Week 4 HW: Protein Design Part I
HW4
Conceptual Questions
Here are my answers to the conceptual questions: [1] How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- ~3x1024 molecules from (500g/(100g/mol))x6.02x1023
[2] Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Proteins are broken into amino acids in the digestive system and reassembled using instructions from human DNA. We reuse the material, but not the entire structure.
[3] Why are there only 20 natural amino acids?
- These 20 amino acids are diverse enough to form all of the complex functional proteins needed throughout evolution. Adding others does not necessarily provide an evolutionary advantage and increases complexity and error rates.
[4] Can you make other non-natural amino acids? Design some new amino acids.
- Yes, you can do this by using engineered ribosomes and system. Some examples of this include adding fluorinated amino acids to increase hydrophobicity and stability and photo-reactive amino acids that respond to light.
[5] Where did amino acids come from before enzymes that make them, and before life started?
- They came from abiotic chemical reactions on early Earth, such as from sources like gases with lightning and UV radiation as energy sources or hydrothermal vents from meteorites.
[6] If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- Natural proteins are made of L-amino acids and form right-handed α-helices. If instead you used entirely D-amino acids you;d make a left-handed α-helix.
[7] Can you discover additional helices in proteins? -Yes, proteins can form multiple helical structures not just the α-helix, some examples are 3₁₀ helices and π-helices.
[8] Why are most molecular helices right-handed?
- Life evolved using L-amino acids, which are right-handed.
[9] Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
- β-sheets form planar structures with backbone hydrogen bond donors and acceptors along their edges. This lets multiple sheets form hydrogen bonds and stack together.
[10] Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
- Skipped
[11] Design a β-sheet motif that forms a well-ordered structure.
- Skipped
Protein Analysis and Visualization - ATP Synthase: P0AB98 · ATP6_ECOLI
Briefly describe the protein you selected and why you selected it.
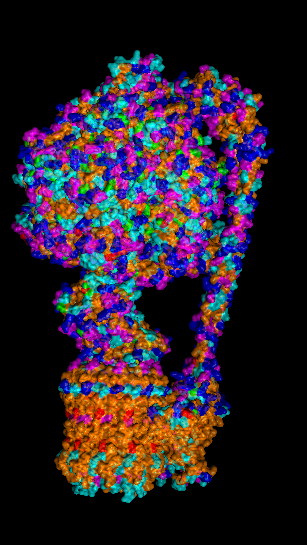
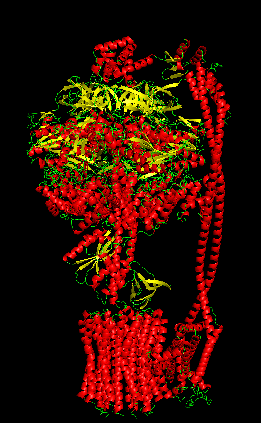
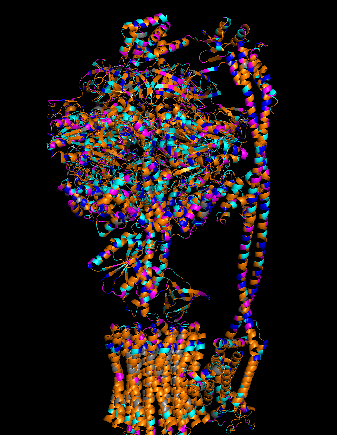
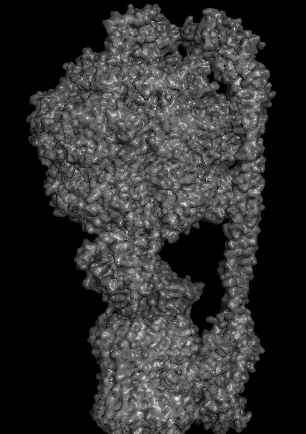
I selected ATP synthase because it is a fascinating molecular machine. As a mechanically driven enzyme, it directly connects structure, motion, and energy conversion. ATP synthase is a large multi-subunit complex composed of two major regions: F₀ (membrane-embedded rotor) and F₁ (soluble catalytic head). The amino acid sequence depends on the organism and subunit. I looked at ATP6_ECOLI which is a critical membrane component of the F₀ motor in E. coli. It has 271 amino acids and is highly hydrophobic with many transmembrane helices. The most frequent amino acid is leucine which appears 45 times.
Here’s the sequence: MASENMTPQDYIGHHLNNLQLDLRTFSLVDPQNPPATFWTINIDSMFFSVVLGLLFLVLFRSVAKKATSGVPGKFQTAIELVIGFVNGSVKDMYHGKSKLIAPLALTIFVWVFLMNLMDLLPIDLLPYIAEHVLGLPALRVVPSADVNVTLSMALGVFILILFYSIKMKGIGGFTKELTLQPFNHWAFIPVNLILEGVSLLSKPVSLGLRLFGNMYAGELIFILIAGLLPWWSQWILNVPWAIFHILIITLQAFIFMVLTIVYLSMASEEH
BLAST searching in UniProt revealed 250 homologs. The protein belongs to the F-type ATPase subunit family, which is common across many life forms because of its role in energy metabolism.
The earliest I could find this structure was solved was back in 2016 but in 2023 it was increased from 6.9 Å to 3.1 Å making it really close to a good quality structure. The solved structures of ATP synthase in E.coli includes multiple proteins and lipid molecules, detergent molecules, and even bound ATP or ADP.
In structural classification systems, subunit a belongs to the membrane protein α-helical bundle class, because it is mostly made of transmembrane α-helices.
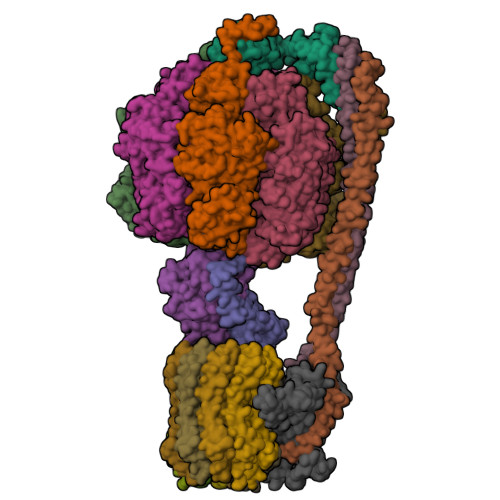

Protein Visualizations: ATP Synthase 6oqw
Group Brainstorm on Bacteriophage Engineering
Computational Engineering of the MS2 Lysis Protein (L) Background. The MS2 L protein is a 75-amino-acid polypeptide that lyses E. coli by an incompletely understood mechanism. Its C-terminal transmembrane (TM) domain inserts into the cytoplasmic membrane and oligomerizes, causing depolarization that triggers host autolytic enzymes to degrade the murein layer. Recessive, conservative missense mutations clustered around a conserved LS dipeptide strongly implies L engages an unidentified host protein target rather than simply disrupting the bilayer. The dispensable N-terminal domain binds chaperone DnaJ (with solved PDB structures), modulating lysis timing. Its removal causes lysis ~20 min earlier. No experimental structure of L exists. Goals. (1) Stabilize L for more robust membrane accumulation. (2) Accelerate lysis by bypassing DnaJ-dependent regulatory timing and improving delivery of functional L to the membrane. Because the downstream lytic target is unknown, we do not attempt to enhance per-molecule toxicity at the point of target engagement; we focus on removing regulatory brakes and increasing the supply of functional protein. Pipeline: Three Tools, Each Non-Redundant
- Clustal Omega (Conservation Map). Align L homologs across Leviviridae (MS2, f2, R17, GA, PP7, AP205, PRR1, M12, KU1, JP34). Conserved C-terminal residues, especially the LS motif, are presumed to mediate the unknown heterotypic interaction and are excluded from mutation. This map constrains all downstream design.
- ESM2 + Deep Combinatorial Scanning (Fitness Oracle). Score every single-point mutation by log-likelihood change: increases at mutable positions indicate stabilizing substitutions (Goal 1). N-terminal scanning identifies mutations that disrupt DnaJ binding (Goal 2). A strict preservation rule applies near the LS motif: mutations are evaluated for maintenance of wild-type fitness, not improvement. The genetics show even conservative changes there cause recessive loss of function. Pairwise combinatorial scanning (about ~2M pairs) captures epistatic synergies at mutable positions. This could be potentially pushed further with enough compute.
- AlphaFold 3 (Structural Filter + Complex Model). Predicts variant structures as a sanity check (does the TM helix survive?) and models the L–DnaJ complex to verify that N-terminal truncations/mutations disrupt the regulatory interface. Used as a filter, not a design engine. PAE matrix identifies confident interface contacts. Ranking. Composite score: ESM2 log-likelihood gain (stability) + conservation preservation (all essential residues intact) + AF3-predicted DnaJ-binding disruption (for timing bypass). Top 10–20 variants advance to experimental validation. Pipeline Schematic

Sources
AI Prompts
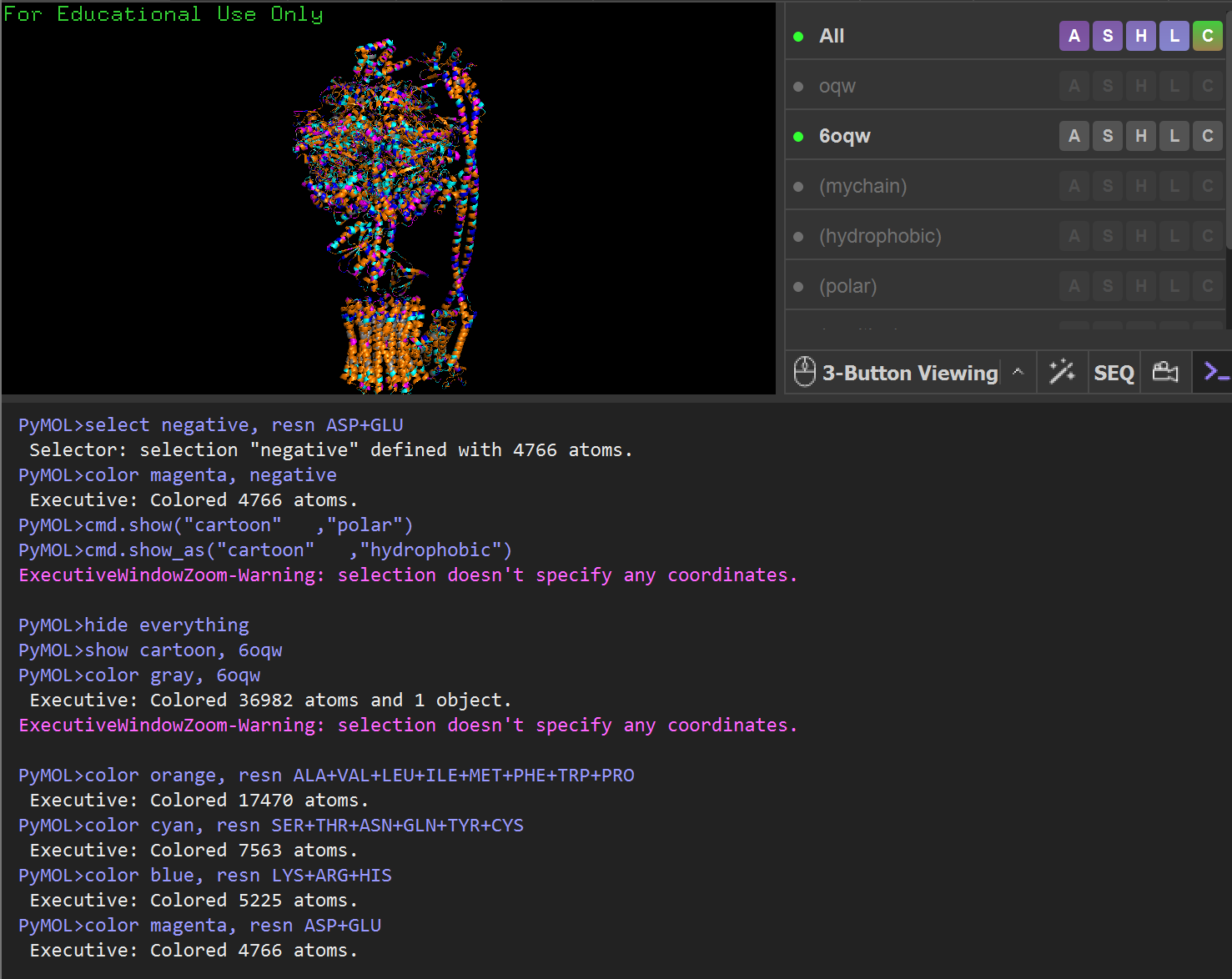
- Write PyMOL code to help me visualize the protein as “cartoon”, “ribbon” and “ball and stick”, color the protein by secondary structure, color the protein by residue type, and visualize the surface of the protein