Week 5 HW: Protein Design Part II
HW5
Part A
Part 1: Generate Binders with PepMLM
I started by getting the SOD1 sequence from UniProt: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Then I added the A4V mutation which changed it to: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
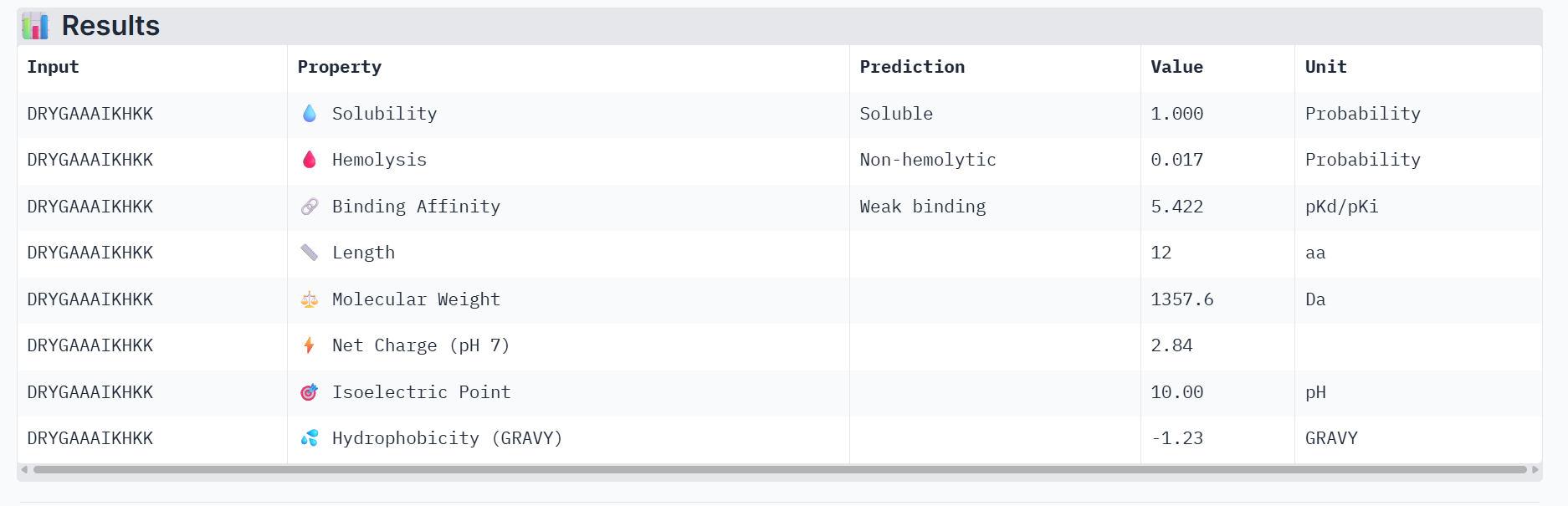
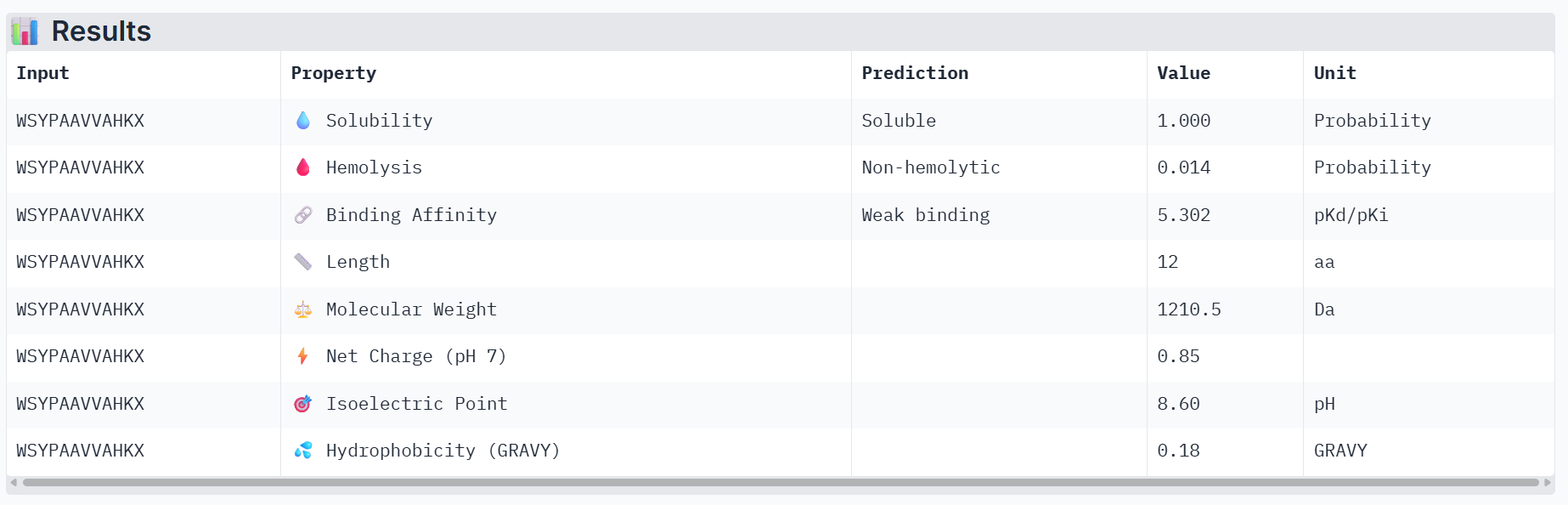
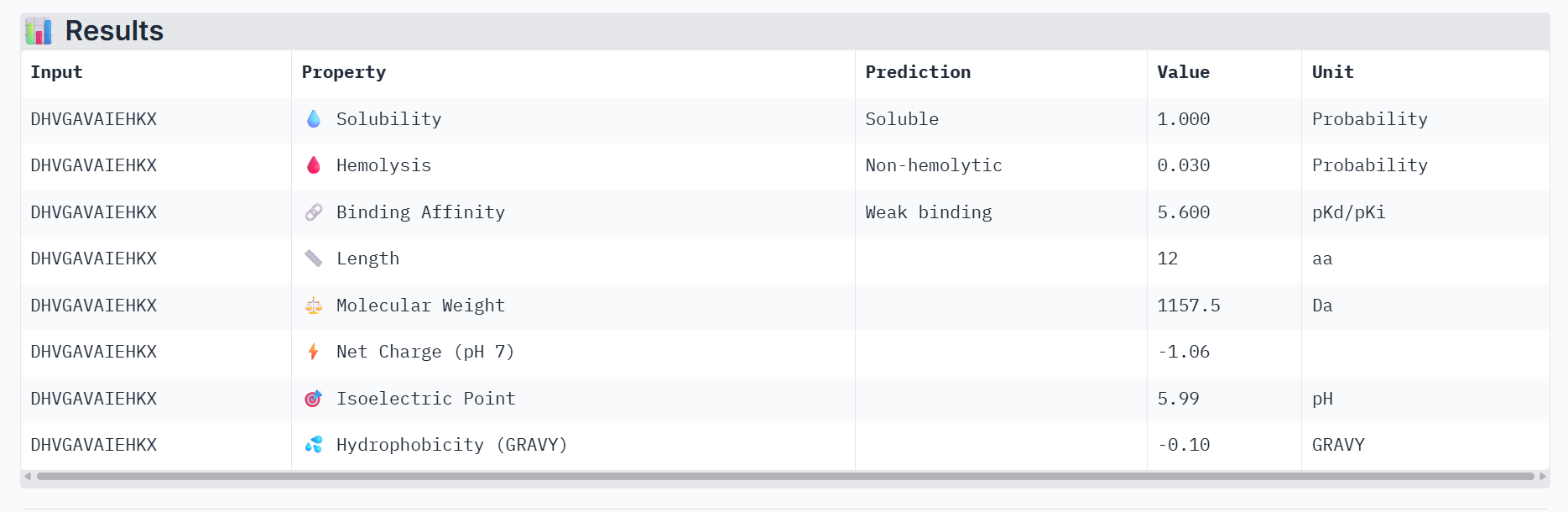
After running PepMLM I got these results: Binder,Pseudo Perplexity DHSYAVVVAWKX,15.47789718276849 DRYGAAAIKHKK,17.832445649262862 WSYPAAVVAHKX,7.216885793300712 DHVGAVAIEHKX,10.94105373182517
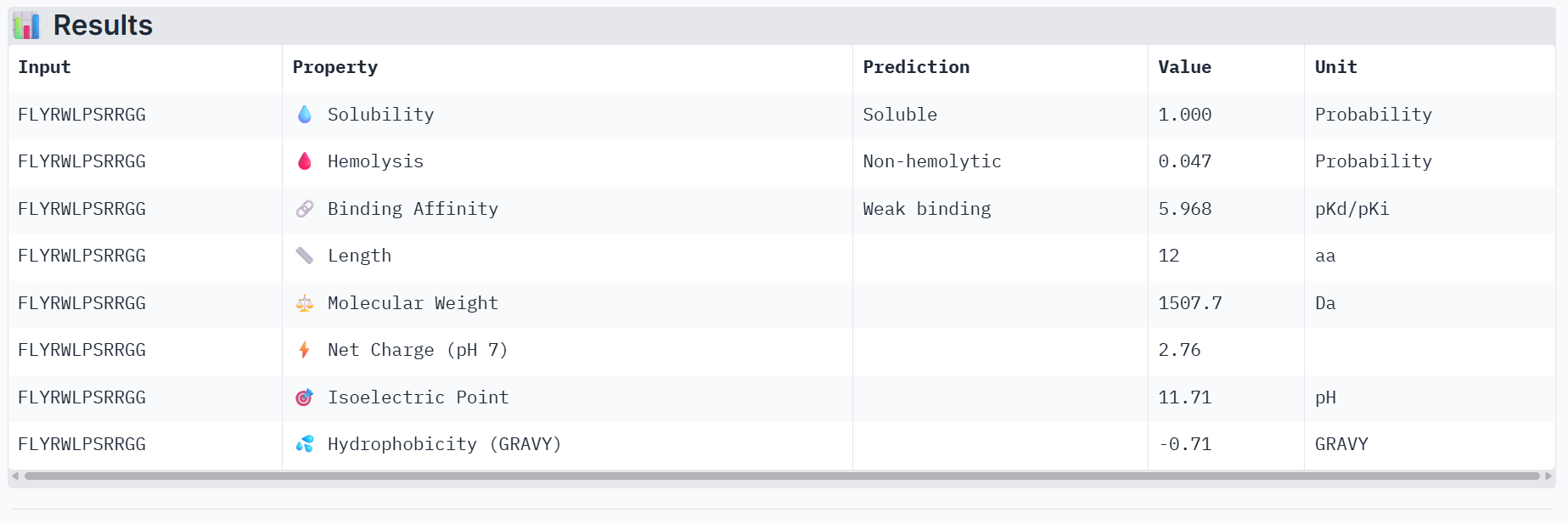
Compared to the known result of: FLYRWLPSRRGG,20.63523127283615
Part 2: Evaluate Binders with AlphaFold3
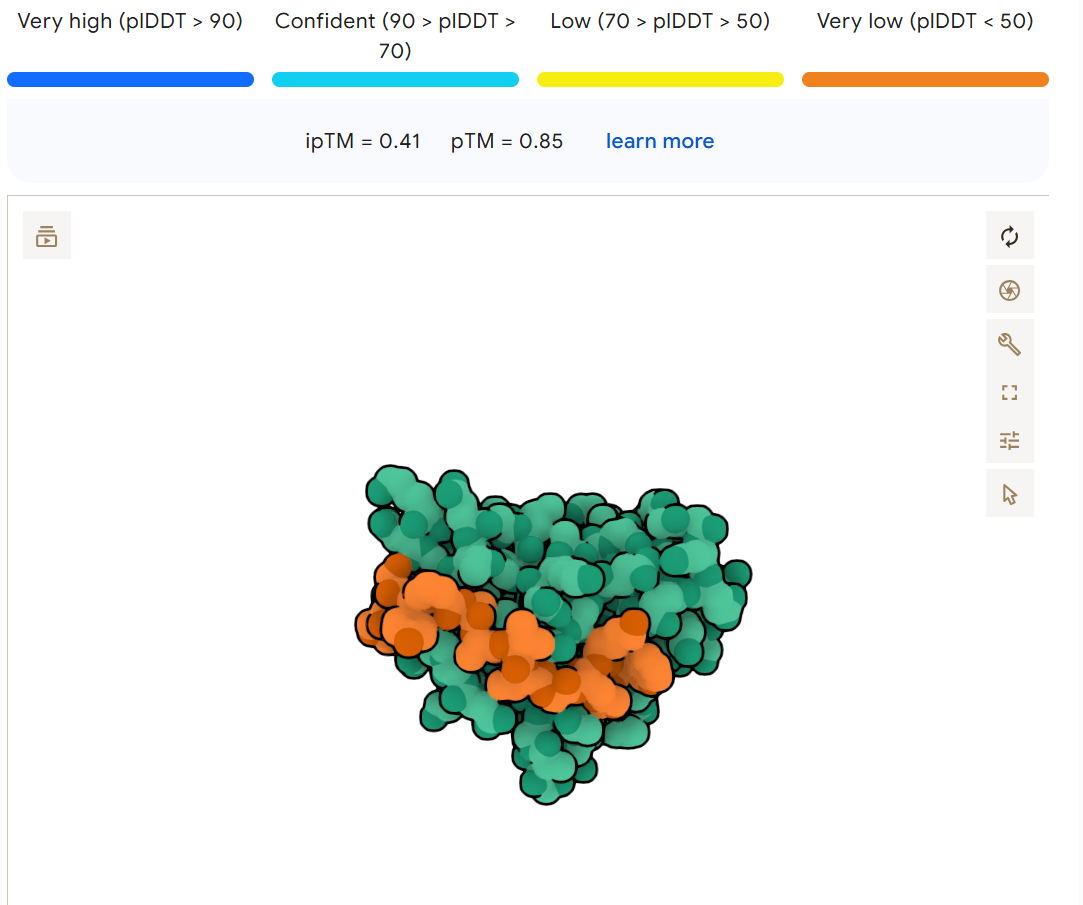
Binder, ipTM, pTM DHSYAVVVAWKX, 0.51, 0.89 DRYGAAAIKHKK, 0.4, 0.87 WSYPAAVVAHKX, 0.41, 0.85 DHVGAVAIEHKX, 0.38, 0.88
Compared to the known result of: FLYRWLPSRRGG, 0.3, 0.82
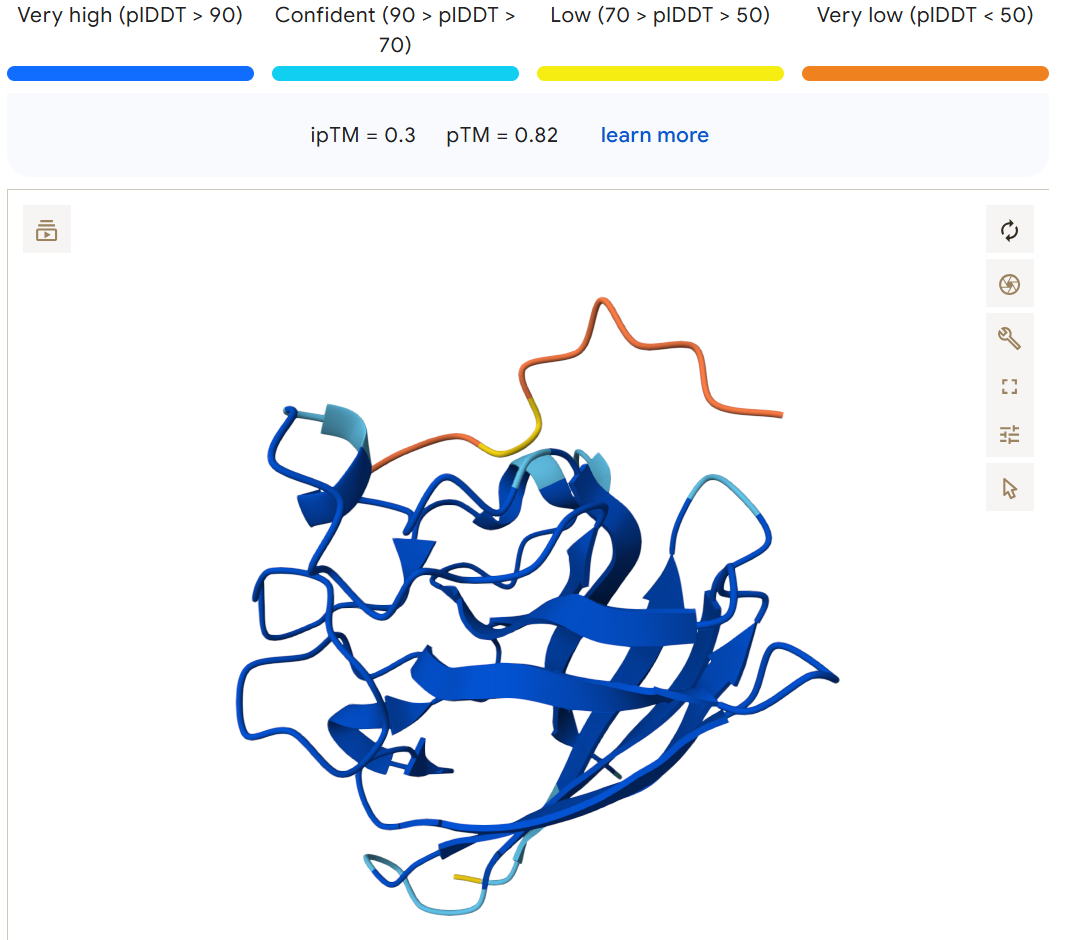
I modeled the complex between A4V mutant Superoxide dismutase 1 and each generated peptide using Alphafold. The PepMLM-generated peptides produced ipTM scores ranging from 0.38 to 0.51, while the known SOD1-binding peptide FLYRWLPSRRGG had an ipTM score of 0.30. The highest scoring peptide, DHSYAVVVAWKX, achieved an ipTM of 0.51 with a pTM of 0.89, suggesting a stronger predicted interaction with the protein compared to the known binder. The other generated peptides (DRYGAAAIKHKK, WSYPAAVVAHKX, and DHVGAVAIEHKX) also showed moderate interaction scores between 0.38–0.41, all exceeding the known peptide’s ipTM. Overall, these results suggest that the PepMLM model was able to generate candidate peptides with predicted binding strengths comparable to or stronger than the previously known SOD1 binder.

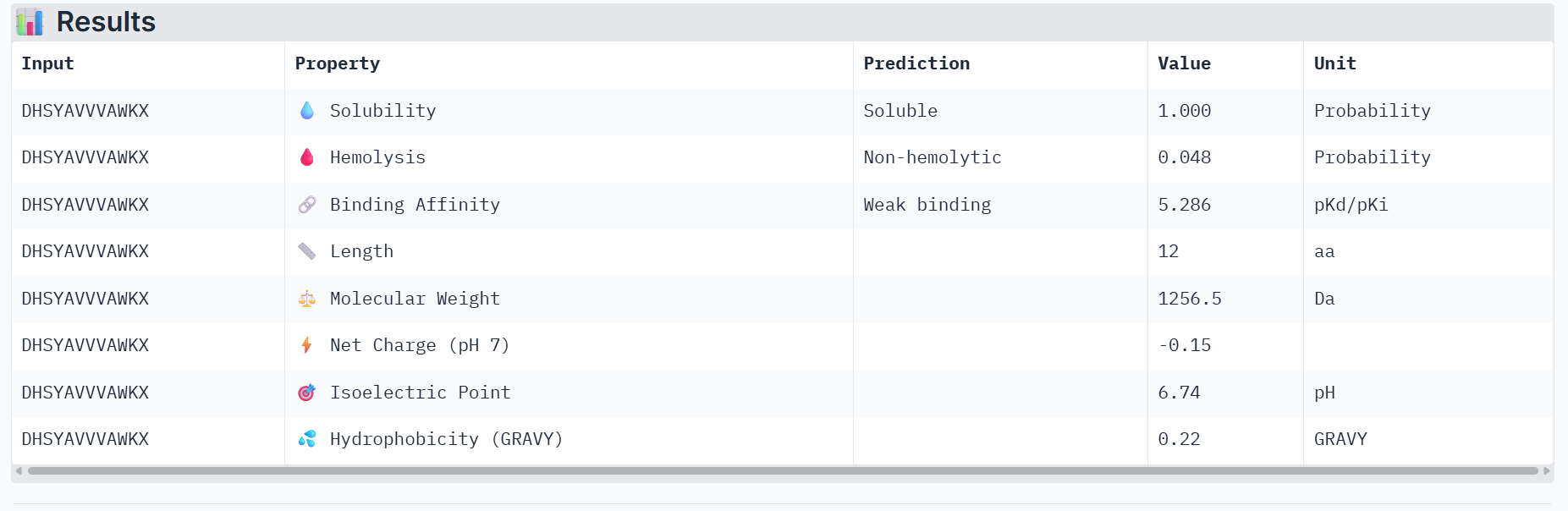
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Part 4: Generate Optimized Peptides with moPPIt
I had issues running the CoLab without a GPU, but here are my predictions. MoPPIt was intended to generate 12-amino-acid peptides that bind to specific residues on the A4V mutant SOD1 while optimizing multiple properties like binding affinity, solubility, and low hemolysis. Unlike PepMLM, which broadly samples possible binders from the target sequence, moPPIt uses guided generation to steer peptides toward a chosen binding site and optimize several objectives simultaneously. As a result, moPPIt peptides would be expected to be more targeted and better optimized for therapeutic properties than the PepMLM peptides.
Before advancing these peptides further, I would first evaluate them computationally by predicting their structures and docking them to the SOD1 binding site to estimate binding strength. I would also check properties like stability, aggregation risk, toxicity, and immunogenicity. The most promising peptides should then be synthesized and tested experimentally with binding assays and cell-based tests to confirm binding, stability, and safety before considering preclinical studies.
Part C: Final Project: L-Protein Mutants
L-Protein Engineering | Option 1: Mutagenesis
To disrupt the interaction between the L-protein and DnaJ, I designed mutations in the soluble region that either change charge, disrupt hydrophobic contacts, or introduce steric changes that could weaken the binding interface. I avoided positions that appear highly conserved in the sequence alignments and focused on residues that are likely surface exposed.
Mutation Set 1: E45K, L48D, Y52A This mutation flips the charge of E45 and introduces a charged residue at L48, which could disrupt electrostatic and hydrophobic interactions at the binding interface.
Mutation Set 2: A60D, V63K, L67D These substitutions introduce charged residues into a region that is likely hydrophobic, which may destabilize the interaction surface with DnaJ.
Mutation Set 3: F72A, Y75A, L78D Replacing aromatic and hydrophobic residues with alanine or charged residues reduces hydrophobic packing and weakens potential binding contacts.
Mutation Set 4: K85E, R88E, L91A This mutation reverses positive charges that may interact with DnaJ and replaces a hydrophobic residue with alanine to reduce interface stability.
Mutation Set 5: V96D, A99K, L102D Introducing charged residues in this region may disrupt local structure or electrostatic interactions that stabilize the L-protein–DnaJ complex.
To evaluate these mutants, I would co-fold each mutant L-protein with DnaJ using AF2-Multimer and compare predicted interface contacts, confidence scores, and binding geometry. Mutants that show reduced interface contacts or lower interaction confidence would be considered more effective at disrupting the interaction.
L-Protein Engineering | Option 3: Random Mutagenesis
Python Function for Random Mutations
AI Prompts
- ‘Help me write python code to create random protein mutations’