Week 2 HW: DNA Read, Write, & Edit
Week 2 Homework
Part 1: Benchling & In-silico Gel Art
| MIT/Harvard students | Required |
| Committed Listeners | Required |
See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis” for details. Overview:
- Make a free account at benchling.com
- Import the Lambda DNA
- Simulate Restriction Enzyme Digestion with the following enzymes:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
- Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks
- You might find Ronan’s website a helpful tool for quickly iterating on designs!
Response
I was able to import all the restriction enzymes into benchling,
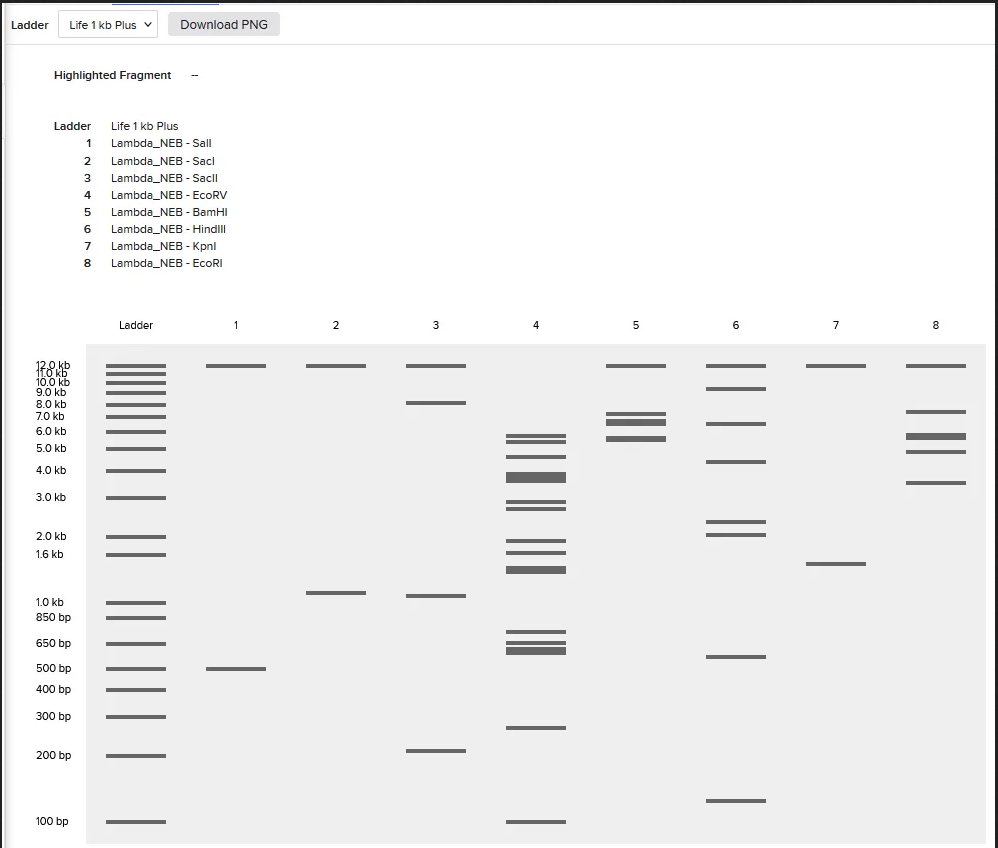
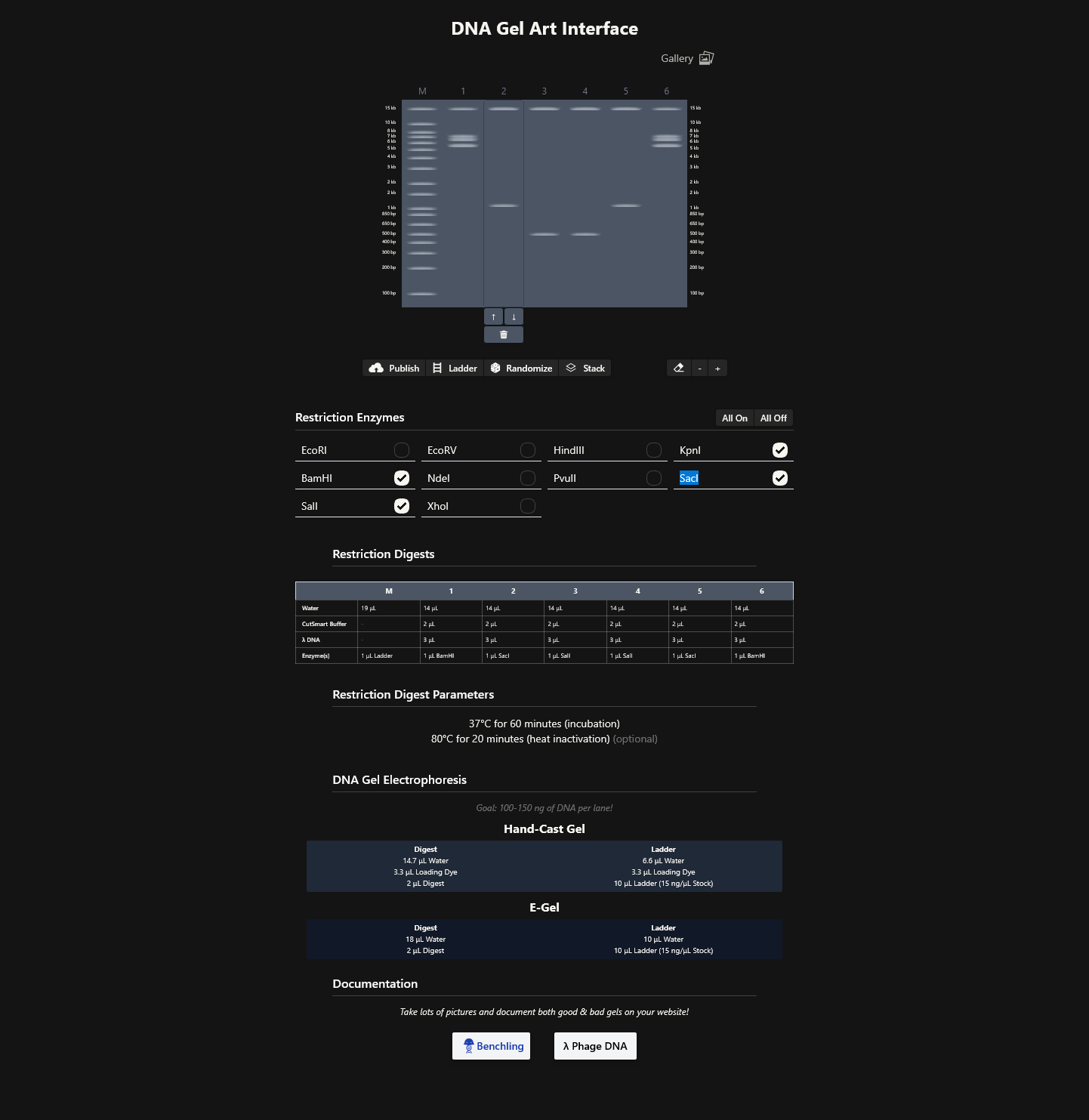

Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
Example from group homework:
>sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
Response
I used UniProt to source Cellulose synthase from Komagataeibacter xylinus as I wanted to investigate growing bacterial cellulose as leather substitute for my final project.
Source: UniProt P19449 — BCSA1_KOMXY
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Example: Get to the original sequence of phage MS2 L-protein from its genome — phage MS2 genome - Nucleotide - NCBI
Lysis protein DNA sequence:
atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
Response
I used the Sequence Manipulation Suite to do the reverse translation :)
Reverse translation of
sp|P19449|BCSA1_KOMXYCellulose synthase catalytic subunit [UDP-forming] OS=Komagataeibacter xylinus — 2262 base sequence of most likely codons:
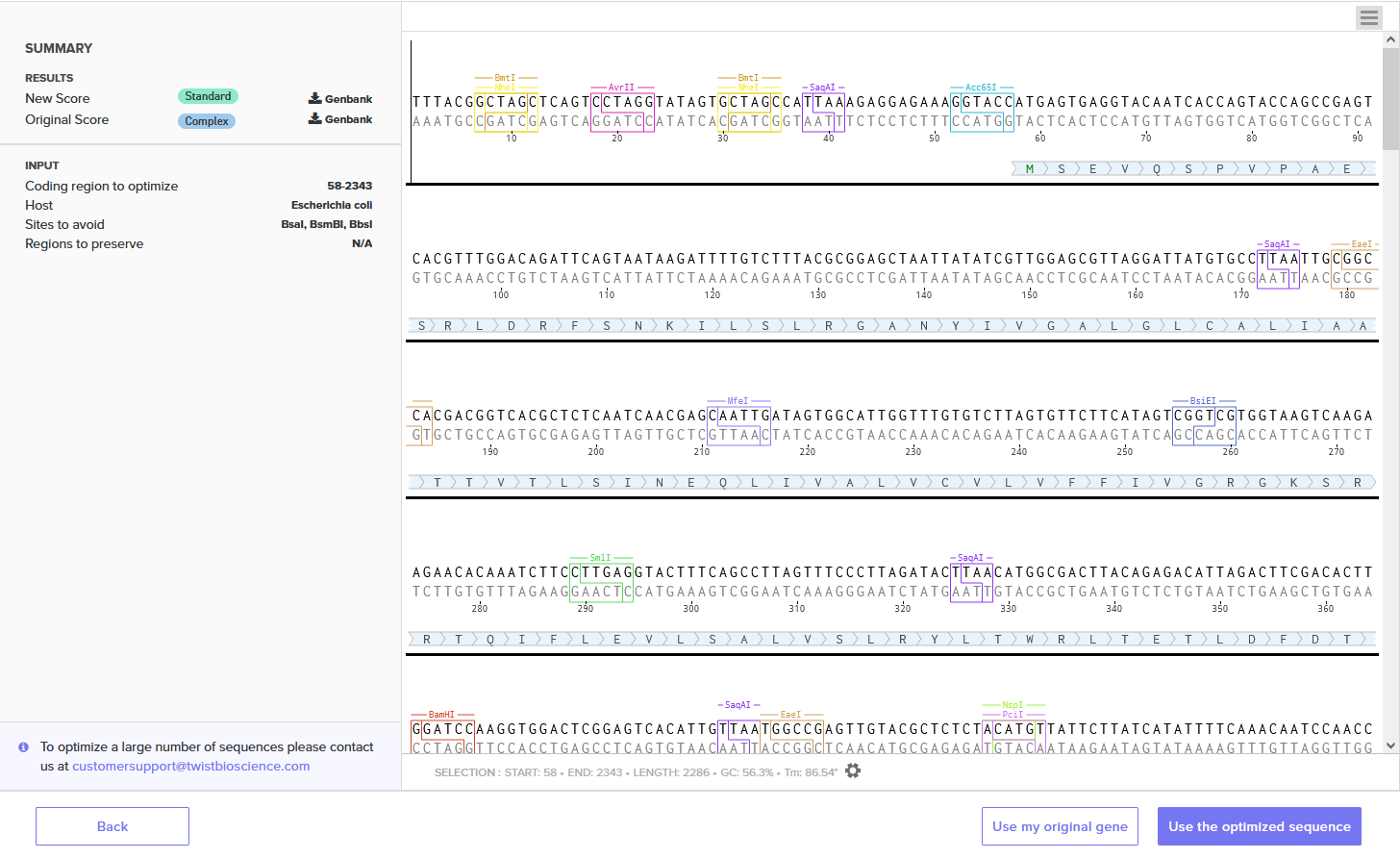
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI
Lysis protein DNA sequence with Codon-Optimization:
ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
Response
Different organisms preferentially use certain codons over others due to varying tRNA availability. By optimizing the BcsA sequence for E. coli, we ensure the ribosomes can read the sequence efficiently and produce high yields of protein. I used Twist Bioscience’s Codon Optimization Tool, optimized for Escherichia coli, avoiding restriction sites BsaI, BsmBI, and BbsI.
Improved DNA[1]: GC=55.06%, CAI=0.93
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Response
To produce cellulose synthase (BcsA) from Komagataeibacter xylinus in a lab setting, we can use a cell-dependent method with E. coli as the host organism. The codon-optimized BcsA gene is inserted into a plasmid, which is then introduced into E. coli via transformation. Once inside the cell, the E. coli transcription machinery reads the DNA and produces a messenger RNA (mRNA) copy of the BcsA gene. The ribosome then translates that mRNA into the BcsA protein by reading each 3-base codon and adding the corresponding amino acid. By cultivating the E. coli at scale, we can produce large quantities of cellulose synthase — and because we codon optimized the sequence for E. coli, this process happens efficiently. This approach also lets us modify the BcsA sequence to potentially engineer cellulose with different material properties for use in bacterial cellulose leather applications.
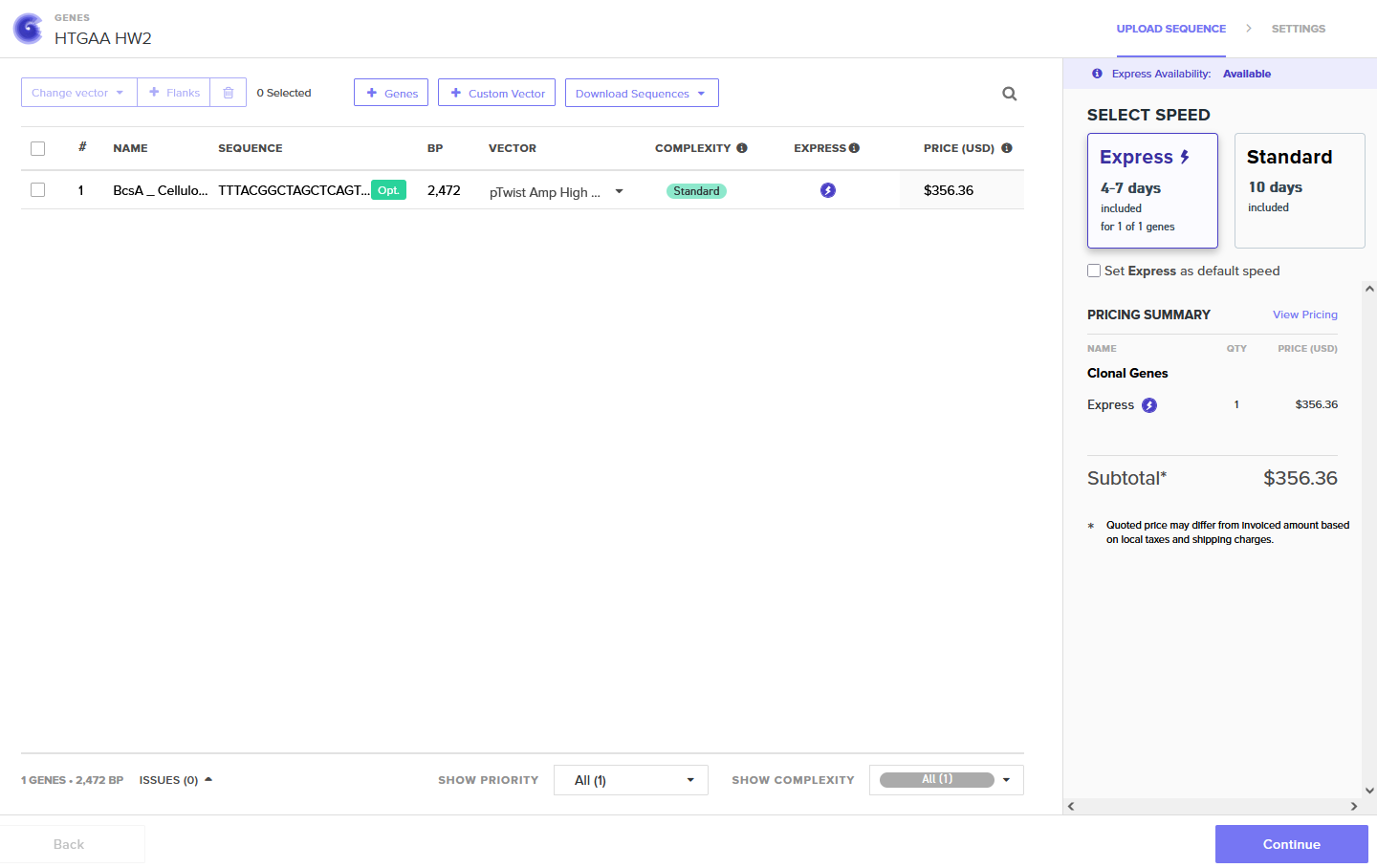
Part 4: Prepare a Twist DNA Synthesis Order
Response
Annotated Benchling insert fragment and Twist order below:
Twist Optimized Cellulose Synthase in Plasmid
Twist Optimized Order
Twist Optimized
Debugging DNA
Download BcsA Cellulose 2nd Try (.gb)
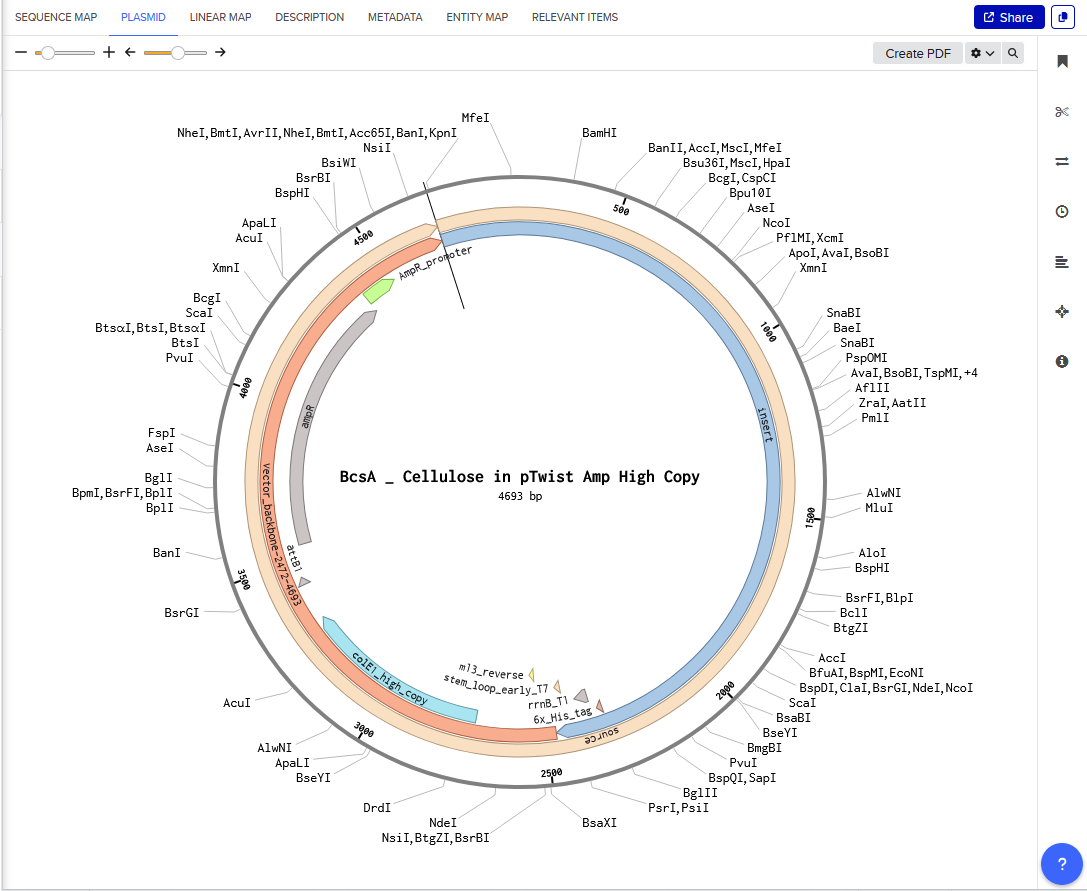
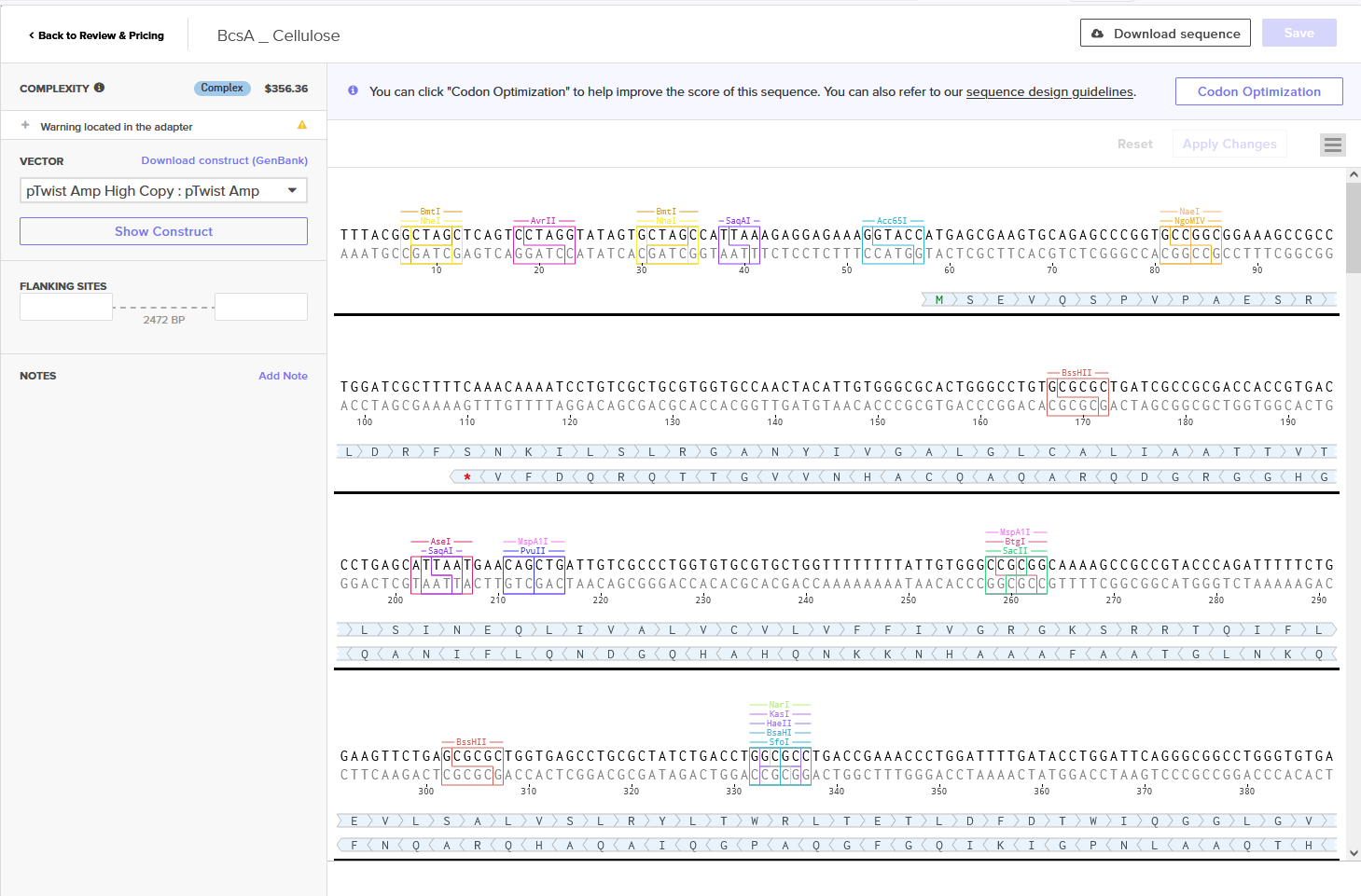
Download BcsA Cellulose Sequence (.pdf)
Part 5: DNA Read/Write/Edit
| MIT/Harvard students | Required |
| Committed Listeners | Required |
5.1 DNA Read
(i) What DNA would you want to sequence and why?
I want to read the sequence off of Komagataeibacter xylinus (the bacteria responsible for cellulose production in kombucha) to pull the cellulose synthase gene for further analysis.
I can also use this to validate my samples from Twist and QA my current batch of E. coli or other model bacteria I’m using.
(ii) What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Oxford Nanopore sequencing to read my DNA.
Is your method first-, second- or third-generation or other? How so? Nanopore sequencing is a third generation technology that lets us read our DNA strands in one shot, without fragmentation or short-read reassembly.
What is your input? How do you prepare your input? List the essential steps.
- Extract plasmid DNA from E. coli colonies
- PCR amplify the insert region
- Attach sequencing adapter molecules to DNA ends via ligation
- Load onto the nanopore flow cell
What are the essential steps of your chosen sequencing technology — how does it decode the bases (base calling)? A protein nanopore sits in a membrane with an electrical current running through it. As a single DNA strand is ratcheted through the pore one base at a time, each base disrupts the current differently — A, T, G, C each produce a characteristic electrical signal. Software decodes this signal into a sequence.
What is the output of your chosen sequencing technology? Nanopore produces long reads up to 100kb+ at high accuracy, which means we can read our entire 2,472 bp plasmid insert in a single pass to verify the cellulose synthase gene transfer worked correctly.
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
I want to write/manufacture the BcsA cellulose synthase expression cassette from Komagataeibacter xylinus, optimized for use in E. coli or yeast. I want to transfer the cellulose-producing capability into a faster but still cheap host organism to iterate on bacterial cellulose leather production.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would perform my synthesis using phosphoramidite chemical synthesis via Twist Bioscience, cloned into the pTwist Amp High Copy vector.
What are the essential steps of your chosen synthesis method?
- Split the DNA sequence into short overlapping oligonucleotides
- Each oligo is synthesized chemically, one base at a time
- Oligos are assembled via PCR-based assembly using overlapping regions
- The assembled sequence is inserted into the plasmid vector
- Colonies are sequence-verified and the correct clone is shipped
What are the limitations of your synthesis method in terms of speed, accuracy, scalability?
- Cost scales up significantly for longer DNA sequences
- Repetitive sequences and extreme GC content can cause synthesis failure — this is why our BcsA sequence was flagged as “Complex” initially
- Error rate is roughly 1 per 500 bp before verification sequencing
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I want to edit the BcsA gene to change how the amino acids control the polymerization of cellulose chains and how they form fibers. By making targeted mutations we could potentially change:
- Fiber diameter and crystallinity (affects texture and tensile strength)
- Production rate (catalytic efficiency)
- Surface chemistry (affects dyeability and water resistance for leather finishing)
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR-Cas9
How does your technology of choice edit DNA?
- Design a guide RNA (gRNA) complementary to the target site in BcsA
- The gRNA directs the Cas9 protein to the exact location in the DNA
- Cas9 makes a double-strand break
- Provide a repair template — a short single-stranded DNA oligo containing the desired mutation with homology arms flanking the cut site
- The cell’s repair machinery integrates the changes automatically
What preparation do you need to do and what are the inputs?
- Cas9 protein or plasmid expressing Cas9
- Guide RNA designed to target the BcsA site (designed in Benchling, checked for off-target sites)
- Repair template containing the desired mutation
- Target cells (E. coli or K. xylinus directly)
What are the limitations of your editing method in terms of efficiency or precision?
- Efficiency is low so we typically only 1-10% of cells receive the edit, requiring selection
- Off-target cuts can introduce unintended mutations elsewhere in the genome
- K. xylinus is significantly harder to work with than E. coli our transformation efficiency is low
- For rapidly iterating BcsA variants, ordering new synthesized sequences from Twist may actually be more practical than CRISPR editing it gives us faster turnaround, no off-target risk, full sequence control