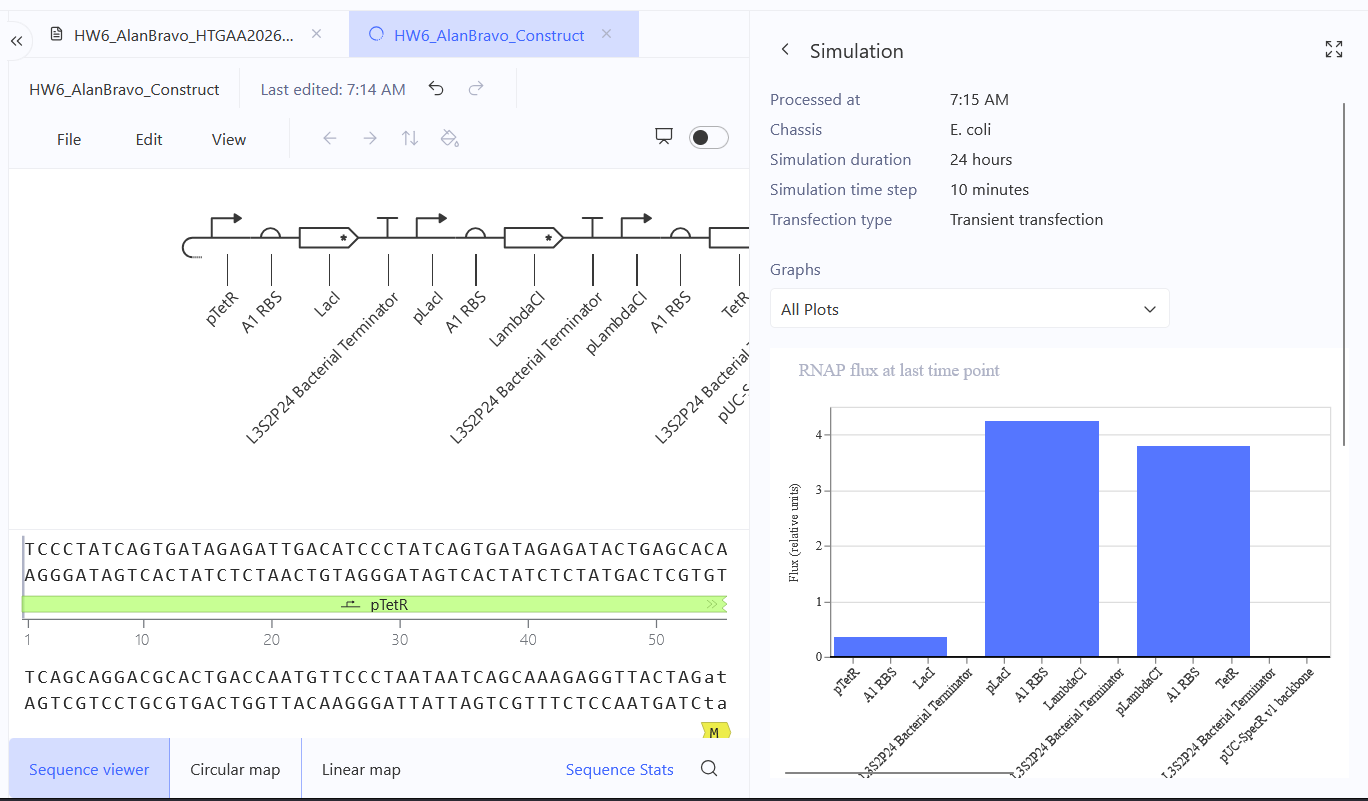
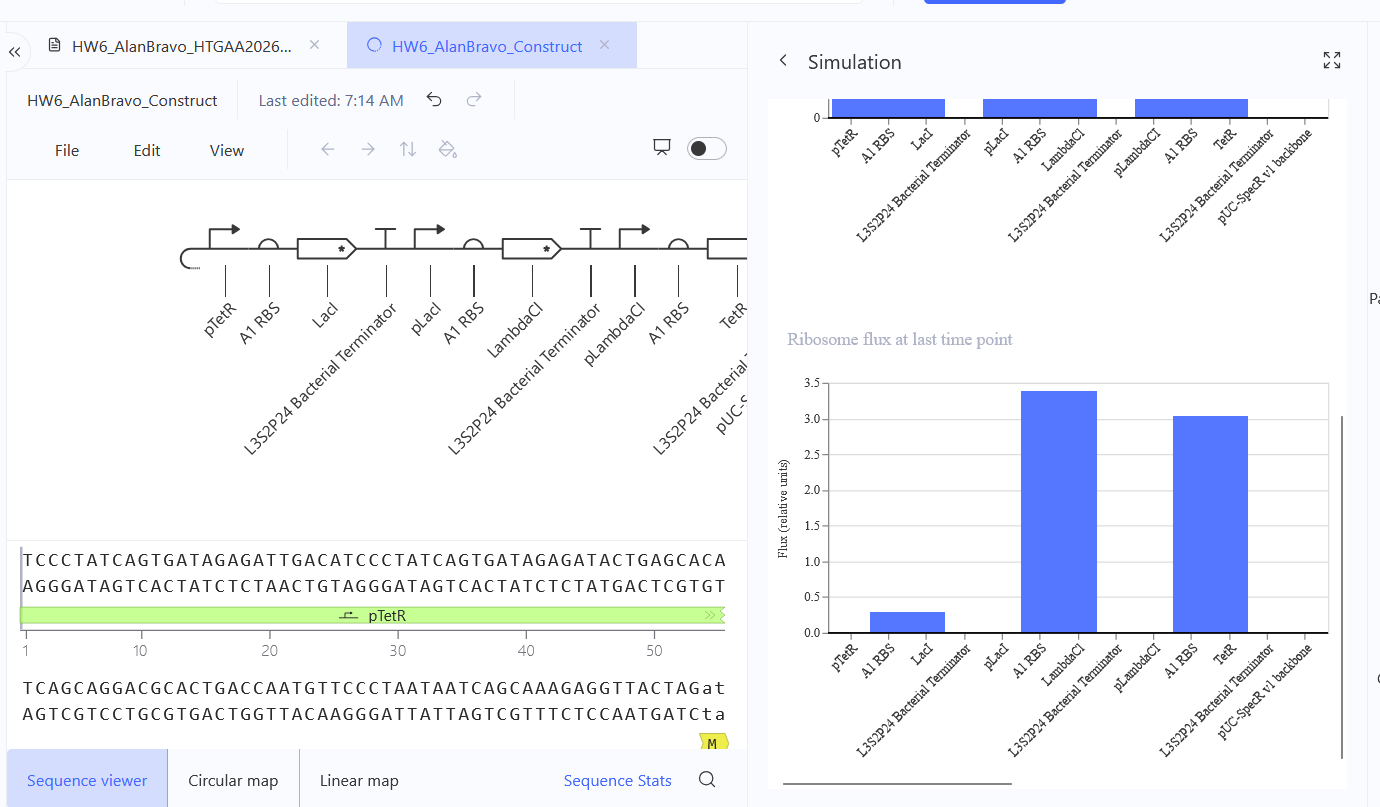
Run the simulator and share your results in the Notebook Entry
(Shared above)
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
It’s odd considering I’ve grabbed the same components and I imitated the same settings, so I believe this has to do with exact sequences, specifically codon optimization, I may have grabbed some sequences that may share the same name, but differ in codon optimization.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Scalability for one, and for second, way more outputs; comparing this to, for example, the lac operon, if I recall correctly it only has a represor, an operator, and the lactose gene. IANNs would have way more repressor, operators, and so way more outputs, and ways said outputs are regulated, by multiple inputs.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
It would be an interesting way to make a diagnosis method by using IANNs, specifically with diseases which may have slight genetic variations; let’s say, there’s a disease that has the same symptoms on a person, but there are 5 variations of it, so the 5 biomarkers are slightly different, with other biomarkers needed too, in order to confirm it really is this disease. So, set of biomarkers needed to be detected, but one of the biomarkers can have 5 variations.
With IANNs, upon receiving the input of all the biomarkers characterized, and the variable biomarker (let’s say, variation n°1), the output could be one fluorescence color. As for, now, variation n°2, same other biomarkers as input, and thanks to this v.n°2, the output will be different, could be luminescence now.
The limitations would definitely be the different responses the are in biology (not all cells will have the exact same response despite having the same genetic components), and personally, defining the threshold needed for the inputs.
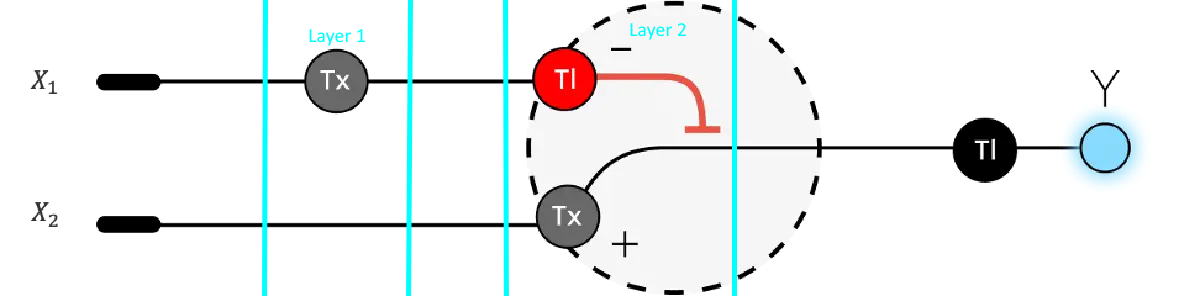
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
My edition, as to my understanding, this is already kind of happening:
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
I know that they’re used for paints, coatings, and textile dyeing! (Venil, 2020) Some of the fungal materials produced are namely: carotenoids, melanins, flavins, phenazines, quinones, and monascins.
As for advantages, they’re not season-dependant like plants, they’re more intense, and even costs are better (Lagashetti et al., 2019)
In literature, there doesn’t seem to be much disadvantages reported.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
For one, the limitation of hyperglicosilation, which is not available to bacteria.
Secondly, the exploitation of pathways that some bacteria do not have, for example, the acetyl-CoA pathway can be taken advantage from, from .S. cerevisiae to avoid competing intermediates, like Yuan and Ching did (2016).
Assignment Part 3: First DNA Twist Order
Review the Individual Final Project documentation guidelines.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS
The first aim of my final project is to genetically engineer autologous hematopoietic stem cells to carry predefined anti-HIV Env broadly neutralizing antibody heavy- and light-chain genes, so that their B-cell progeny express an HIV-specific broadly neutralizing B-cell receptor, by utilizing CRISPR-based gene editing, donor DNA design for bnAb heavy- and light-chain insertion, ex vivo HSC culture, and downstream differentiation and molecular validation assays
The following is what I’ve submitted in the Google Form.
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Lagashetti, A. C., Dufossé, L., Singh, S. K., & Singh, P. N. (2019). Fungal Pigments and Their Prospects in Different Industries. Microorganisms, 7(12), 604. https://doi.org/10.3390/microorganisms7120604
Venil, C. K., Velmurugan, P., Dufossé, L., Devi, P. R., & Ravi, A. V. (2020). Fungal Pigments: Potential Coloring Compounds for Wide Ranging Applications in Textile Dyeing. Journal of fungi (Basel, Switzerland), 6(2), 68. https://doi.org/10.3390/jof6020068
Yuan J., Ching C. B. (2016). Mitochondrial acetyl-CoA utilization pathway for terpenoid productions. Metab. Eng. 38, 303–309. 10.1016/j.ymben.2016.07.008
Week 9 — Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis not only is a robust and efficient system compared to in vivo methods, in which yields are comparatively lower, but if we go in terms of flexibility and experimental variables, there’s a lot of control to be had here given that we only use the components that are essential, this means that variables such as metabolism, possible toxicity, and laborous work such as transformation, transfection, just the whole genetically enginering part (and the assays that come with it, such as, verifying if my organism is indeed genetically modified!) is something to not worry about anymore. Now, because of all the previously mentioned, we do have more control too on elements needed for protein synthesis, such as, nucleotides, salts, DNA concentration, pH; said elements would typically be directed towards other cellular processes, but not here because there is no cell.
Now, as for two cases where it is more beneficial, one of the most obvious ones is the production of proteins that are normally toxic to the host. Since there’s no host, there’s no stopping in the production (hopefully). A second case would be when we need a quick check of a construct for something like a fluorescent protein, like I mentioned earlier, the protocols for genetic engineering do not need to be followed and so it is a huge time save.
Describe the main components of a cell-free expression system and explain the role of each component.
Kind of like PCR, there are components that are a must, such as:
DNA template
An energy system
Salts and buffers
Aminoacids
A cell lysate that comes with transcription and translation components such as enzymes and transcription factors
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Without energy, the machinery stops, all things in biology (unless I missed an exception just like biology always likes to have) do require energy.
The method I would use is glucose and phosphate to supply ATP, like the one in the article by Anderson, et al., (2015).
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic are cheaper first and foremost, so I would choose a GFP for example.
Now, everything changes depending on what you need, so, let’s say I need antibodies or I need a glicosilated protein, which can’t be done by prokaryotic systems; then I’d choose an eukaryotic system here, for.. let’s choose denosumab, If I recall correctly it is an antibody that targets osteoblasts and slows down osteoporosis.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins and their folding problems thanks to the lack of membrane environment.. that’s the thing I’d tackle, the environment, so I’d add liposomes!
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Poor transcription: I’d check codon optimization or even promoter or DNA concentration (of course, I’d check all of these 1 by 1 otherwise I’m gonna be real confused at which one is it really)
Protein misfolding: I’d add chaperones
ATP running out: Add more components of my ATP regeneration system, or possibly just switch it to a different one
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
A biosensor for, let’s say mercury.
What would your synthetic cell do? What is the input and what is the output?
It would detect the presence of mercury! The input would be mercury, the output would be a fluorescent signal such as GFP.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, it could be. Encapsulation’s preferred though because of stability.
Could this function be realized by genetically modified natural cell?
Not by most, definitely. Because there are bacteria and fungi that are resistant to mercury thanks to detoxification proteins (Golysheva et al., 2025). But for the purpose of a system like this, it’d be optimal to not have to employ these proteins in the first place.
Describe the desired outcome of your synthetic cell operation.
Fluorescence when mercury is present.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
Phospholipids like POPC + cholesterol
What would you encapsulate inside? Enzymes, small molecules.
Ribosomes, a DNA construct with a promoter that responds to mercury for GFP, aminoacids, an ATP regeneration system cell lysate components I’ve previously mentioned.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
It could be just E. coli because there’s no eukaryotic PTMs needed
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The mercury ions could diffuse in through the membrane
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
The intensity of GFP (it would be also neat to measure the intensity to different levels of mercury)
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
A smart textile biosensor patch with embedded freeze-dried cell-free reactions that detects cancer proteins that are secreted through sweat.
How will the idea work, in more detail? Write 3-4 sentences or more.
The textile would contain freeze-dried cell free reactions integrated into this wearable patch and when it comes in contact with cancer proteins that come from sweat, the sweat would activate a reaction in the cell-free system, which would be the expression of a reporter, such as the generation of a visible color like dye or fluorescence.
What societal challenge or market need will this address?
Personal health monitoring, specifically, early cancer detection.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Sweat contains water (I think) or, well, it’s a liquid so that should be enough to activate the patch. It might be a one-time use so, I suppose multiple once every few months would be a good routinary way of going about it.
This idea was inspired by my molecular biology teacher, Dr. Azael Adrián Cavazos Jaramillo, who has made a biosensor with electrochemistry for cancer. Very interesting stuff!
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Radiation in space: it can damage DNA in long missions. In a distant future, this’ll be a problem because future exploration will depend on the safety, more like stability of biological materials that have DNA in it, without addressing this, DNA damage could happen and gene expression would change. A cell-free system here is useful because BioBits can express proteins without the need of living ccells, and the P51 viewer can detect fluorescent outputs quite simply.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Multiple GFP-coding DNA sequences exposed to potential DNA-damaging conditions. Let’s let it sit on the moon for some time, or mars (simulating it’s under human skin)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
If the GFP gene is damaged, the BioBits system may produce less fluorescent protein, or just simply, none at all, and so it makes it a simple reporter for seeing how DNA integrity (as in how much the DNA sequence remains unaltered) affects expression in a spacial context. Thanks to the function of the P51 viewer, then this would be a pretty straightforward readout, whether the damaged DNA still functions, or not. It’d also be interesting to sequence the genes once in Earth. Maybe in space if that’s possible too.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
My hypothesis is that the DNA exposed to possible damaging conditions in space will produce a lower GFP expression or none at all in hte BioBits cell free system than intact DNA. I would expect intact GFP DNA to give a stronger, “default” fluorescent signal, while damaged DNA will give a weaks ingla because the sequence may not be able to be transcribed and translated as efficiently, or it simply just won’t be able to. The goal is to study the sensitivity of gene expression in DNA that’s in a spacial context. The relevance comes from one of humanity’s challenges: understanding space’s effect on DNA stability.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I would prepare the BioBits reactions with two samples: intact GFP DNA sequences and damaged GFP DNA sequences. The intact DNA would be a positive control and the damaged one would be a negative control, miniPCR would be used to amplify before adding it ot the BioBits reaction. And after expression, the fluorescence of both controls would be measured with the P51 Molecular Fluorescence Viewer. The data collected would be fluorescence intensity, and also a sequencing of both controls.
Homework Part B: Individual Final Project
We’d like students to start exploring their final project in depth this week! Of your three Aims, for this week you should have at least Aim 1 decided and written down.
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1:
MIT/Harvard/Wellesley ONE FINAL PROJECT IDEA
Committed Listener ONE FINAL PROJECT IDEA
Submit this Final Project selection form if you have not already.
Begin planning how you will write your final project documentation based on these guidelines
Prepare your first DNA order and put it in the “Twist (MIT)” or “Twist (Nodes)” tab of the 2026 HTGAA Ordering: DNA, Reagents, Consumables spreadsheet, as appropriate.
First Twist order deadline for MIT/Harvard/Wellesley students is Friday, April 3 at 11PM ET
First Twist order deadline for Committed Listeners is Friday, April 10 at 11PM ET. (Your Node Lead will place the Twist order, so please work with them to finalize your constructs and ordering decisions.)
Done in my final project page!
References
Anderson, M. J., Stark, J. C., Hodgman, C. E., & Jewett, M. C. (2015). Energizing eukaryotic cell-free protein synthesis with glucose metabolism. FEBS letters, 589(15), 1723–1727. https://doi.org/10.1016/j.febslet.2015.05.045
Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Homework: Final Project
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
The integration of the VRC01 sequence in the hematopoietic cells.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
VRC01 sequence (bnAbs that target Env) in the hematopoietic cells
A GFP reporter gene
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Flow cytometry we can get to know how many cells have been genetically modified.
PCR can be done too, targetting the construct we’ve inserted with primers designed just for that.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
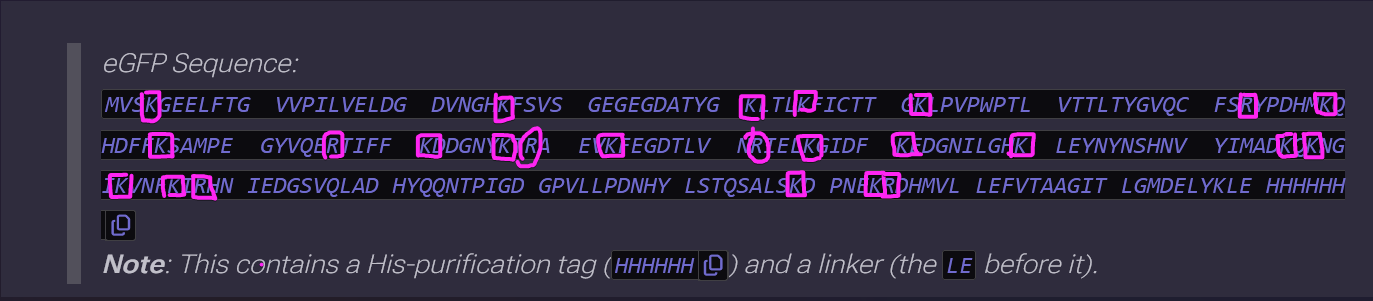
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
The molecular weight is 28006.60
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
1. Determine for each adjacent pair of peaks using (n,n + 1):
Substitution of the values
z = 875.4421 / (903.7148 - 875.4421)
z = 875.4421 / (28.2727)
z = 30.96, but we’ll round it up to 31
2. Determine the MW of the protein using the relationship between m/zn, MW and z
Now, MW = zn (m / zn - H) is the formula we’re gonna use
MW = 31 (903.7148 − 1)
MW = 31 (902.7148)
MW = 27,984.15Da
MW = 27.98KDa
Quite close to 28
3. Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Accuracy = (MW experiment - MW theory) / MW theory
Substituting these would be
Accuracy = |(27,984.15 - 28,000)| / 28,000
Accuracy = 15.85 / 28,000
Accuracy = 0.000566, as in, error of 0.000566%
Homework: Waters Part II — Secondary/Tertiary structure
Optional, so I’ll skip on it for now. I do have plans on coming back to it but thesis and meetings in the laboratory + classes have kept me busy!
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
There’s 21 Lysines (K) and 5 Arginines (R)
How many peptides will be generated from tryptic digestion of eGFP?
27, and it’s not 26 because, let’s say there’s only 1 lysine 0 arginines; we’d have 2 peptides in the end.
1. Navigate to https://web.expasy.org/peptide_mass/
2. Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
3. Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
4. Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Oh, 19 peptides. I was wrong.
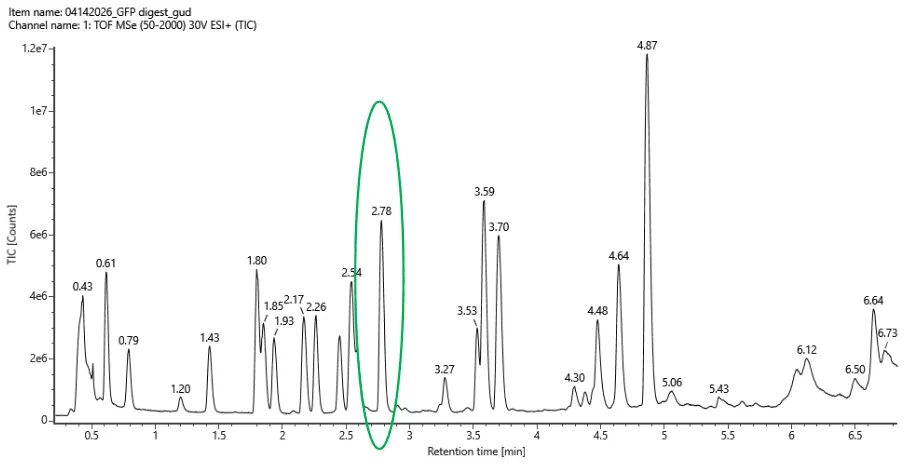
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
23 peaks
Figure5a
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Well, based on my first answer (27), there’s less. Based on the real answer (19), there’s more, which is 23. There are more peaks in he chromatogram.
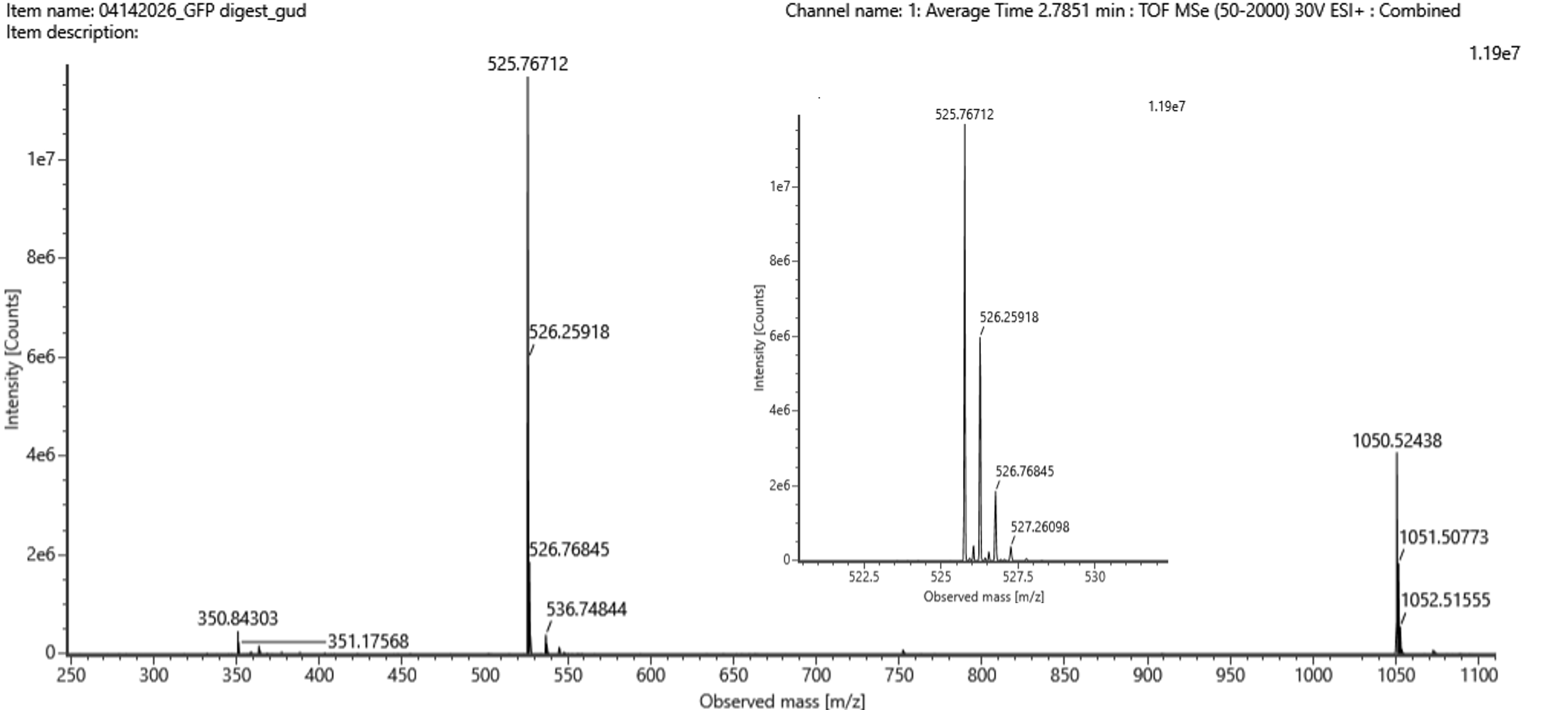
Figure5b
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z
and z
Most abundant peak is 525.76712,
The ones besides are at a seperation of:
526.25918 - 525.76712 = 0.49206
526.76845 - 526.25918 = 0.50927, so both answers at around 0.50
△m/z = 1/z, which would also be 1/0.5 = 2
So then, 525.76712 = [M+2(1.0073)] / 2
(2)(525.76712) = [M+2(1.0073)]
1051.53424 = (M+2.0146)
M+2.0146=1051.53424
M=1049.51964
So finally, (M+H)+=M+1.0073
(M+H)+= 1050.52694
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that Accuracy = |MWexperiment - MWtheory| / MWtheory)
So it would be MWexperiment = 1050.52694
And for MWtheory = 1050.5214
So, replacing values in the formula it would be:
Accuracy = 1050.52694 - 1050.5214 / 1050.52694
Accuracy = 5.27e-6, as in, error of 0.000000527%
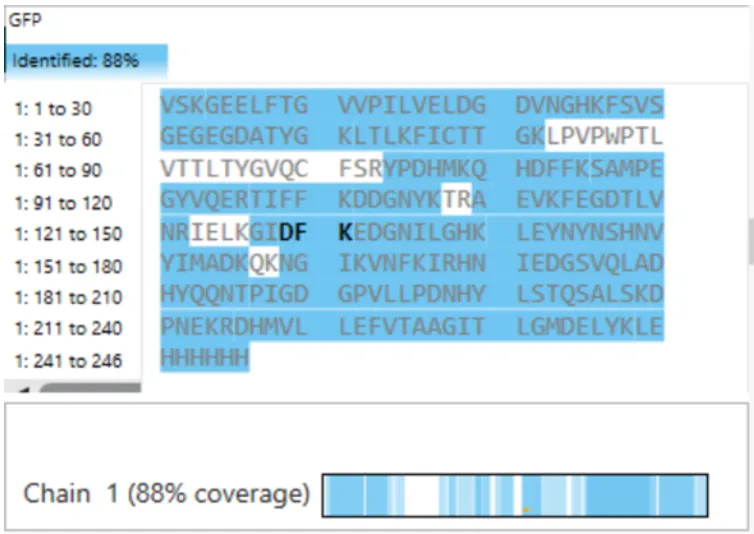
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Figure 6
It’s 88%
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Theoretical (kDa)
Observed / Measured (kDa)
PPM Mass Error
Molecular weight (kDa)
28.006
27.982
857
Week 11 — Bioproduction & Cloud Labs
Homework
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Assignees for this section
MIT/Harvard students Required
Committed Listeners Required
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
If I recall correctly, I was helping on the HTGAA spelling in the bottom left corner!
what you liked about the project, and
what about this collaborative art experiment could be made better for next year.
I love the inspiration from r/place, I actually have participated in r/place twice!
I think we could work with more space (if, physically possible). That’d be neat.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
It is for the conversion of RNA to DNA via retrotranscription
Salts/Buffer
Potassium Glutamate
A co-factor that provides a source of potassium ions for the enzyme
HEPES-KOH pH 7.5
This is the buffer, and it sets the pH to an optimal one for the enzyme
Magnesium Glutamate
A co-factor that provides a source of magnesium ions for the enzyme
So these two tend to go together as buffering salts (they too help with the pH), and as phosphate sources (as this helps with ATP regeneration systems)
Energy / Nucleotide System
Ribose
Ribose is essential because of Deoxyribose, from Deoxyribose Nucleic Acid (aka DNA), so, it’s the backbone of nucleotides.
Glucose
Glucose is the energy source, some CFPS platforms use glucose-6-phosphate too
AMP
While this is an indicator of low ATP, this too when coupled with inorganic polyphosphate, polyphosphate and AMP phosphotransferase, it can be regenerated from AMP to ADP (Itoh et al., 2006).
CMP
This is a precursor to cytidine triphosphate (CTD), which is a nucleotide
GMP
This is a precursor to guanosine triphosphate (GTP), which is required by the ribosome
UMP
This is a precursor to uridine triphosphate (UTP), one of 4 ribonucleotides required for transcription
Guanine
This is a precurstor to both GMP and GTP!
Translation Mix (Amino Acids)
17 Amino Acid Mix
Aminoacids are very much needed by the ribosome in order to synthesize proteins
Tyrosine
It is another aminoacid, however, at pH 12 it is more concentrated
Cysteine
This is yet another aminoacid, however, this one’s very reactive and oxidizes very quickly
Additives
Nicotinamide
Precursor for NAD+ and NADP+
Backfill
Nuclease Free Water
Well the water needs to be free of nucleases in order for the nucleotides bonds to remain intact
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
They can be be differentiated from the energy components, because these will dictate also how long these reactions will keep going. The 1-hour one uses PEP and nucleoside triphosphates, so they’re going for fast yields, while the 20-hour one uses simple precursors and glucose for ATP regeneration
Bonus question: How can transcription occur if GMP is not included but Guanine is?
I would assume that guanine would be able to then later become GMP with an added enzyme (or enzymes) that do(es) this
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
sfGFP: it is reported to mature in just 13.6 minutes, so fluorescence appears quite quickly.
mRFP1: in here we can see quite the opposite, fluorescence appears pretty slowly given that the maturation time is 60 minutes
mKO2: we can see the same here in mKO2, except that the maturation time is doubled, with 108 minutes until maturation. These last 2 proteins are already expressed but it'll take a while until fluorescence (and consequently, readouts) happens.
mTurquoise2: mTurquoise2 has a pretty decent readout time thanks to its maturation time which is 33.5 minutes.
mScarlet_I: comparted to mRFP1, its maturation time is almost tripled with a 174 minute maturation time.
Electra2: a very bright protein! since it has a reported brightness of 61.48, maturation time however is unknown.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
We’re gonna go with mScarlet_I
In an article by Liu et al., (2021), it is mentioned that mSCarlet_I is pretty sensitive to pH changes. So that’s what we’re going to tackle here
So if we’re going to go for a 36-hour incubation, we have to keep in mind the pH changes that could happen, and to avoid them, I’d focus on buffering strength with HEPES-KOH and keeping the potassium phosphate & glutamate balance near neutral pH. I’d expect this sensitive protein to hopefully last around the incubation period of 36 hours.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Potassium Glutamate 312.56 mM
HEPES-KOH pH 7.5 61.25 mM
Magnesium Glutamate 15.72 mM
Potassium phosphate dibasic 5.63 mM
Potassium phosphate monobasic 5.63 mM
17 Amino Acid Mix 4.06 mM
Tyrosine pH 12 4.06 mM
Cysteine 4.00 mM
Nicotinamide 3.13 mM
AMP 625.00 uM
CMP 375.00 uM
UMP 375.00 uM
Guanine 156.25 uM
Ribose 11.625 g/L
Glucose 1.250 g/L
Nuclease-Free Water 1.325 uL
This is what I’ve come up with! As I previously mentioned, HEPES-KOH is great to keep for a protein like the mScarlet_I, and I also noticed there’s not a whole lot MgGlutamate compared to KGlutamate, so I tweaked that as well.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
If given the chance, I’ll do it, because it looks really cool! If this page doesn’t change by the deadline though, you may assume I wasn’t able to do it in time.
Anyways, as this is the end, please, feel free to go to my final project. Cheers!
Bak, D. W., Bechtel, T. J., Falco, J. A., & Weerapana, E. (2019). Cysteine reactivity across the subcellular universe. Current opinion in chemical biology, 48, 96–105. https://doi.org/10.1016/j.cbpa.2018.11.002
Calhoun, K. A., & Swartz, J. R. (2005). Energizing cell-free protein synthesis with glucose metabolism. Biotechnology and bioengineering, 90(5), 606–613. https://doi.org/10.1002/bit.20449
Itoh, H., Kawazoe, Y., & Shiba, T. (2006). Enhancement of protein synthesis by an inorganic polyphosphate in an E. coli cell-free system. Journal of microbiological methods, 64(2), 241–249. https://doi.org/10.1016/j.mimet.2005.05.003
Liu, A., Huang, X., He, W., Xue, F., Yang, Y., Liu, J., Chen, L., Yuan, L., & Xu, P. (2021). pHmScarlet is a pH-sensitive red fluorescent protein to monitor exocytosis docking and fusion steps. Nature Communications 2021 12:1, 12(1), 1413-. https://doi.org/10.1038/s41467-021-21666-7