Lab Documentation Pipetting Lab Objective: Practice accurate pipetting techniques and proper laboratory handling.
Procedure:
Identified pipettes (P20, P200, P1000) and their volume ranges. Set desired volumes and aspirated/dispensed liquids correctly. Changed tips between samples to avoid contamination. Held pipettes vertically during aspiration and at a slight angle during dispensing. Challenges and Fixes:
First, I created an account on Benchling. Then, I uploaded the Lambda sequence and introduced the restriction digestive enzymes. By combining them, I obtained the following result: 2. Unfortunately, I did not have access to a laboratory equipped with all the necessary materials to perform the experiment. 3. 3.1 The human oxytocin (OXT) gene is located on chromosome 20 and encodes a precursor protein (prepropeptide) of 125–126 amino acids, which is subsequently processed into the active 9-amino acid hormone (nonapeptide: Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly) and neurophysin I. The cDNA sequence for the human OXT precursor (NM_000915.4) consists of a 378-base pair open reading frame. I used UniProt to identify the oxytocin sequence:
I have completed the required sections in Python and structured the protocol for automation as requested.
Post-Lab Questions Part I – Published Paper Utilizing Automation A notable example is Rauch et al., 2020 – Open-Source Robotic Platform for SARS-CoV-2 Testing Using the Opentrons OT-2. In this study, researchers used the Opentrons OT-2 to automate RT-qPCR testing for COVID-19. Automation enabled RNA extraction, sample transfer, and reaction setup in 96-well plates with high reproducibility and minimal human intervention. This approach demonstrated that low-cost robotic systems can perform clinically relevant diagnostics, increase throughput, and reduce variability, making automation accessible to laboratories with limited resources.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? Meat is approximately 20% protein. • 500g of meat $\times$ 0.20 = 100g of protein. • Average molecular weight of an amino acid $\approx$ 100 Daltons (g/mol). • 100g / 100 g/mol = 1 mole of amino acids. • Using Avogadro’s number, you consume approximately $6.022 \times 10^{23}$ molecules of amino acids.
PART A: Therapeutic Peptide Design for SOD1 (ALS) 1. Objective The goal of this section was to design a synthetic peptide binder capable of stabilizing the mutant SOD1 (A4V) protein, a primary cause of Amyotrophic Lateral Sclerosis (ALS). We aimed to create a “molecular shield” to prevent toxic protein aggregation.
Subsections of Homework
Week 1 HW: Principles and Practices
Lab Documentation
Pipetting Lab
Objective: Practice accurate pipetting techniques and proper laboratory handling.
Procedure:
Identified pipettes (P20, P200, P1000) and their volume ranges.
Set desired volumes and aspirated/dispensed liquids correctly.
Changed tips between samples to avoid contamination.
Held pipettes vertically during aspiration and at a slight angle during dispensing.
Challenges and Fixes:
Initially inconsistent volumes due to air bubbles and hand instability.
Fixed by slowing plunger release and maintaining steady posture.
Reflection:
Accurate pipetting is critical in biological experiments; small mistakes can significantly affect results.
Class Assignment: Ethics, Governance, and Biotechnology
1. Biological Engineering Application
I am interested in developing patient-derived cancer organoids from stem cells to support personalized oncology treatment. These organoids replicate a patient’s tumor properties and can be used to test multiple therapies in vitro before applying them clinically. This approach aims to reduce trial-and-error in cancer treatment, lower side effects, and improve outcomes.
2. Governance and Policy Goals
Primary Goal: Ensure that organoid technologies are used ethically, safely, and equitably.
Sub-goals:
Protect sensitive genetic data.
Ensure informed consent for current and future uses.
Prevent unethical commercialization of patient materials.
Promote equitable access to treatments.
3. Governance Actions
Option 1: Expanded Informed Consent
Purpose: Patients fully understand long-term use of their organoids.
Design: Clear consent forms, IRB oversight, opt-in for secondary research.
Assumptions: Patients can understand complex biomedical information.
Risks: Consent fatigue; complexity may discourage participation.
Option 2: Genetic Data Governance
Purpose: Protect patient genetic data from misuse.
Design: Secure storage, anonymization, restricted access, audits.
Assumptions: Institutions maintain strong security systems.
Risks: Higher administrative cost; smaller labs may struggle.
Option 3: Regulation of Commercial Use
Purpose: Prevent profit without patient consent.
Design: Rules for academic-industry partnerships; transparency in commercialization.
Assumptions: Regulations balance ethics and innovation.
Risks: Slower translation to clinic; reduced private investment.
5. Recommended Governance Strategy
I recommend combining Option 1 (Expanded Informed Consent) and Option 2 (Genetic Data Governance). This ensures patient protection and ethical use of organoids without impeding research. Option 3 can be selectively applied to prevent unethical commercialization. Audience: academic medical centers, hospital ethics committees, and national health regulators.
Ethical Reflection
Organoids contain personal genetic information, raising privacy and ownership concerns. Ethical responsibility extends beyond technical feasibility. Ensuring proper consent, secure data handling, and equitable access is essential to maintain trust in biotechnological innovation.
Week 2 Lecture Preparation
Professor Jacobson
Polymerase error rate: ~1 error per 10⁹–10¹⁰ nucleotides after proofreading.
Human genome size: ~3 × 10⁹ bp.
Biological solution: DNA proofreading and mismatch repair mechanisms.
DNA coding variability: Codon redundancy exists, but factors like codon bias, mRNA stability, and translation efficiency limit practical coding options.
Dr. LeProust
Oligo synthesis method: Solid-phase phosphoramidite.
Difficulty >200 nt: Accumulated errors and incomplete coupling.
Why 2000 bp gene can’t be made directly: Error rate too high; must assemble from shorter oligos.
George Church
Essential amino acids: His, Ile, Leu, Lys, Met, Phe, Thr, Trp, Val (Arg conditionally).
Reflection: Essential amino acids highlight vulnerabilities that influence engineering decisions.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
1
2
• By helping respond
2
1
2
Foster Lab Safety
• By preventing incident
1
1
1
• By helping respond
2
1
2
Protect the environment
• By preventing incidents
1
1
1
• By helping respond
1
1
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
2
• Feasibility?
2
2
2
• Not impede research
2
3
2
• Promote constructive applications
1
1
2
Week 2 HW: DNA Read, Write and Edit
1. First, I created an account on Benchling. Then, I uploaded the Lambda sequence and introduced the restriction digestive enzymes. By combining them, I obtained the following result:
2. Unfortunately, I did not have access to a laboratory equipped with all the necessary materials to perform the experiment.
3.
3.1 The human oxytocin (OXT) gene is located on chromosome 20 and encodes a precursor protein (prepropeptide) of 125–126 amino acids, which is subsequently processed into the active 9-amino acid hormone (nonapeptide: Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly) and neurophysin I. The cDNA sequence for the human OXT precursor (NM_000915.4) consists of a 378-base pair open reading frame.
After determining the nucleotide sequence encoding oxytocin, codon optimization is necessary to ensure efficient expression in the chosen host organism. Although the genetic code is universal, different organisms prefer certain codons over others. This phenomenon is known as codon usage bias. If a gene contains codons that are rarely used in the host organism, translation efficiency may decrease, resulting in low protein yield or incomplete translation.
By optimizing codon usage, the DNA sequence is modified to incorporate codons that are more frequently used in the selected organism, without altering the amino acid sequence of oxytocin (CYIQNCPLG). This process improves translation efficiency, increases protein production, enhances mRNA stability, and reduces the risk of ribosome stalling.
4.1 I created an account.
4.2
5.
5.1 (i) What DNA would I sequence and why?
I would sequence tumor DNA obtained from the patient as well as DNA from patient-derived organoids. The objective is to identify somatic mutations responsible for cancer development and progression.
Key cancer-associated genes that may be analyzed include TP53, KRAS, BRCA1, and EGFR.
Additionally, I would sequence germline DNA extracted from the patient’s blood in order to compare normal and tumor DNA. This comparison allows the distinction between inherited (germline) variants and tumor-specific (somatic) mutations.
The overall purpose is to identify actionable mutations and evaluate personalized therapeutic strategies using patient-derived organoids before administering treatment to the patient.
5.2 (i) What DNA would I want to synthesize and why?
I would synthesize a genetic construct encoding a tumor-specific CAR (Chimeric Antigen Receptor) or a CRISPR-based gene editing system that can be tested in patient-derived organoids.
Option 1: CAR construct (for personalized immunotherapy)
Target gene example: EGFR
Synthetic construct would include:
Promoter (e.g., CMV promoter)
scFv domain targeting tumor antigen
Transmembrane domain
Intracellular signaling domains (CD28/CD3ζ)
PolyA signal
Purpose:
Engineer immune cells or organoids to test personalized immunotherapy
Evaluate response outside the patient’s body
Optimize treatment before clinical use
Option 2: CRISPR therapeutic construct
Components to synthesize:
Cas9 coding sequence
Guide RNA targeting a mutated gene (e.g., TP53 mutation)
Target gene example: TP53
5.3 (i) What DNA would I want to edit and why?
In my project, I would edit tumor-derived stem cells or patient-derived organoids to:
Correct oncogenic mutations
For example, restoring normal function of TP53.
Knock out oncogenes
Such as mutated KRAS or EGFR.
Introduce therapeutic modifications
Insert suicide genes
Enhance immune recognition
Increase sensitivity to chemotherapy
Goal:
Test gene correction strategies in organoids
Evaluate personalized therapies
Develop safer and more effective cancer treatments
5.3 (ii) What technology would I use?
I would use CRISPR-Cas9 genome editing technology.
CRISPR is:
Precise
Relatively simple to design
Efficient in human cells
Widely used in research and clinical trials
How does CRISPR-Cas9 edit DNA?
CRISPR-Cas9 works by creating a double-strand break (DSB) at a specific DNA sequence.
Essential steps:
Guide RNA (gRNA) binds to the target DNA sequence.
Cas9 enzyme creates a double-strand break.
The cell repairs the break via:
Non-homologous end joining (NHEJ) → gene knockout
Homology-directed repair (HDR) → precise gene correction using a repair template
Preparation and input
Design steps:
Identify target mutation
Design specific guide RNA
Design repair template (if correction is needed)
Input materials:
Cas9 protein or Cas9-expressing plasmid
Guide RNA
Repair template DNA (for HDR)
Patient-derived stem cells or organoids
Delivery methods:
Electroporation
Lipid nanoparticles
Viral vectors
Limitations
Off-target effects
Cas9 may cut unintended DNA regions.
Efficiency issues
HDR (precise correction) is less efficient than NHEJ.
Mosaic editing
Not all cells are edited equally.
Delivery challenges
Efficient and safe delivery into primary human cells can be difficult.
Conclusion
By using CRISPR-Cas9 to edit cancer-related genes in patient-derived organoids, we can:
Study mutation function
Test gene correction strategies
Develop personalized cancer therapies in a controlled ex vivo system
Week 3 HW: Lab Automation
I have completed the required sections in Python and structured the protocol for automation as requested.
Post-Lab Questions
Part I – Published Paper Utilizing Automation
A notable example is Rauch et al., 2020 – Open-Source Robotic Platform for SARS-CoV-2 Testing Using the Opentrons OT-2. In this study, researchers used the Opentrons OT-2 to automate RT-qPCR testing for COVID-19. Automation enabled RNA extraction, sample transfer, and reaction setup in 96-well plates with high reproducibility and minimal human intervention. This approach demonstrated that low-cost robotic systems can perform clinically relevant diagnostics, increase throughput, and reduce variability, making automation accessible to laboratories with limited resources.
Part II – Final Project Proposal
The goal of my project is to use automation to generate and screen patient-derived cancer organoids from stem cells for personalized oncology treatment. Organoids better replicate in vivo tumor structure and heterogeneity, but manual handling is prone to variability. Automation ensures reproducible organoid formation, precise drug delivery, and scalable high-throughput testing.
Core Automation Steps
Organoid Seeding
Dispense controlled numbers of stem cells per well
Deliver extracellular matrix at consistent volumes
Maintain uniform droplet size to ensure reproducibility
pipette.pick_up_tip()
for well in plate.wells():
pipette.aspirate(cell_volume, stem_cell_source)
pipette.dispense(cell_volume, well)
pipette.aspirate(matrix_volume, ecm_source)
pipette.dispense(matrix_volume, well)
pipette.drop_tip()
Automated Drug Screening
Generate serial dilutions of chemotherapeutics
Distribute drugs across 96–384 wells
Test multiple treatment combinations and replicates
for drug in drug_panel:
for concentration in dilution_series:
pipette.transfer(drug_volume, drug_stock, target_well)
High-Throughput Design
Automation allows multiple conditions per experiment, consistent organoid formation, and reproducible drug exposure, which are difficult to achieve manually.
Potential Cloud Integration
Platforms like Ginkgo Bioworks Nebula could enable remote design of patient-specific constructs, automated screening, and iterative data analysis for precision therapy selection.
Why Automation Is Critical
Organoids are sensitive to cell density, matrix volume, and handling. Automation provides:
Precise volumetric control
Consistent organoid size and viability
Scalable screening of multiple treatments
High reproducibility suitable for personalized medicine
By automating seeding, drug delivery, and screening, this project aims to bridge experimental cancer modeling and individualized treatment selection.
I have also completed the presentation section with 3 independent ideas for my final project:
Standardized Organoid Platform – Automated organoid formation with reproducible size and structure.
High-Throughput Functional Screening – Parallel testing of multiple drugs and combinations with automated readouts.
Microenvironment-Enhanced Organoids – Including stromal or immune cells to mimic tumor microenvironment and identify new therapeutic targets.
Week 4 HW: Protein Designe Part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat?
Meat is approximately 20% protein.
• 500g of meat $\times$ 0.20 = 100g of protein.
• Average molecular weight of an amino acid $\approx$ 100 Daltons (g/mol).
• 100g / 100 g/mol = 1 mole of amino acids.
• Using Avogadro’s number, you consume approximately $6.022 \times 10^{23}$ molecules of amino acids.
Why do humans eat beef but do not become a cow, or eat fish but do not become fish?
During digestion, enzymes break down foreign proteins into their individual “bricks” (amino acids). Our body then absorbs these amino acids and uses our own DNA “instruction manual” to reassemble them into human-specific proteins. We share the same bricks, but we build a different house.
Why are there only 20 natural amino acids?
This is likely an “evolutionary frozen accident.” These 20 amino acids provide enough chemical diversity (charge, size, and polarity) to fold into almost any functional shape required for life. While more exist in nature, these 20 were sufficient for the ancestor of all life.
Can you make other non-natural amino acids? Design some.
Yes. Scientists use “Expanded Genetic Code” techniques to create hundreds of non-natural amino acids (ncAAs).
• Design Example: p-azidophenylalanine. It contains an azide group that allows for “Click Chemistry,” letting us attach fluorescent dyes or drugs directly to a specific spot on a protein.
Where did amino acids come from before enzymes and before life started?
They were created through abiotic synthesis. Experiments like the Miller-Urey experiment showed that lightning, heat, and UV radiation acting on a primitive atmosphere (methane, ammonia, water) can spontaneously create amino acids. They have also been found on meteorites, suggesting they can form in space.
If you make an $\alpha$-helix using D-amino acids, what handedness would you expect?
Natural L-amino acids form right-handed $\alpha$-helices. Therefore, D-amino acids would form a left-handed helix due to the mirrored geometry of the side chains.
Can you discover additional helices in proteins?
Yes. Beyond the standard $\alpha$-helix (3.6 residues per turn), there are:
• $3_{10}$ helix: Tighter and more elongated.
• $\pi$-helix: Wider and shorter.
Why are most molecular helices right-handed?
Because life is “chiral” and almost exclusively uses L-amino acids. For L-amino acids, the right-handed twist is energetically more stable because it minimizes physical clashing (steric hindrance) between the side chains and the protein backbone.
Why do $\beta$-sheets tend to aggregate? What is the driving force?
$\beta$-sheets have “sticky” edges where hydrogen bonds are exposed. The primary driving force is the Hydrophobic Effect and inter-strand hydrogen bonding. They aggregate to hide their “greasy” hydrophobic parts from water, snapping together like Lego bricks
Part B. Protein Analysis (KRAS)
Protein Selection and Description
• Protein Selected: KRAS (Kirsten Rat Sarcoma Virus).
• Selection Rationale: I selected KRAS because it is a fundamental “molecular switch” in human cells. It controls signaling pathways for cell growth and survival. Mutations in KRAS, particularly at the G12 position, are responsible for approximately 25% of all human cancers, making it a primary target for modern drug design and AI-driven structural analysis.
Amino Acid Sequence and Frequency
• Sequence (from PDB 4DS1): MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSEDVPMVLVGNKCDLPSRTVDTKQAQDLARSYGIPFIETSAKTRQGVDDAFYTLVREIRKHKEK
• Length: This specific structure (catalytic domain) is 169 amino acids long.
• Most Frequent Amino Acid: Valine (V) and Leucine (L). Using the Colab frequency counter, Leucine and Valine appear most often, as they are essential for packing the hydrophobic core of the Rossmann fold.
Homologs and Family
• Number of Homologs: A UniProt BLAST search reveals over 5,000 homologs. KRAS is highly conserved across eukaryotes, from yeast to humans.
• Protein Family: It belongs to the Ras family of small GTPases.
RCSB Structure Details
• RCSB Page: 4DS1
• Solved Date: The structure was solved in 2012.
• Quality/Resolution: The resolution is $1.60\text{ \AA}$. This is an excellent quality structure (well below the $2.70\text{ \AA}$ limit), providing near-atomic detail of the binding pocket.
• Other Molecules: In addition to the protein, the structure contains GDP (Guanosine-5’-Diphosphate), a Magnesium ion (MG) which is crucial for catalysis, and Water (HOH) molecules.
Structural Classification
• Structure Classification Family: KRAS is classified as having a Rossmann fold architecture (Alpha/Beta). It consists of a central 6-stranded $\beta$-sheet surrounded by 5 $\alpha$-helices.
3D Molecule Visualization (PyMol)
A. Visualization Styles
• Cartoon: show cartoon; hide everything else
• Ribbon: show ribbon
• Ball and Stick: show sticks; set stick_radius, 0.2
B. Secondary Structure
• Observation: KRAS features a balanced mix of helices and sheets. It has 5 main $\alpha$-helices and 6 $\beta$-strands that form the central “floor” of the protein.
• PyMol Command: color red, ss h; color yellow, ss s; color green, ss l+’'
C. Residue Type (Hydrophobic vs. Hydrophilic)
• Observation: When colored by residue type, you can see a clear hydrophobic core (red) where amino acids like Valine and Leucine are tucked away from water. The hydrophilic residues (blue) are spread across the surface to interact with the cellular environment.
• PyMol Command: color red, hydrophobic; color blue, hydrophilic
D. Surface and Binding Pockets
• Surface: show surface
• Binding Pockets: KRAS has a very prominent “hole” or binding pocket where the GDP molecule and the Magnesium ion sit. This pocket is formed by the P-loop and the two “Switch” regions (Switch I and Switch II). These switches change shape when KRAS is active, allowing it to interact with other proteins.
Part C. Using ML-Based Protein Design Tools
Selected Protein: KRAS (Kirsten Rat Sarcoma Virus)
PDB ID: 4DS1
In this section, I explored the capabilities of modern AI models in analyzing and designing the KRAS protein. The computational experiments were conducted in a Google Colab environment utilizing a T4 GPU for efficient model inference.
C1. Protein Language Modeling (ESM2)
Deep Mutational Scans (DMS)
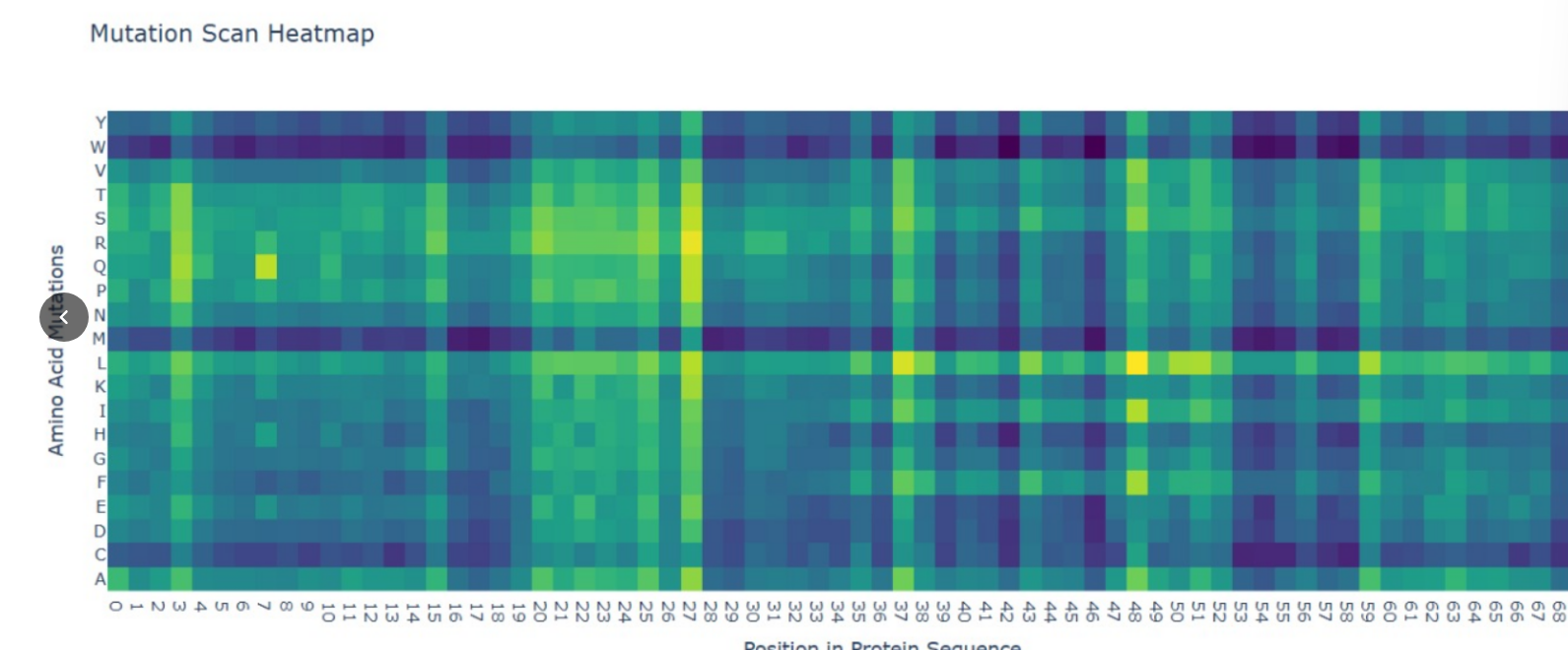
• Methodology: I used the ESM2 language model to generate an unsupervised deep mutational scan of the KRAS catalytic domain.
• Key Observation: The heatmap reveals a significant mutational “cold spot” at Glycine 12 (G12), where almost all substitutions are predicted to be highly unfavorable (dark blue/purple).
• Biological Significance: This aligns with clinical reality; G12 is a critical residue in the P-loop, and mutations here lock KRAS in an active state, driving tumor growth. The AI correctly predicted these structural constraints without being trained on cancer data.
Latent Space Analysis
• Visualizing the “Grammar”: Using t-SNE, I projected protein sequence embeddings into a 3D space to see how the model organizes biological information.
• Clustering: My KRAS sequence was grouped within a cluster of other small GTPases, demonstrating that the model understands functional similarity based purely on sequence patterns.
C2. Protein Folding (ESMFold)
Folding the Oncogene
• Result: I used ESMFold to predict the 3D structure of the KRAS sequence.
• Accuracy: The predicted ribbon model shows a high-confidence Rossmann fold architecture, consisting of a central $\beta$-sheet and surrounding $\alpha$-helices, matching the experimental PDB structure.
• Resilience Testing: I tested the fold’s resilience by introducing mutations. While the surface was tolerant, mutations in the hydrophobic core significantly reduced folding confidence, illustrating the delicate balance required to maintain the KRAS structural scaffold.
C3. Protein Generation (ProteinMPNN)
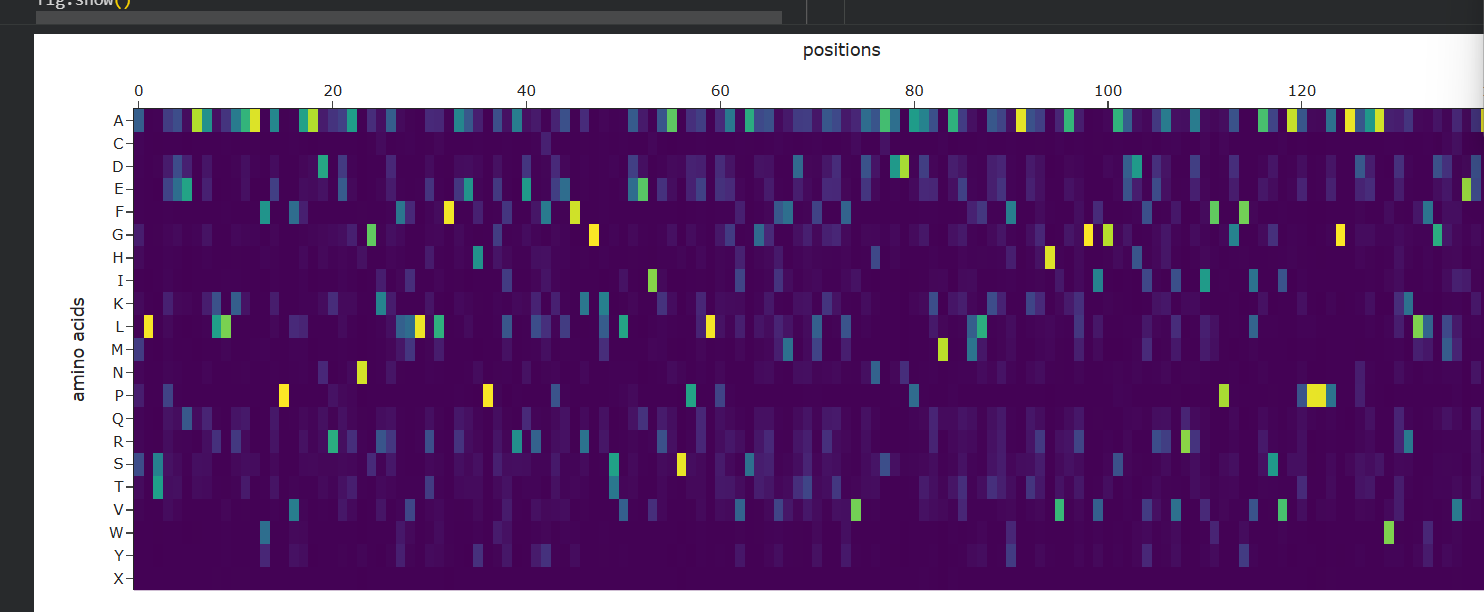
Inverse Folding and Sequence Redesign
• Approach: Using the 3D backbone of KRAS as a fixed template, I used ProteinMPNN to design new sequences that could fold into the same shape.
• Probability Analysis: The resulting matrix shows bright yellow spots for residues that the AI considers essential for the fold’s stability. Many of these correspond to the internal $\beta$-strands that form the structural floor of the protein.
• Validation: Re-folding these AI-generated sequences with ESMFold confirmed that they maintain the characteristic KRAS topology, proving the effectiveness of the inverse-folding pipeline for designing stable variants.
Part D. Group Brainstorm on Bacteriophage Engineering
Project Title: Optimizing Phage Lysis Protein Stability using a KRAS-inspired AI Pipeline.
The Sub-problem: Thermal Instability of Lysis Proteins
Bacteriophages are promising alternatives to antibiotics, but many therapeutic phages are sensitive to environmental stress, such as heat or pH changes, which causes their proteins to denature. We chose to focus on the Lysis Protein, which is responsible for rupturing the bacterial cell wall during the phage life cycle.
Proposed Computational Approach
We propose applying the exact AI-driven workflow used for KRAS in Part C to design a “super-stable” version of the lysis protein:
• ESM-2 for Mutation Scanning: Just as we identified the critical G12 residue in KRAS, we will use ESM-2 to generate a Deep Mutational Scan (DMS) of the lysis protein to find residues that can be mutated to increase thermodynamic stability without losing function.
• ProteinMPNN for Sequence Redesign: Following the KRAS “inverse-folding” logic, we will use ProteinMPNN to redesign the protein’s hydrophobic core. The goal is to maximize the probability of a stable fold while maintaining the specific 3D geometry needed to attack the bacterial wall.
• ESMFold for Structural Validation: Every new AI-generated sequence will be folded in silico. We will compare the predicted structures to the wild-type to ensure the active site remains intact.
Why These Tools?
• Efficiency: Traditional laboratory “trial and error” for protein stabilization can take years. AI tools like ESMFold can provide structural insights in seconds.
• Evolutionary Logic: Language models like ESM-2 capture the “grammar” of protein sequences, ensuring that our designed mutations are biologically plausible.
Potential Pitfalls
• Activity-Stability Trade-off: Increasing the stability (rigidity) of a protein can sometimes reduce its enzymatic activity. A lysis protein that is too stable might not be flexible enough to function properly.
• AI Hallucinations: AI models can sometimes predict sequences that look good on screen but fail to express or fold correctly in a real bacterial cell.
Schematic of the Pipeline
Input: WT Lysis Sequence $\rightarrow$ ESM-2 (Stability Map) $\rightarrow$ ProteinMPNN (Sequence Redesign) $\rightarrow$ ESMFold (3D Validation) $\rightarrow$ Output: Optimized Candidate for Synthesis.
Week 5 HW: Protein Design Part II
PART A: Therapeutic Peptide Design for SOD1 (ALS)
1. Objective
The goal of this section was to design a synthetic peptide binder capable of stabilizing the mutant SOD1 (A4V) protein, a primary cause of Amyotrophic Lateral Sclerosis (ALS). We aimed to create a “molecular shield” to prevent toxic protein aggregation.
2. Methodology & Results
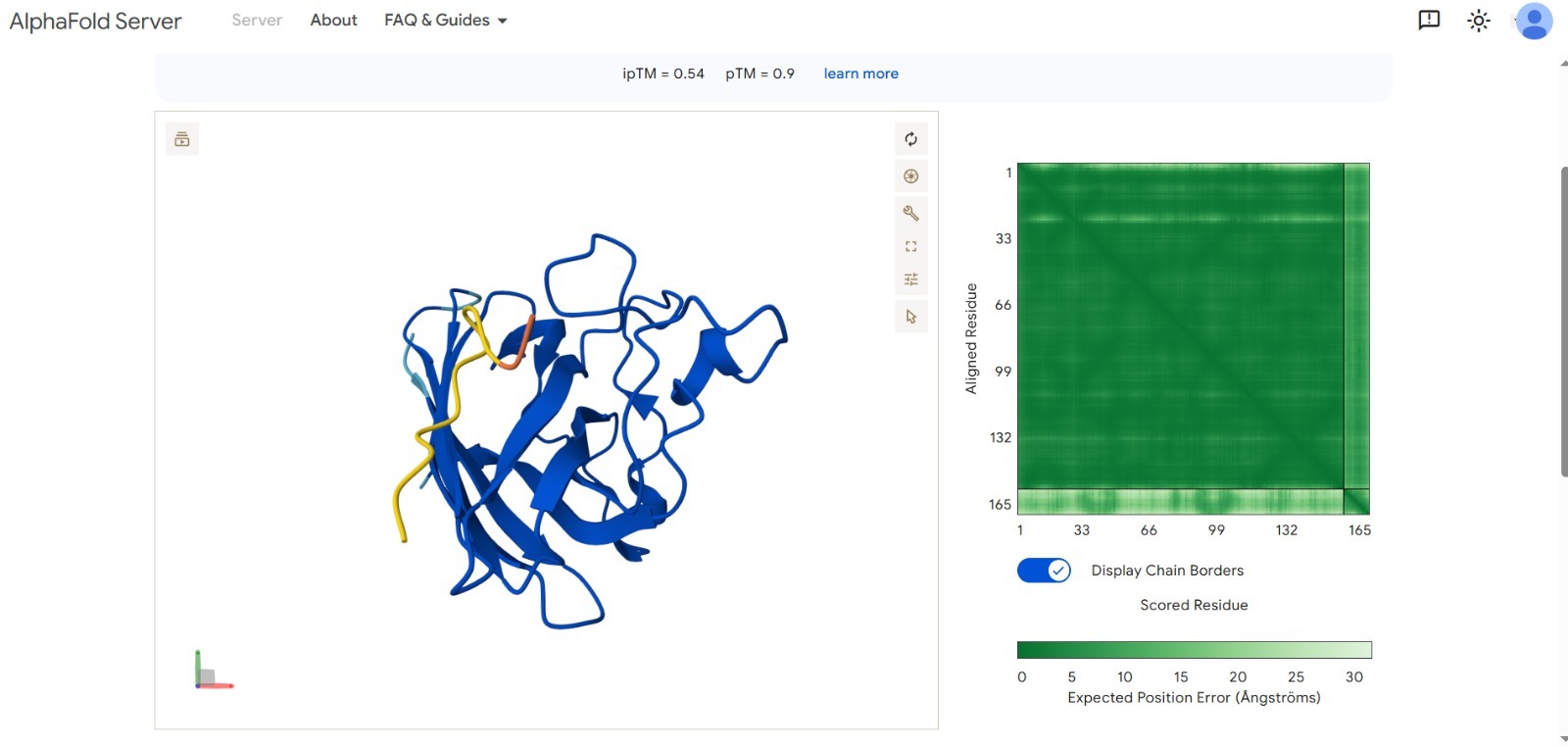
We used AI-driven models to generate and validate peptide candidates. Our best result achieved a significant binding confidence score.
Best Score:ipTM 0.54 (Optimized via targeted design).
Structural Validation: The AlphaFold visualization shows the peptide (in yellow/orange) effectively docking onto the SOD1 surface.
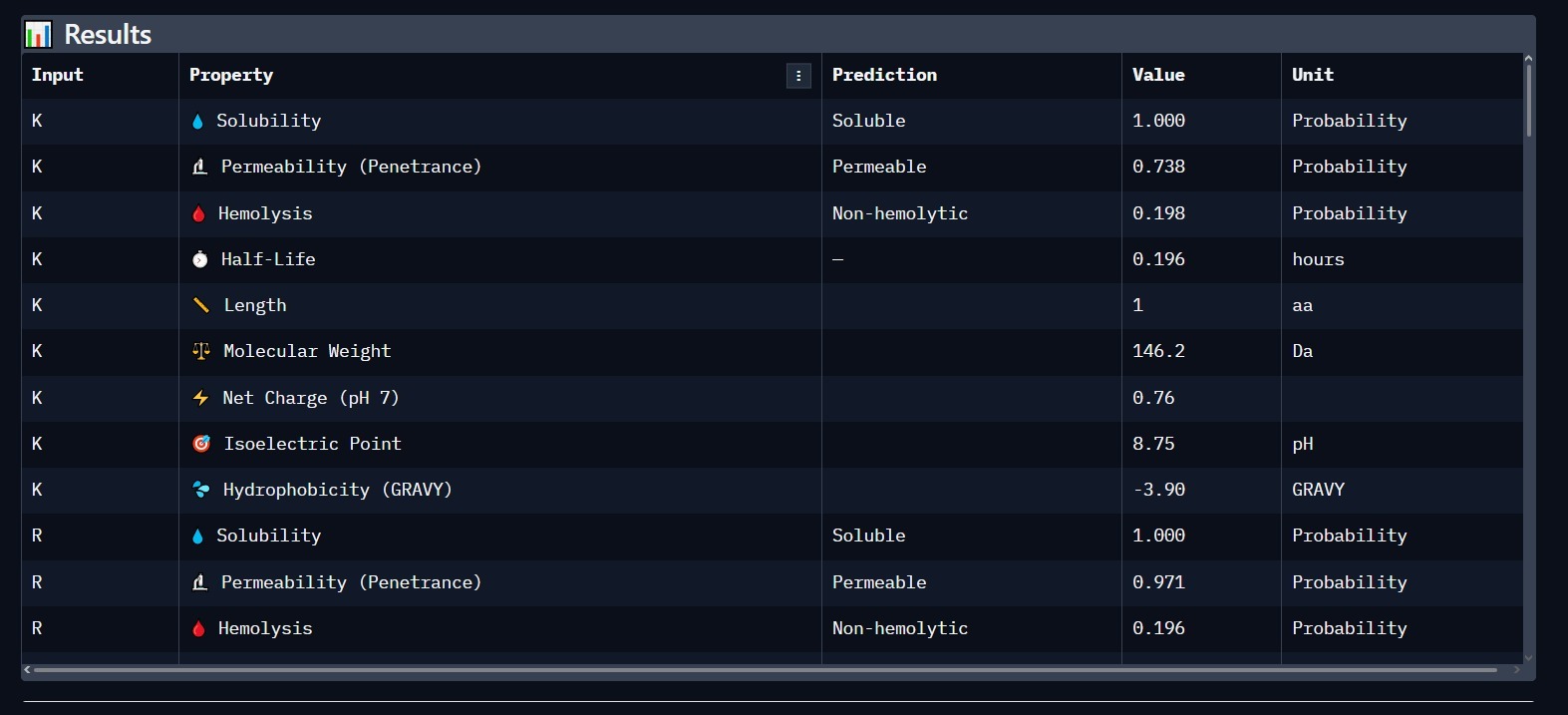
3. Physicochemical & Safety Profile
Using PeptiVerse, we analyzed the lead candidate for clinical viability. The results confirm that the peptide is highly soluble and safe for biological systems.
Solubility: 1.000 (Highly Soluble).
Hemolysis: Non-hemolytic (Safe for blood contact).
PART C: Phage Lysis Protein Engineering
1. The Challenge: Overcoming E. coli Resistance
Bacteriophages use the L-Protein to lyse (kill) bacteria. E. coli develops resistance by mutating the DnaJ chaperone, which prevents the L-protein from folding correctly. Our objective was to engineer L-protein mutants that fold independently of DnaJ.
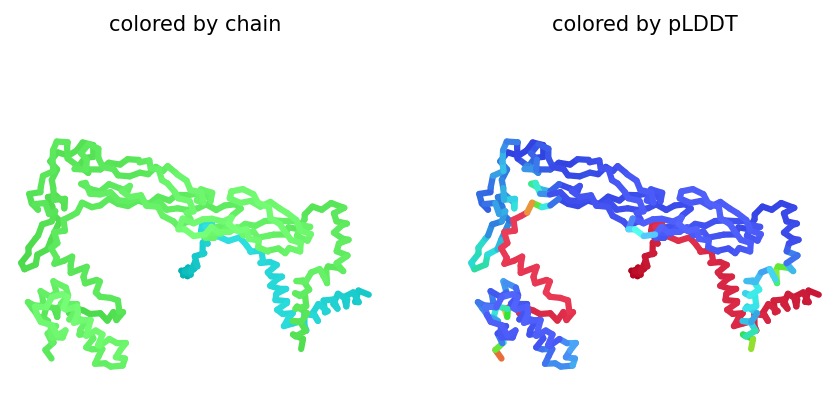
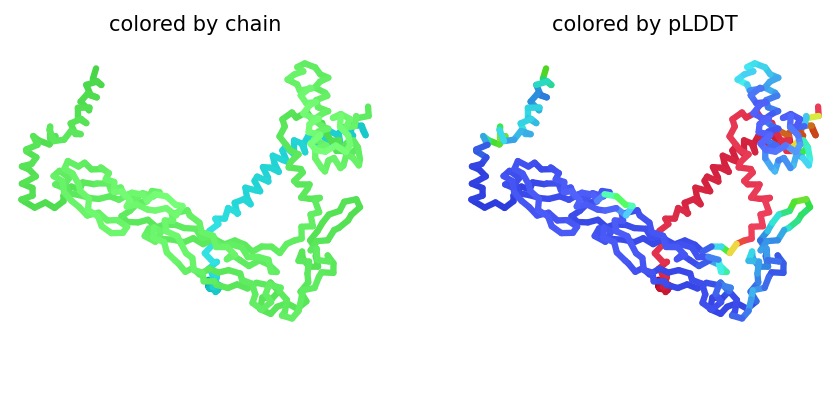
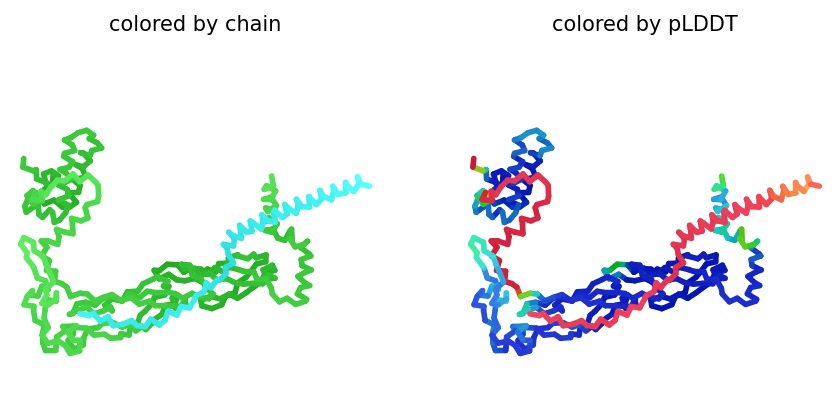
2. Structural Analysis (Wild-Type)
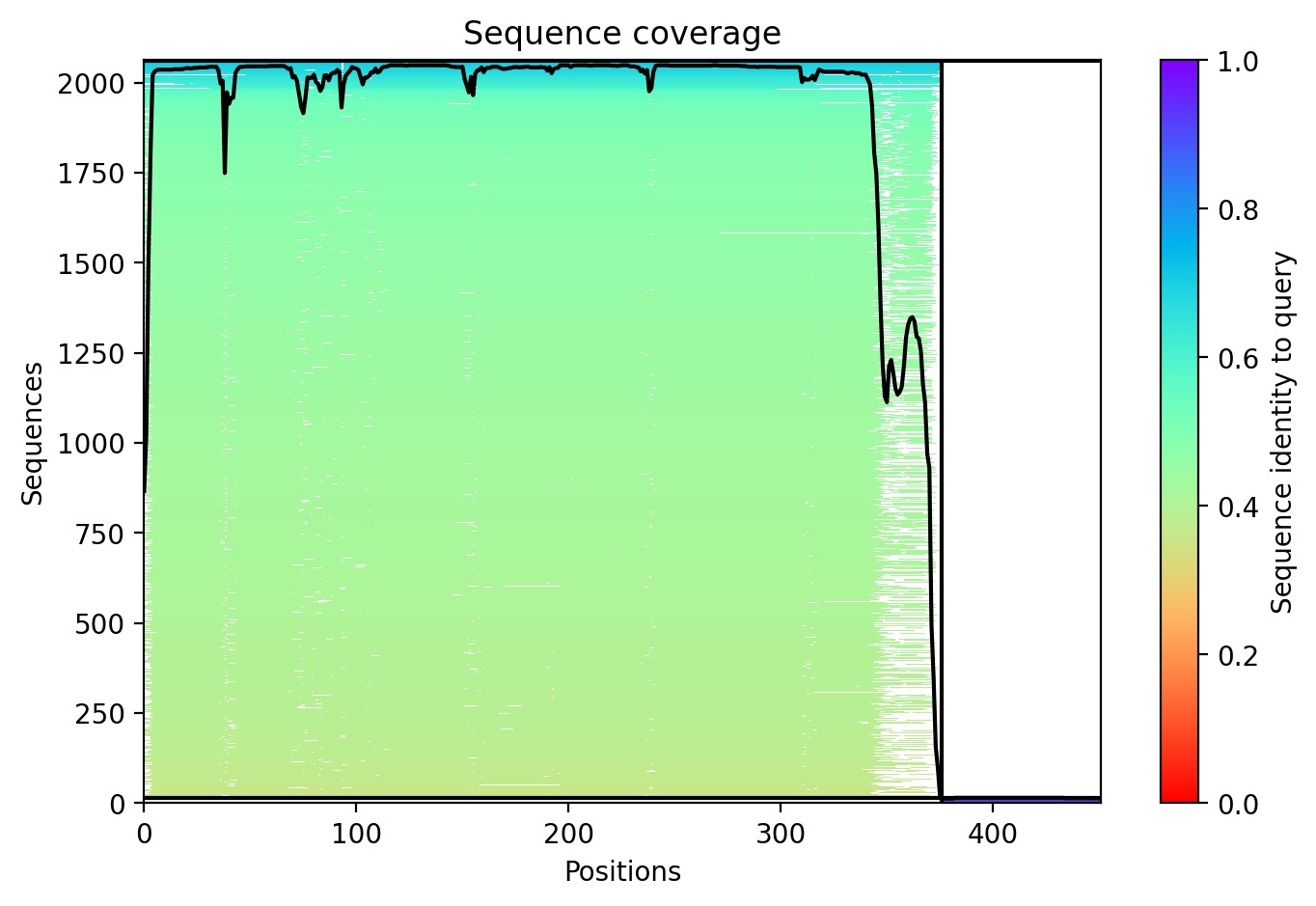
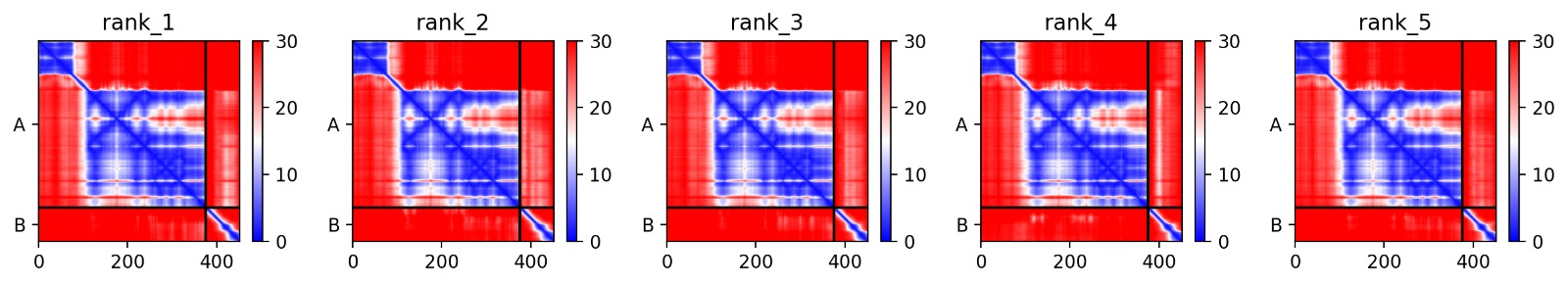
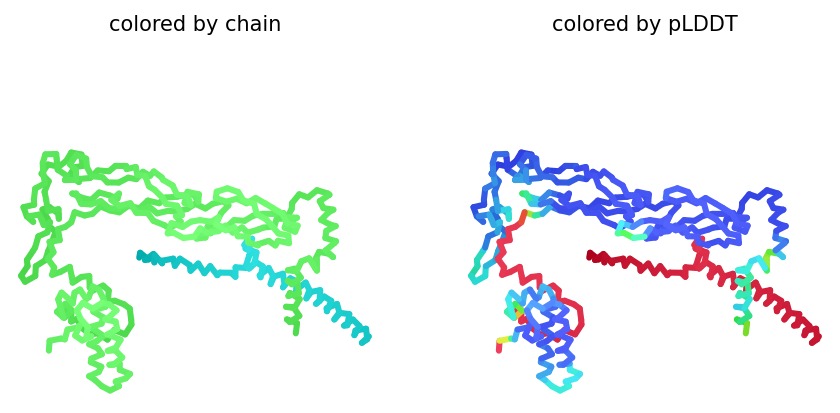
Using AlphaFold2 Multimer, we simulated the interaction between DnaJ and the L-Protein to identify structural vulnerabilities.
Sequence Coverage: Analysis shows high coverage for DnaJ but very low evolutionary conservation for the L-protein region (positions 380+), indicating it is a unique viral protein.
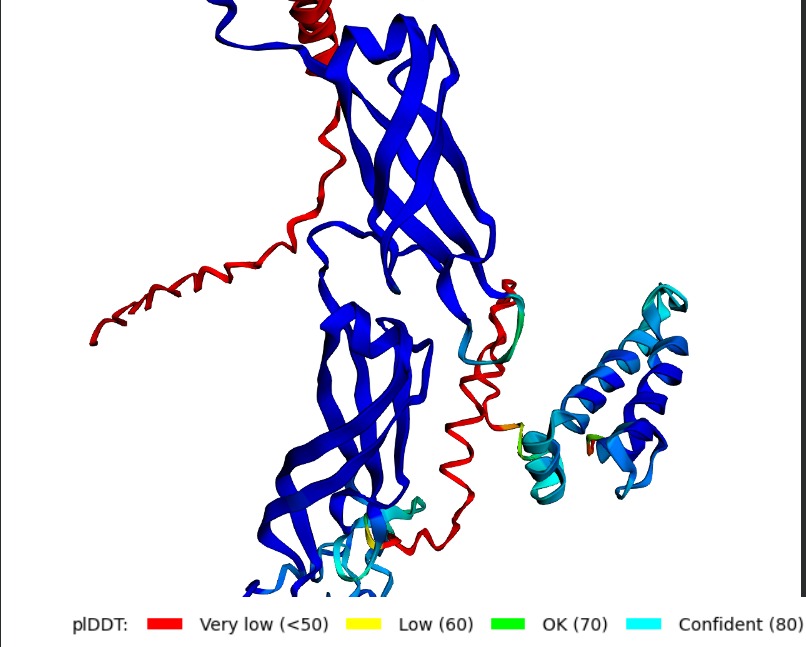
Structural Stability (pLDDT): The L-protein (Chain B) exhibits very low pLDDT scores (red color), confirming it is structurally unstable without the DnaJ chaperone.
Interaction Matrix (PAE): The PAE plots show the specific residues where the L-protein contacts DnaJ. This data was used to guide our mutation strategy.
3. Proposed Engineering Strategy (5 Lead Mutants)
Based on the structural data, I have designed 5 mutations to enhance autonomous folding and lysis efficiency:
#
Mutation
Region
Engineering Goal
1
L25P
Soluble
Increase structural autonomy (DnaJ independence).
2
R14G
Soluble
Stabilize the domain responsible for folding.
3
F52L
Transmembrane
Enhance membrane insertion for faster killing.
4
W45A
Transmembrane
Optimize pore stability in the bacterial wall.
5
S31A
Soluble
Stabilize the N-terminal soluble domain.
4. Defining Success
A successful mutant is defined by its ability to maintain a high Plaque Forming Unit (PFU) count on resistant bacterial strains. Computationally, success is marked by an improved pLDDT score, indicating the protein has gained structural independence.
Final Synthesis
This project demonstrates a complete workflow in protein engineering: from designing “molecular shields” for neurodegenerative diseases (SOD1) to engineering viral proteins to bypass antibiotic resistance. Computational tools like AlphaFold and moPPIt allow us to solve critical biological challenges with high precision.