First, describe a biological engineering application or tool you want to develop and why Bioengineered watercress salad plants with nanoparticles enhancing conductivity and emitting light (phluorescence or phosphorecence). Along with Venus flytrap plant which naturally performs electrophysiological reactions closer to those in animals these research objects will be studied at Open BioArt Lab course on bioart practices related with plant electrophysiology. The course will be based on collaboration of Art&Science Center and Hybrid nanophotonics lab at ITMO University (Russia).
Part 1: Benchling & In-silico Gel Art Simulation of Lambda genome Restriction Enzyme Digestion Using the same enzymes I tried to perform Gel Art. Perhaps, columns would have been closer in real life, but this preview reminds me sunset (if look on spare place).
Assignment: Python Script for Opentrons Artwork Being a CL at Designer Cells Node, I had an opportunity for my image being printed!
Here’s my code which was written with assistance of multiple AI agents, Gemini was not sufficient to correct all my mistakes.
I used this Art Nouveau-ish ornament with Ginkgo leaves as a reference, manually assigning printing dots via Ronan’s website.
Conceptual Questions How many amino acid molecules are in 500 g of meat? Average amino acid ≈ 100 Da = 100 g/mol. 500 g meat (assuming mostly protein for estimation) ≈ 500 g protein ≈ 500 g / (100 g/mol) = 5 mol amino acids. Number of molecules = 5 × 6.02×10²³ ≈ 3.0 × 10²⁴ amino acid molecules (approximate).
SOD1 Binder Peptide Design (From Pranam) Generate Binders with PepMLM Human SOD1 sequence:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutant SOD1 sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I inserted mutant sequence and set 12 as peptide length in PepMLM Google Colab code. To compare FLYRWLPSRRGG binding peptide pseudo perplexity, I added another code cell into Google Colab:
DNA Assembly Components in Phusion High-Fidelity PCR Master Mix and their purpose Phusion High-Fidelity PCR Master Mix typically contains a high-fidelity DNA polymerase (such as Phusion polymerase), dNTPs, Mg²⁺ (often as MgCl₂), and a reaction buffer. The high-fidelity polymerase has proofreading (3’→5’ exonuclease) activity, which reduces DNA replication errors.
Part 1: Intracellular Artificial Neural Networks (IANNs) Advantages of IANNs over traditional Boolean genetic circuits Intracellular Artificial Neural Networks (IANNs) differ from traditional genetic circuits because they process information in a graded, weighted, and adaptive manner rather than using only binary ON/OFF logic.
Part A: General and Lecturer-Specific Questions Advantages of cell-free protein synthesis (CFPS) over in vivo expression Greater experimental control Open reaction environment Faster design-build-test cycles Toxic protein production Better access to non-natural chemistry Easier monitoring and automation Cases where cell-free expression is more beneficial than cell production Case 1: Toxic proteins Example:
Final Project Aspects of the project that will be measured Several aspects of the nanoparticle-mediated plant transfection system will be measured to evaluate delivery efficiency, functional gene expression, and electrophysiological response.
The primary measurable parameters include:
efficiency of nanoparticle-mediated DNA delivery into plant tissues expression of fluorescent reporter proteins functional activity of Channelrhodopsin-2 electrophysiological responses after light stimulation physicochemical properties of peptide-based nanoparticles (size and charge) Additional measurements may include comparison between magnetic nanoparticles and peptide-based nanoparticles in terms of transfection efficiency and signal intensity.
Subsections of Homework
Week 1 HW: Principles and Practices
First, describe a biological engineering application or tool you want to develop and why
Bioengineered watercress salad plants with nanoparticles enhancing conductivity and emitting light (phluorescence or phosphorecence).
Along with Venus flytrap plant which naturally performs electrophysiological reactions closer to those in animals these research objects will be studied at Open BioArt Lab course on bioart practices related with plant electrophysiology. The course will be based on collaboration of Art&Science Center and Hybrid nanophotonics lab at ITMO University (Russia).
Governance/policy goals
Safety and security: the course will be oriented on non-professionals in Biology and take place in the public university with some access limitation. So, prior to final student’s list announcing, there should be Lab safety training (written test? video course? in-person at lab?). I also will suggest some introductory courses on some biological concepts to potential participants in case it will improve their experience.
Accessibility and impact for community: the main goal of the course is to open-up the curtain of research world and engage people to participate in creative, even artistic, practices, which could contribute to modern scientific and philisophical discourse either. Another aim is in promoting more ethic and compassionate attitude towards plants in line with “plant turn” tendency. Hence, from open call announcement till the classes themselves this philosophy should be clearly stated.
Interaction with other nodes of bioart, bio-hack, open-bio etc. communities for mutual enrichment our discourse and practices
Ethics and non-harmfulness: ensure that nanoparticles injections won’t harm plants and cause unnecessary molecular and cellular responses. Propper ways are either via stomata or root system.
Interaction with the faculty administration for coordination and presentation of key course goals and ideas which correspond to the University’s goals. It will increase chances for financial support and dealing with all the concerns for their approval.
Purpose: education + art + bioengineering. Accuiring new skills allong with proposing new ideas and their implementation
Design: collaboration of biologists, nanochimists, and bioartists as teachers with approval of University administration and support of bio-enthusiasts’ community. Participation of St. petersburg bio- and bioart-interested community as listeners and contributors. Potential funding sources: University, federal grants, and industrial partnership (non-profit organisations functioning is down-regulated in this jurisdiction)
Assumptions: acc. to my experience, heterogenity of potential listeners could either improve experience of all the participants or over-complicate the working process
Failure: complicated interaction with University, failure in experiments, troubles with finding and decrease in quality of research and the course.
Success: impact to University public appearance, some course projects can be developed as student thesis or art projects, new collaborations, impact to more ethic and emphatic attitude towards plants
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
• By helping respond
Foster Lab Safety
• By preventing incident
• By helping respond
Protect the environment
• By preventing incidents
• By helping respond
Other considerations
• Minimizing costs and burdens to stakeholders
• Feasibility?
• Not impede research
• Promote constructive applications
Professor Jacobson
What is the error rate of polymerase? 1:106
How does this compare to the length of the human genome? Human genome is 3-3.2*109 bp, hence, 3000 bp of human genome could be wrong
How does biology deal with that discrepancy? There are DNA repare systems: MutS, MutH, and MutL among prokaryotes, MSH and MLH in eukaryotes
How many different ways are there to code (DNA nucleotide code) for an average human protein? Average human protein consists of 300-400 AA. There are 20 types of proteinogenic AA which are coded by 61 codons in total. Considering code degeneracy, there could be up to 6 synonymous substitutions per some AA. Considering this, there could be up to 10200 theoretical sequences.
In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? There are conservative domains providing mRNA and/or folding stability, some functional patterns, zones marking exons/introns, start/termination of translation etc. So some sequences won’t give chemically stable, functioning or translation apropriate proteins.
Dr. LeProust
What’s the most commonly used method for oligo synthesis currently? Next Generation (Chip Based) Oligo Nucleotide Synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis? Yield decrease with further synthesis steps, lower fidelity + error accumulation, hairpin / dimers / cloggs formation
Why can’t you make a 2000bp gene via direct oligo synthesis? Direct oligo synthesis is step-by-step base addition to the chain. With this technology, the yield of the full-length product decreases exponentially with each added base. Even if synthesize exact 2000 bp oligo, it would be hard to purify from, for instance, 1990 bp oligo by gel electrophoresis.
George Church
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”? There is 9 essential amino acids: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine. There is pyrrolysine also, wich occurs only in some organisms, so it could be considered as 10th essential one. Lysine Contigency in “Jurassic Park” movie was presented as “engineered” lack of dinosaurs’ ability to produce lysine amed to tie them to the park therritory where they could get needed supplements. As we can see, almost all vertebrates share this disability, so the movie creators should’ve used alanine, for instance
Week 2 HW: DNA read, write, and edit
Part 1: Benchling & In-silico Gel Art
Simulation of Lambda genome Restriction Enzyme Digestion
Using the same enzymes I tried to perform Gel Art. Perhaps, columns would have been closer in real life, but this preview reminds me sunset (if look on spare place).
Part 3: DNA Design Challenge
3.1. Choose your protein
I chose one of the Anthozoan chromoproteins — spisPINK from Stylophora pistillata. These proteins are actively studied for last decades due to their potential as markers in imaging, reporters in genetic engineering, and a source for synthetic biology. There were data of successful expression of this protein by E.coli, so it’s interesting to get the strain capable of it.
In this article, authors describe spisPink’s structure, hybridization, and dimerization tendency. Interestingly,
In phylogenetic analyses of anthozoan chromoproteins and related fluorescent proteins that have a common ancestor, gfasPurple, amilCP and eforRED are within the same larger clade with ‘dsRed-like’ red fluorescent proteins, whereas spisPINK belongs to a sister clade containing predominantly blue and green fluorescent proteins.
Also, it is really pink!
The sequence I worked with uploaded by the article authors.
Using Reverse Translate Tool from bioinformatics.org, I received the following sequences:
reverse translation of pdb|7SWU|D Chain D, Chromoprotein spisPINK to a 666 base sequence of most likely codons.
atgagccatagcaaacaggcgctggcggataccatgaaaatgacctggctgatggaaggcagcgtgaacggccatgcgtttaccattgaaggcgaaggcaccggcaaaccgtatgaaggcaaacagagcggcacctttcgcgtgaccaaaggcggcccgctgccgtttgcgtttgatattgtggcgccgaccctgnnntttaaatgctttatgaaatatccggcggatattccggattattttaaactggcgtttccggaaggcctgacctatgatcgcaaaattgcgtttgaagatggcggctgcgcgaccgcgaccgtggaaatgagcctgaaaggcaacaccctggtgcataaaaccaactttcagggcggcaactttccgattgatggcccggtgatgcagaaacgcaccctgggctgggaaccgaccagcgaaaaaatgaccccgtgcgatggcattattaaaggcgataccattatgtatctgatggtggaaggcggcaaaaccctgaaatgccgctatgaaaacaactatcgcgcgaacaaaccggtgctgatgccgccgagccattttgtggatctgcgcctgacccgcaccaacctggataaagaaggcctggcgtttaaactggaagaatatgcggtggcgcgcgtgctggaagtg
This is the best for synthetic gene design for high expression, and PCR primers. Alternatively, the tool offers consensus sequence, which is better for degenerate PCR primer design, where accounting for multiple potential codons is necessary:
reverse translation of pdb|7SWU|D Chain D, Chromoprotein spisPINK to a 666 base sequence of consensus codons.
atgwsncaywsnaarcargcnytngcngayacnatgaaratgacntggytnatggarggnwsngtnaayggncaygcnttyacnathgarggngarggnacnggnaarccntaygarggnaarcarwsnggnacnttymgngtnacnaarggnggnccnytnccnttygcnttygayathgtngcnccnacnytnnnnttyaartgyttyatgaartayccngcngayathccngaytayttyaarytngcnttyccngarggnytnacntaygaymgnaarathgcnttygargayggnggntgygcnacngcnacngtngaratgwsnytnaarggnaayacnytngtncayaaracnaayttycarggnggnaayttyccnathgayggnccngtnatgcaraarmgnacnytnggntgggarccnacnwsngaraaratgacnccntgygayggnathathaarggngayacnathatgtayytnatggtngarggnggnaaracnytnaartgymgntaygaraayaaytaymgngcnaayaarccngtnytnatgccnccnwsncayttygtngayytnmgnytnacnmgnacnaayytngayaargarggnytngcnttyaarytngargartaygcngtngcnmgngtnytngargtn
3.3. Codone Optimization
In case of using pUC19 plasmid backbone (have it in lab) for cloning spisPINK gene in E. coli, I’d consider E. coli codon preferences and avoid cleavage sites of NheI + XhoI enzymes, becaise they i) cut once in the vector MCS, ii) do NOT cut inside spisPINK gene according to Benchling, and iii) produce incompatible sticky ends. I used Codon Optimization Tool from Vector Builder and here’s improved sequence:
3.4. What technologies could be used to produce this protein from your DNA?
Using high-copy plasmid + adding strong promoter to the gene sequence is reliable and well-known technology for receiving strain which constitutively express smth. However, ligation percentage could be low and it is not seamless. Golden Gate assembly overcomes this troubles, and it is indeed elegant method—in case of higher DNA design skills. Besides considering E. coli codon preferences, BsaI and BsmBI sites should be removed and overhang to be designed. Thus, in my case of inserting only one gene sequence, high-copy plasmid + adding strong promoter should be OK.
Part 4: Prepare a Twist DNA Synthesis Order
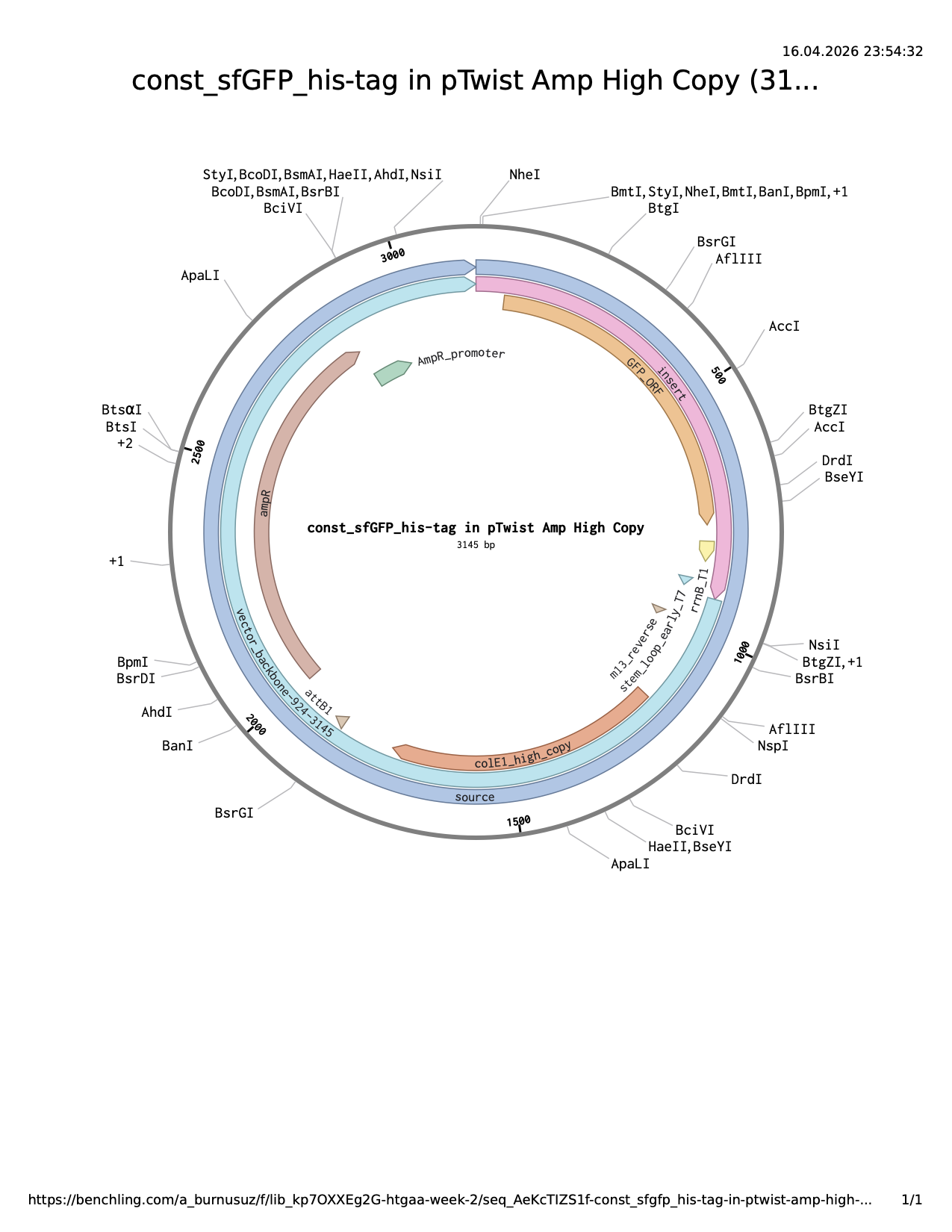
I used sfGFP sequence from example. All the parts together:
Then, I uploaded it to Clonal Genes order section at Twist using pTwist Amp High Copy (both my lab and my node have ampicillin). The result plasmid sequence and map are attached.
Part 5: DNA Read/Write/Edit
5.1. DNA Read
What DNA would you want to sequence (e.g., read) and why?
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
1)
2)
3)
4)
5.2. DNA Write
What DNA would you want to synthesize (e.g., write) and why?
What technology or technologies would you use to perform this DNA synthesis and why?
1)
2)
5.3. DNA Edit
Week 3 HW: Lab Automation
Assignment: Python Script for Opentrons Artwork
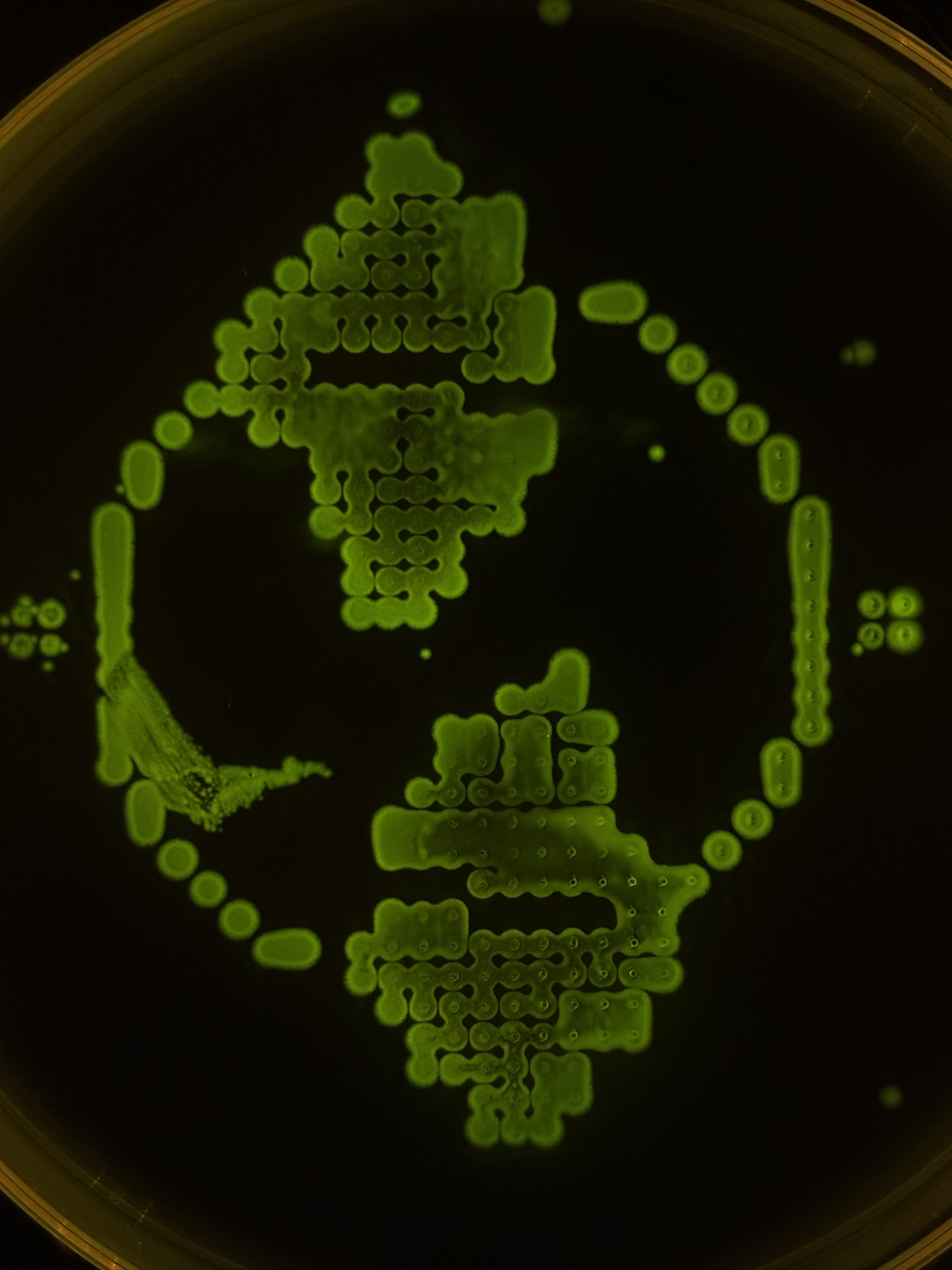
Being a CL at Designer Cells Node, I had an opportunity for my image being printed!
Here’s my code which was written with assistance of multiple AI agents, Gemini was not sufficient to correct all my mistakes.
I used this Art Nouveau-ish ornament with Ginkgo leaves as a reference, manually assigning printing dots via Ronan’s website.
Only red and green fluorescent E. coli strains wera available at my Node at the moment, and that matched perfectly with the picture. I like the resulting bio-print, although I should’ve selected lesser drop volume to avoid dots merging.
Opentrons in Published Research
I found a paper entitled “Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT-2 and Flex” surprizingly starting with “slowpoke” and ending with “flex”… (cough) The paper introduces Slowpoke, an easy-to-use automated system for DNA assembly that streamlines the cloning process using popular liquid-handling robots. It includes a free graphical interface for users and has been tested successfully with various DNA toolkits, making it a flexible solution for synthetic biology labs.
The study found that the Slowpoke automated workflow successfully created DNA constructs, achieving high assembly efficiencies with different genetic toolkits across two liquid-handling platforms, the OT-2 and Flex. Specifically, all 17 attempts resulted in successful colonies with one toolkit on the OT-2, 11 out of 12 were successful on Flex, and 8 out of 13 were successful using another toolkit on the OT-2. Additionally, when testing combinations of parts, 55 out of 57 resulted in correct DNA constructs, demonstrating that the workflow is effective and adaptable for various applications in synthetic biology. To make the Slowpoke tool easier to use, the developers created an online version with a simple interface, allowing users to generate experimental protocols with just one click. Users need to provide specific input files in CSV format, which outline the genetic components and their arrangements for tasks like Golden Gate cloning. This setup includes a standard toolkit map and a custom parts map, making it flexible for users to combine different genetic parts for their experiments, while being guided through the process both online and offline. Slowpoke allows researchers to prepare up to 96 Golden Gate assemblies and perform colony PCR reactions at the same time, streamlining laboratory work. In a typical setup using the Opentrons OT-2 robot, one thermocycler is used for both assembly and transforming E. coli cells, but it takes up a lot of space. To make better use of the available slots on the robot, researchers can switch to standard benchtop thermocyclers, allowing more flexibility and increasing the number of experiments that can be conducted simultaneously.
Slowpoke stands out from other DNA-assembly automation tools by being affordable and user-friendly. While tools like AssemblyTron and DNA-BOT require coding skills or focus on less commonly used methods, Slowpoke offers a complete solution that covers various processes like assembly, transformation, plating, and colony PCR without needing any programming knowledge. This makes Slowpoke accessible for more users in synthetic biology, particularly due to OT-2 and Flex relatively low price. Although Slowpoke has many advantages for automating DNA assembly, it still has some limitations that require human involvement, such as sealing PCR plates and transferring tubes between machines. Colony picking remains the most labor-intensive task because current Opentrons systems don’t fully support this step. However, new open-source solutions, like the one developed by Marburg iGEM 2019 team, are working to automate this process using 3D-printed technology and neural networks, which could help make Slowpoke even more efficient by reducing the need for human intervention.
Final Project Ideas + Lab Automation Tools
At the current stage, I elaborate some ideas related with Mgnetic or Protein-Based Nanoparicles (MNP and PBNP respectively) as DNA cargo for transfection. An Opentrons OT-2 liquid handling robot could be programmed to automate preparation of nanoparticle–DNA complexes by dispensing plasmid DNA, peptide solutions, and buffer components at controlled ratios (e.g., optimized N/P charge ratios for PBNP). This would reduce pipetting variability and allow rapid screening of different nanoparticle compositions and concentrations.
Python-based workflow scripting in Google Colab could be used to generate automated experimental layouts, calculate reagent volumes, and export OT-2-compatible protocols. For example, scripts could automatically produce dilution matrices for testing different peptide:DNA ratios or different nanoparticle formulations. Pseudocode example:
Regarding cargo DNA, computational tools such as Ginkgo Nebula could also be used for construct design and sequence organization, including codon optimization, annotation of plasmid features.
Week 4 HW: Protein Design Part I
Conceptual Questions
How many amino acid molecules are in 500 g of meat?
Average amino acid ≈ 100 Da = 100 g/mol.
500 g meat (assuming mostly protein for estimation) ≈ 500 g protein ≈ 500 g / (100 g/mol) = 5 mol amino acids.
Number of molecules = 5 × 6.02×10²³ ≈ 3.0 × 10²⁴ amino acid molecules (approximate).
Why humans do not become what they eat
Food proteins are broken down into amino acids during digestion. These amino acids are then reassembled into human-specific proteins according to human DNA instructions. The body does not preserve the original organism’s structure: instead, it uses the building blocks to synthesize its own molecules.
Why only 20 natural amino acids
The standard 20 amino acids represent an evolutionarily optimized set balancing chemical diversity, metabolic cost, and translational accuracy. This set provides sufficient structural and functional diversity for proteins while remaining compatible with ribosomal synthesis and tRNA charging systems.
(That’s not a serious answer, but one of my professors joked thet there are only two laws in biology: i) it is the way it supposed to be, ii) it happened so. This is probably a second-law case)
Can we design non-natural amino acids?
Yes. Non-natural amino acids can be synthesized by modifying side chains or backbone structures. Examples include fluorinated amino acids (to increase stability), N-methyl amino acids (to restrict flexibility), and photo-reactive amino acids (e.g., azido groups for crosslinking). These are widely used in protein engineering and synthetic biology.
Origin of amino acids before life
Amino acids likely formed through prebiotic chemistry such as lightning-driven reactions (Miller–Urey experiment), hydrothermal vent chemistry, and extraterrestrial delivery via meteorites (e.g. Murchison meteorite). These processes produced simple organic molecules before enzymes existed.
Handedness of α-helix with D-amino acids
L-amino acids form right-handed α-helices, therefore, D-amino acids form left-handed α-helices.
Can we discover additional helices in proteins?
Yes. Beyond α-helices, proteins contain 3₁₀ helices, π-helices, and less common distorted helices. Computational modeling and structural biology continue to reveal new helical geometries, especially in membrane proteins and engineered peptides.
For instance, ε-Azido-lysine ((–N₃) on ε-amino group): azide interacts via click chemistry, such as strain-promoted azide-alkyne cycloaddition, enabling site-specific labeling, crosslinking, and drug conjugation. At the same time, it is inert to “natural” functional groups.
Why are most molecular helices right-handed?
Because life is built primarily from L-amino acids, and L-amino acids energetically favor right-handed α-helices due to steric constraints and optimal hydrogen bonding geometry. This chirality bias became fixed through evolutionary selection.
Why do β-sheets aggregate?
β-sheets expose alternating side chains that can form strong intermolecular hydrogen bonds. This allows extended stacking between sheets. Hydrophobic residues further drive aggregation by minimizing exposure to water, making aggregation energetically favorable.
Why do amyloid diseases form β-sheets? Can they be used as materials?
Under stress, mutation, aging, or chemical imbalance, some proteins fail to fold correctly. When misfolded proteins interact, they often rearrange into β-sheet-rich structures because β-sheets allow many strong hydrogen bonds between neighboring protein chains, maximize packing efficiency, cooperative stabilization between many molecules. So once aggregation begins, it becomes energetically favorable for more proteins to join the growing fibril
Misfolded protein strands
→ Alignment side-by-side
→ Intermolecular hydrogen bonding
→ Formation of extended β-sheets
→ Stacking into fibrils (amyloid fibers)
This stability is not always harmful. Amyloid β-sheets can be engineered as biomaterials, such as nanofibers, hydrogels, and scaffolds, though toxicity must be controlled. Their potential is promissing, since they are self-assembling nanoscale ordered structures.
β-sheet motif design
A stable β-sheet motif should include alternating hydrophobic and hydrophilic residues to promote proper side-chain packing.
Example sequence pattern:
Val–Thr–Val–Thr–Val–Thr (repeating)
Hydrophobic residues (Val) stabilize sheet core
Polar residues (Thr) face solvent
Multiple strands align antiparallel
Interstrand hydrogen bonding forms stable sheet
Edge residues capped to prevent uncontrolled aggregation
Protein Analysis and Visualization
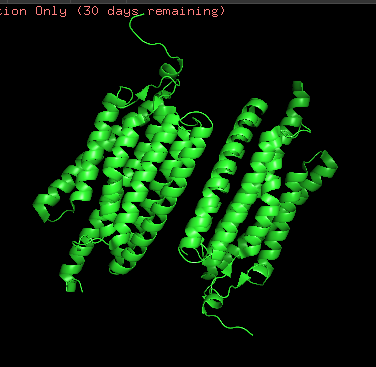
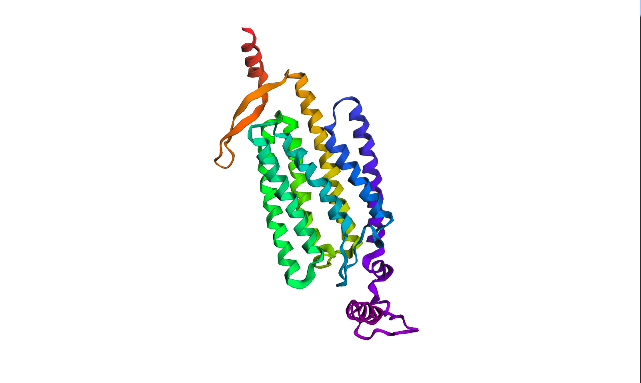
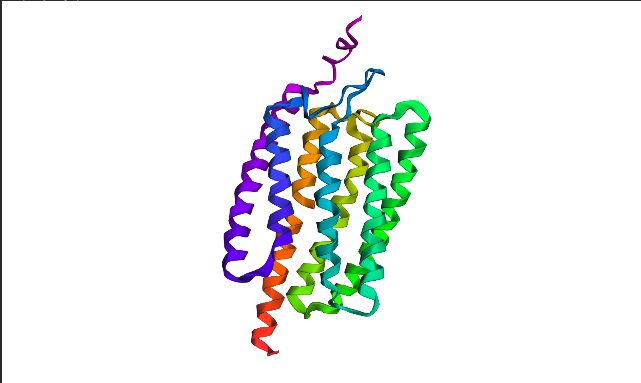
I chose Channelrhodopsin-2 (encoded by ChR2 gene) from Chlamydomonas reinhardtii. ChR2 is a light-gated ion channel and photoreceptor originally isolated from this green alga. When illuminated with blue light (maximally around 470 nm), ChR2 undergoes a conformational change that opens a pore, allowing positively charged ions (like sodium, calcium, and protons) to flow into the cell. Currently, I do a little research for my individual final project idea, and ChR2 might be relevant.
ChR2 consists of 315 AA rsidues, 35 residues of which are leucine and 34 belong to glycine, making them the most frequent.
6EID_1|Chains A, B|Archaeal-type opsin 2|Chlamydomonas reinhardtii (3055)
MDYGGALSAVGRELLFVTNPVVVNGSVLVPEDQCYCAGWIESRGTNGAQTASNVLQWLAAGFSILLLMFYAYQTWKSTCGWEEIYVCAIEMVKVILEFFFEFKNPSMLYLATGHRVQWLRYAEWLLTCPVILIHLSNLTGLSNDYSRRTMGLLVSDIGTIVWGATSAMATGYVKVIFFCLGLCYGANTFFHAAKAYIEGYHTVPKGRCRQVVTGMAWLFFVSWGMFPILFILGPEGFGVLSVYGSTVGHTIIDLMSKNCWGLLGHYLRVLIHEHILIHGDIRKTTKLNIGGTEIEVETLVEDEAEAGAVNKGTGK
This protein belongs to archaeal/bacterial/fungal opsin family. Uniprot BLAST finds 250 homologous sequences with identity range from 19.5% to 100%. 162 similar sequences belong to Archaea domain, 17—to Bacteria, 69 are found in distinct algae taxons, and just 2 belong to Fungi taxon.
Wild-type ChR2 3D structure at RCSB was released on 2017-12-06 and resolved at 2.39 Å scale.
Besides protein polymer, resolved structure includes 3 ligands: OLC ((2R)-2,3-dihydroxypropyl(9Z)-octadec-9-enoate), EDT [(BIS-CARBOXYMETHYL-AMINO)-ETHYL]-CARBOXYMETHYL-AMINO-ACETIC ACID), and phosphate.
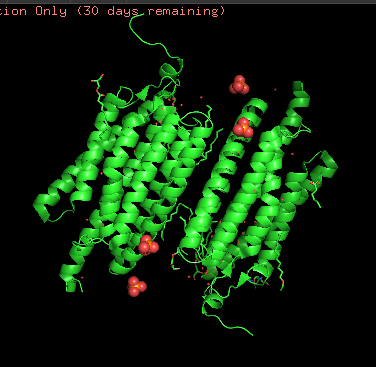
Structurally, this protein belongs to 7-transmembrane (7TM) α-helical membrane proteins.
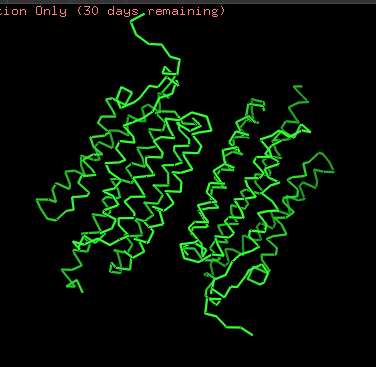
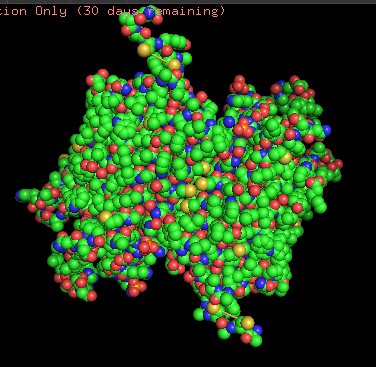
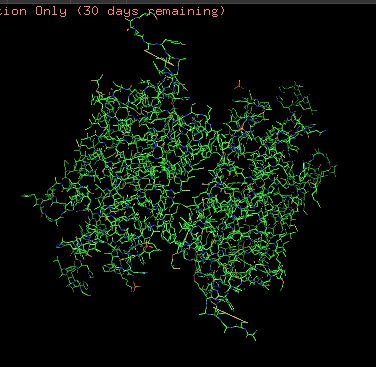
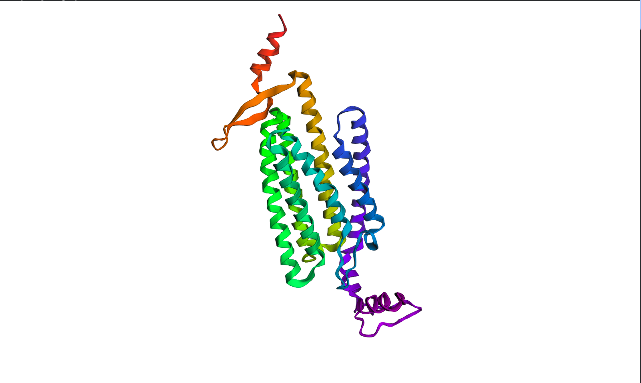
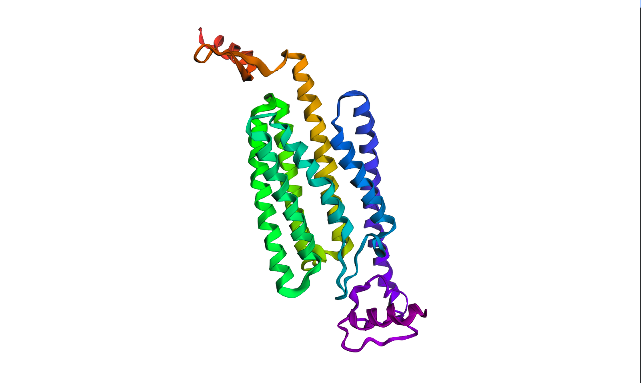
Protein structure visualized as “cartoon”, “ribbon”, and “balls & spheres (with different diameter)” respectively:
Coloring the protein by secondary structure. As it follows from structure family name, it has many α-helices
Coloring the protein by residue type. Being a transmembrane protein, Chr2 has hydrophobic residues. Hydrophobic residues are highly abundant and clustered in long continuous regions, consistent with transmembrane helices. Hydrophilic residues are more exposed on the surface and likely participate in solvent interactions. Hydrophobic vs. hydrophilic residues indication is shown below
ChR2 surface visualization. It doesn’t actually show any obvious pockets, but contains narrow central channel and ligand binding sites
Using ML-Based Protein Design Tools
Protein Language Modeling
Deep Mutational Scans
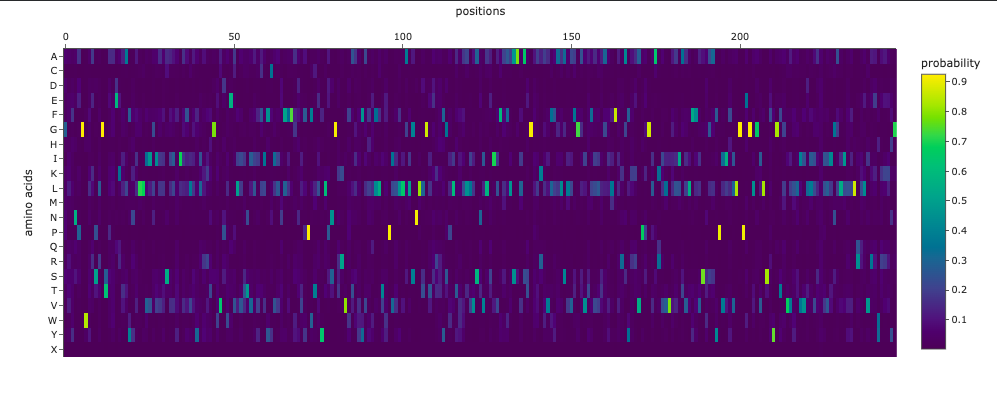
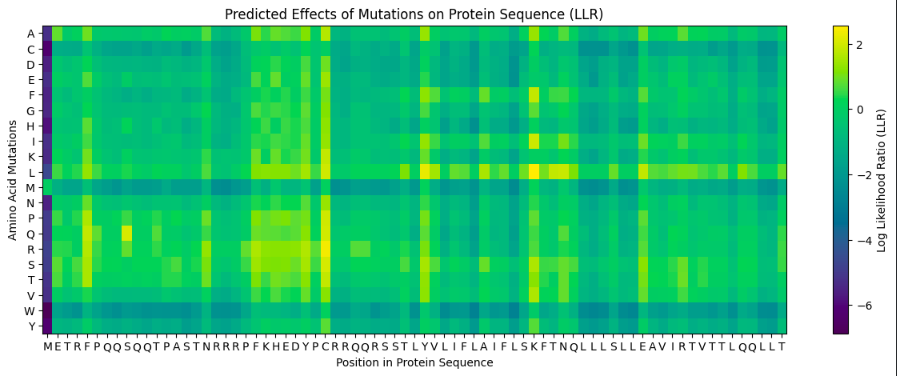
The resulting heatmap shows:
Many bright yellow spots in leucine row at various positions. L is the most frequent residue
Multiple bright yellow spots at position 27, predominantly regarding P, Q, R, S, T amino acids
Almost all the W-row is blue or dark blue
Mostly blue columns at 16-18, 53-58, 71-72 positions
The heatmap ends on position 72, although there are 315 AA
Leucine (L) is strongly favored in hydrophobic environments, stabilizing protein cores and helices. It is very common in membrane proteins and buried regions, so it’s expected.
Multiple tolerated substitutions at position 27. Probably, this position is structurally permissive or surface-exposed. Since bright yellow spots are concentrated in P, Q, R, S, T rows, it is likely on the protein surface or in a loop region. P (Proline) introduces kink suggests flexibility allowed. Q (Glutamine) is polar, surface-friendly. R (Arginine) is charged, usually solvent-exposed. S/T (Serine/Threonine) are small polar, often in loops.
W (Tryptophan) is a very large bulky aromatic residue, sterically expensive, requiring space and hydrophobic packing. So, this is relevant for many proteins and not surprising.
16-18, 53-58, 71-72 columns represent likely structurally constrained positions. It could mean both protein core / folding nucleus and transmembrane helix core.
It seems like ether an tokenizer is not aligned with amino-acid sequence length—which is doubtful—OR the algorythm reads only one of two polypeptide chain. Previous visualization show that ChR2 is a dimer.
Latent Space Analysis
Protein Folding
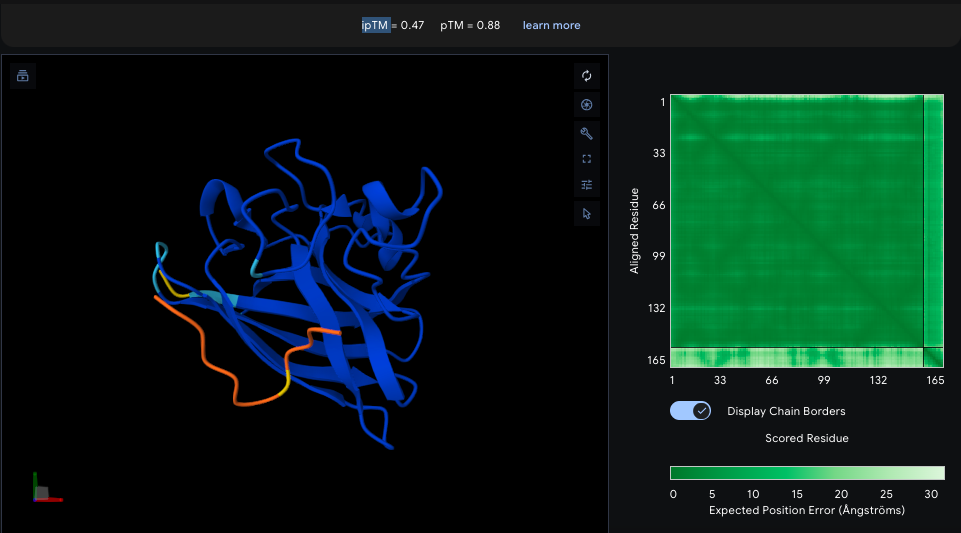
ESMFold visualized only a half of the functional protein, only one polypeptide chain. However, the chain itself looks pretty much like in PyMOL visualizations. The model is rather confident with ptm score of 0.751 and plddt of 75.483.
I inserted Lysine (K)—positively charged AA—into region that is supposed to be conservative according to Mutation Heatmap (near 50s positions). A51K point mutation sequence:
Probably, initially moderate confidence of the model does not allow to catch specific mutation induced folding destabilization. Alternatively, even this mutation did not affect critical region.
Protein Generation
Using ProteinMPNN (model v_48_020, sampling temperature 0.1), I performed ChR2 (PDB ID: 6EID) inverse folding. The resulting sequence recovery score is 0.4024. meaning 40% of the original amino acids were reproduced by the model. That could mean many ChR2 positions are not strictly sequence-specific, its structure can tolerate multiple amino acids, and the backbone constraints are more important than exact residues.
The new sequence consists of only 247 AA in contrast to original ChR2. Visualization looks pretty much similar, althoug one of the terminal regions was noticeably dropped. However, new ptm is 0.866 and plddt is 84.774 vs. 0.751 and 75.483 of original sequence. ProteinMPNN has probably replaced “hard-to-fit” residues with those that are more structurally compatible to “general vision” of this protein.
I inserted mutant sequence and set 12 as peptide length in PepMLM Google Colab code. To compare FLYRWLPSRRGG binding peptide pseudo perplexity, I added another code cell into Google Colab:
Here are the downloaded results rounded to hundredths:
Binder
Pseudo Perplexity
KRYYVTGVRLKK
30.16
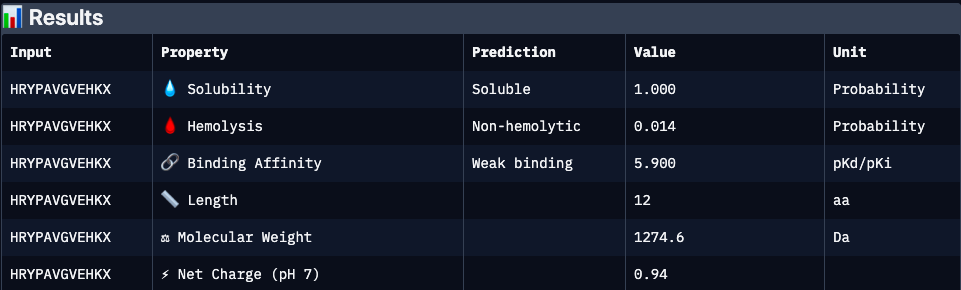
HRYPAVGVEHKX
15.33
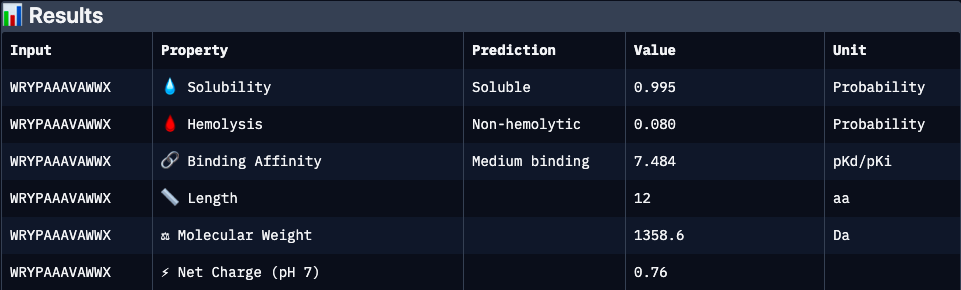
WRYPAAAVAWWX
10.03
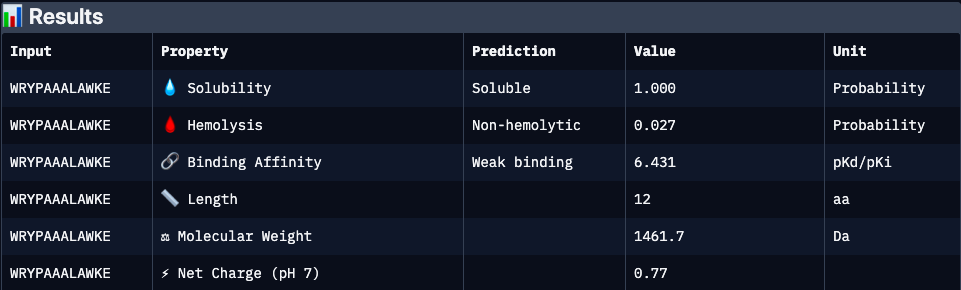
WRYPAAALAWKE
11.92
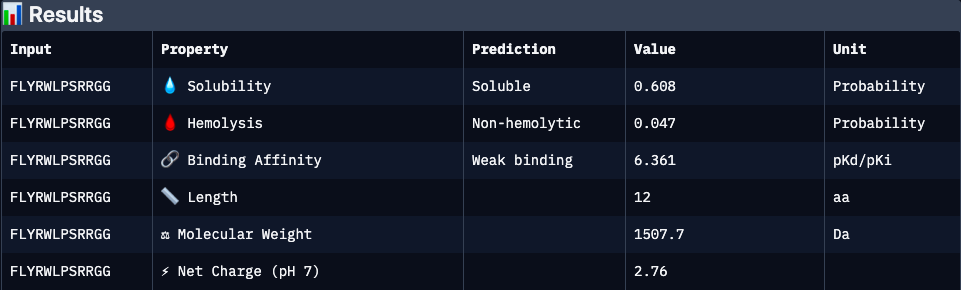
FLYRWLPSRRGG
20.64
WRYPAAAVAWWX showed the lowest perplexity (10.03) upon this peptide-generation session, meaning highest plausibility. Interestingly, experimentaly approved FLYRWLPSRRGG showed the second lowest perplexity, whereas “the best” variant contains X (undefined) amino acid making it should be carefully interpreted. As the current LLMs’ outputs in general.
Evaluate Binders with AlphaFold3
Binders 2(HRYPAVGVEHKX) and 3 (WRYPAAAVAWWX, with the lowest pseudo perplexity) were excluded from the analyzis because of invalid “X” character.
Evaluate Properties of Generated Peptides in the PeptiVerse
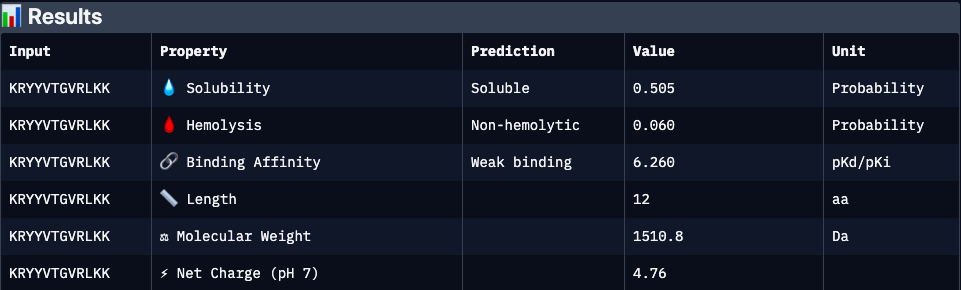
Binder 1 (KRYYVTGVRLKK):
Binder 2 (KRYYVTGVRLKK):
Binder 3 (WRYPAAALAWKE):
Binder 4 (WRYPAAALAWKE):
Binder 5, experimentaly verified (FLYRWLPSRRGG):
Binder
ipTM
Binding Affinity
Solubility
Hemolysis
Net Charge (pH 7)
Molecular Weight
Interpretation
KRYYVTGVRLKK
0.47
6.26
0.51
0.06
4.76
1510.8
Moderate structural binding support; strongly cationic peptide may enhance target interaction but raises risk of nonspecific electrostatic binding and moderate cytotoxicity. Solubility is acceptable but not optimal
HRYPAVGVEHKX
N/A
5.9
1.00
0.01
0.94
1274.6
Excellent developability profile with very high solubility and minimal hemolysis; however, absence of structural validation (ipTM unavailable) and weaker predicted affinity make binding confidence uncertain
WRYPAAAVAWWX
N/A
7.48
0.99
0.08
0.76
1358.6
Strongest predicted affinity among candidates and highly soluble, but elevated hydrophobic/aromatic content may contribute to increased hemolytic potential and aggregation risk. Requires structural validation
Experimentally validated binder with moderate predicted affinity and cationic character. Lower predicted solubility and interface confidence may reflect dynamic or partially disordered binding interactions rather than absence of activity
Generate Optimized Peptides with moPPIt
moPPIt was run targeting hydrophobic motif positions 3–9 of A4V SOD1 (KVVCVLK). 5 peptide samples of 12 residues were generated considering Hemolysis, Solubility, Affinity, and Motif parameters optimization.
Peptide
Hemolysis
Solubility
Binding Affinity
Motif
Interpretation
CTSGVNVGPGVP
0.04
0.99
6.11
0.63
Strong developability profile with very low predicted hemolysis and excellent solubility. Moderate motif alignment suggests partial but potentially indirect engagement of the hydrophobic KVVCVLK region; likely acts via peripheral or transient interaction rather than deep motif mimicry
ADSEIKAPSSGH
0.08
1.00
5.52
0.67
Highly favorable physicochemical properties with maximal solubility and low hemolysis risk. Motif similarity is moderate, suggesting a more electrostatically driven or scaffold-like interaction rather than direct hydrophobic motif mimicry
RSKYQWVPYHVT
0.04
1.00
6.31
0.49
Balanced candidate with strong predicted affinity and good solubility, but lower motif score indicates weaker structural motif mimicry. Likely binds through mixed hydrophobic–aromatic interactions rather than targeted motif recognition
SFAGICNVEQQT
0.05
1.00
5.95
0.75
Best overall motif alignment among the set, suggesting strongest intended targeting of the KVVCVLK hydrophobic patch. Favorable solubility and low hemolysis support developability, with potential for more direct motif-driven binding
QEPCEELQFNHF
0.02
0.51
6.26
0.66
Strong predicted affinity and acceptable motif similarity, but reduced solubility may limit practical usability. Likely engages target through distributed polar–aromatic interactions rather than strict hydrophobic motif complementarity
Final Project: L-Protein Mutants
The goal of this project is to design mutant MS2 lysis protein (L-protein) to reduce its interaction with the bacterial chaperone DnaJ. Since DnaJ is important for proper folding and processing of the L-protein, weakening this interaction may help the phage remain functional even if bacteria modify DnaJ. To estimate this interaction, co-folding predictions were performed using AlphaFold2 Multimer with both proteins entered together. In AlphaFold2, both sequences are inserted into a single input field separated by a colon “:”. Since transmembrane domain affects the L-protein lysis activity, mutations were intoduced in N-terminal region.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
DNA Assembly
Components in Phusion High-Fidelity PCR Master Mix and their purpose
Phusion High-Fidelity PCR Master Mix typically contains a high-fidelity DNA polymerase (such as Phusion polymerase), dNTPs, Mg²⁺ (often as MgCl₂), and a reaction buffer. The high-fidelity polymerase has proofreading (3’→5’ exonuclease) activity, which reduces DNA replication errors.
Factors determining primer annealing temperature
Primer annealing temperature depends mainly on:
primer melting temperature (Tm), which is influenced by GC content (stronger hydrogen bonding)
primer length
nucleotide sequence composition
kation concentration, such as Mg²⁺, also affects primer binding stability
primer design factors such as self-complementarity, secondary structures (hairpins), and primer-dimer formation
PCR vs restriction enzyme digestion
Feature
PCR
Restriction Enzyme Digestion
Purpose
Amplifies a specific DNA fragment
Cuts DNA at specific recognition sites
Mechanism
Uses primers and DNA polymerase through thermal cycling
Uses restriction enzymes that recognize and cleave specific sequences
Input requirement
Requires primers designed for target sequence
Requires existing DNA containing restriction sites
Output
Many copies of a defined DNA fragment
One or more DNA fragments of defined lengths
Specificity control
Determined by primer design
Determined by enzyme recognition sites in DNA
Equipment needed
Thermocycler
Usually incubator/water bath
Flexibility
High (can create custom fragments)
Limited to naturally occurring or engineered restriction sites
Typical use case
Amplifying genes for cloning, sequencing, diagnostics
Cutting plasmids or genomic DNA for cloning or mapping
Advantages
Fast amplification, highly specific, can introduce mutations or tags
Predictable cuts, simple workflow, widely used in traditional cloning
Limitations
Requires careful primer design, risk of amplification errors
Requires suitable restriction sites, can leave unwanted “scar” sequences
Ensuring DNA fragments are suitable for Gibson cloning
To ensure compatibility with Gibson assembly, DNA fragments must have overlapping homologous ends (typically 20–40 bp) that match adjacent fragments. These overlaps are usually added through PCR primer design. PCR products should be purified to remove enzymes, primers, and nucleotides that could interfere with assembly. For restriction-digested fragments, ends must be designed or processed to create compatible overlaps (often via PCR or adapter sequences). Additionally, sequences should be checked for unwanted secondary structures, repeats, or mismatches in overlap regions to ensure efficient annealing and correct assembly.
How plasmid DNA enters E. coli during transformation
Plasmid DNA (outside cell)
→ Cell made competent (CaCl₂ treatment or electroporation)
→ Negative charges on DNA + cell membrane partially neutralized
→ DNA brought close to bacterial membrane
→ Heat shock (chemical method) OR electric pulse (electroporation)
→ Temporary pores form in membrane
→ Plasmid DNA enters cytoplasm
→ Membrane reseals
→ Plasmid is maintained and can replicate inside E. coli
Golden Gate Assembly
Golden Gate Assembly is a molecular cloning method that uses Type IIS restriction enzymes, such as BsaI, which cut DNA outside of their recognition sites. This creates custom-designed overhangs that allow multiple DNA fragments to be ligated together in a specific order. The reaction occurs in a single tube containing both the restriction enzyme and DNA ligase, cycling between digestion and ligation steps. Because the recognition sites are removed during assembly, the final construct is seamless and does not retain unwanted restriction sequences. This method is highly efficient for assembling multiple fragments simultaneously, making it useful for constructing complex genetic circuits or multi-gene plasmids. It is often preferred over Gibson assembly when precise, standardized overhangs are desired and when high-throughput cloning is needed.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks (IANNs)
Advantages of IANNs over traditional Boolean genetic circuits
Intracellular Artificial Neural Networks (IANNs) differ from traditional genetic circuits because they process information in a graded, weighted, and adaptive manner rather than using only binary ON/OFF logic.
Analog signal processing
Traditional Boolean circuits treat inputs as discrete states (0 or 1). IANNs can respond continuously to varying molecular concentrations. It allows finer control, tunable responses, and probabilistic decision-making.
Ability to integrate many inputs
Boolean circuits become increasingly complex as the number of inputs grows because each logical relationship requires additional gates. IANNs naturally combine many weighted inputs similarly to biological signaling pathways, which enables scalable intracellular computation.
Learning-like behavior and adaptability
Traditional logic circuits are static once constructed. IANNs can theoretically mimic neural-network properties such as weighting, feedback adaptation, memory, nonlinear classification etc.
Noise tolerance
Boolean circuits are often fragile under noise because they depend on strict thresholds, while biological systems are noisy.
Compact implementation of complex behaviors
Complex Boolean functions often require many genes and regulatory parts. A multilayer IANN can sometimes implement the same classification behavior with fewer regulatory interactions, reusable regulatory motifs, and hierarchical processing.
Release a therapeutic payload only when confidence is high
Inputs (molesular factors):
High lactate concentration
Hypoxia marker
miRNA associated with tumors
Elevated TGF
Inflammatory cytokine pattern
The hidden layer processing serves for weighted integration, since tumor detection goes not after definig “Is marker A AND marker B present?” but according to molecular pattern recognition where general context affects each factor importance.
Outputs
Each Input could regulate transcription factors, CRISPR effectors, RNA regulators, riboswitches etc. If the weighted activation exceeds a threshold, the output gene is expressed, e.g.:
Apoptosis-inducing protein
CAR-T activation signal
Cytokine release
Fluorescent reporter
Drug synthesis enzyme
The output could also be graded.
Since cancer biomarkers are noisy, heterogeneous, and overlapping with healthy tissue in its molecular signature, Boolean logic risks to fail. However, there are several limitations:
Biological noise (affects precision)
Activation Response Crosstalk (regulatory molecules may unintentionally interact with endogenous pathways)
Limited orthogonal components (which coexist without interference)
Metabolic burden (utilizing of ATP, ribosomes, and transcriptional capacity)
Evolutionary instability (probability of synthetic circuits silencing over time)
Intracellular multilayer perceptron diagram
Layer 1
Input X1 encodes an endoribonuclease (for example Csy4-like). The endoribonuclease processes or represses an intermediate RNA regulator
X1 DNA ──Tx/Tl──> Endoribonuclease E1
│
▼
Cleaves/regulates intermediate RNA
Layer 2
The processed intermediate regulates translation of a fluorescent protein output
Intermediate RNA ──Tx/Tl──> Fluorescent protein Y
In other words,
Input Layer
X1 → Endoribonuclease E1
X2 → Regulatory RNA R1
Hidden Layer
E1 modifies R1
R1 acts as weighted regulatory signal
Output Layer
R1 regulates translation of fluorescent protein Y
↓
Fluorescence output
Part 2: Fungal Materials
Existing fungal materials, uses, advantages, and disadvantages
Fungal materials are typically made from mycelium, the filamentous root-like network of fungi. Mycelium can grow through agricultural waste and form lightweight, biodegradable composite materials.
Mycelium packaging
Companies grow mycelium around agricultural byproducts (corn husks, hemp hurds, sawdust) to produce protective packaging.
Uses
Replacement for Styrofoam
Shipping protection
Insulation
Advantages over plastic foam
Biodegradable
Compostable
Renewable
Low-energy manufacturing
Lower carbon footprint
Disadvantages
Less water resistant
Lower durability over long periods
Can deform under moisture or heavy loads
Shorter shelf life
Mycelium leather
Fungal biomass is processed into leather-like sheets.
Uses:
Fashion products
Shoes
Bags
Upholstery
Advantages over animal leather:
No animal agriculture
Lower greenhouse gas emissions
Faster production
Reduced water usage
Potentially customizable texture
Disadvantages:
Usually less durable than high-quality leather
May require synthetic coatings
Mechanical properties still improving
Scaling production remains expensive
Fungal food products
Fungi are already widely used as biomaterials in food.
Examples:
Quorn™ mycoprotein
Tempeh fermentation fungi
Mushroom-derived proteins
Uses:
Meat substitutes
Protein supplements
Advantages:
High protein yield
Lower environmental impact than livestock
Efficient land use
Disadvantages:
Texture/flavor limitations
Allergen concerns
Consumer acceptance barriers
In comparison with traditional counterparts, fungal materials are more ecologically sustainable, require relatively few resources, are capable of self-assembly and biodegradation. Growth conditions can alter their density, flexibility, porosity, and texture.
However, many fungal composites are less resistant than plastics, metals, or concrete. Relatively high fungal water absorption could also be detrimental in some contexts. Finally, mycellium grows relatively slow and not totally reproducible, which is a disadvantage in terms of industrial mass production.
Why genetically engineer fungi?
Fungi are extremely versatile organisms that already produce enzymes, antibiotics, pigments, organic acids, structural biomaterials etc.
Improved biomaterials, such as:
stronger mycelium
water-resistant composites
elastic or flexible materials
conductive biomaterials
Why?
This could replace:
plastics
synthetic foams
petroleum-derived textiles
Drug production, such as:
penicillin
cyclosporine
statins
Why?
lower manufacturing costs
faster drug discovery
sustainable bioproduction
Environmental remediation, by degrading:
plastics
oil pollutants
pesticides
toxic chemicals
Why?
Fungi can access environments difficult for bacteria to colonize
Biosensors to detect
toxins
explosives
pathogens
heavy metals
Fungi vs. bacteria in synthetic biology
Advantages of fungal synthetic biology over bacteria:
Ability to grow large multicellular structures
Superior secretion capabilities
Complex post-translational modifications
Better degradation of complex substrates
Filamentous growth
Higher tolerance for some harsh environments
Disadvantages:
Slower growth
More complex genetics
Difficult scaling and control
Fewer standardized tools
Part 3: First DNA Twist Order
Week 9 HW: Cell-Free Systems
Part A: General and Lecturer-Specific Questions
Advantages of cell-free protein synthesis (CFPS) over in vivo expression
Greater experimental control
Open reaction environment
Faster design-build-test cycles
Toxic protein production
Better access to non-natural chemistry
Easier monitoring and automation
Cases where cell-free expression is more beneficial than cell production
Case 1: Toxic proteins
Example:
pore-forming membrane proteins
antimicrobial peptides
viral toxins
In vivo expression may kill the host, reduce growth, and cause plasmid instability.
Case 2: Rapid prototyping of genetic circuits
CFPS allows same-day testing without cloning into cells:
DNA or mRNA template: promoter, ribosome binding site, coding sequence, terminator
Amino acids
Energy source: phosphoenolpyruvate (PEP), creatine phosphate, glucose, maltodextrin
Nucleotides
Salts and cofactors
Optional additives: chaperones, detergents, liposomes, disulfide bond catalysts, protease inhibitors
Why energy regeneration is critical
Protein synthesis is extremely energy intensive. Each peptide bond formation requires ATP, GTP, tRNA charging, and ribosome translocation. Energy sources are rapidly depleted during protein synthesis, and accumulation of inorganic phosphate due to their cleavage additionally inhibits reactions.
Example method for continuous ATP supply
Phosphoenolpyruvate (PEP) regeneration system
PEP + ADP → Pyruvate + ATP
(requires pyruvate kinase)
This is a simple method for fast ATP regeneration commonly used in CFPS. Although it is relatively expensive and does not solve phosphate accumulation-induced inhibition of synthesis.
Prokaryotic CFPS are ideal for proteins with simple folding and no glycosylation needed, which also express highly in E. coli: e.g. GFP. In this case, there are no obstacles for choosing this inexpensive and fast method.
Human monoclonal antibodies, such as IgG, require disulfide bonds, glycosylation and complex folding. Eukaryotic CFPS provide ER-like folding conditions, post-translational modifications and better assembly.
Designing a CFPS experiment for membrane protein expression
Membrane proteins are difficult because they tend to aggregate, misfold, and precipitate outside membranes. Also, they contain hydrophobic domains.
Choose suitable CFPS system: E. coli lysate with nanodiscs or eukaryotic lysate with microsomes
Optimize lipid composition: proportions of phosphatidylcholine + phosphatidylglycerol + cholesterol
Include chaperones
Control translation rate: lower temperature, reduced magnesium, weaker promoters (?)
Challenge
Solution
Aggregation
Add detergents/nanodiscs
Misfolding
Add chaperones
Poor insertion
Use lipid vesicles
Low solubility
Optimize ionic conditions
Instability
Add protease inhibitors
Low protein yield: reasons and troubleshooting
Reason
Solution
Poor template quality
Verify DNA integrity, optimize promoter/RBS, purify template, increase template concentration
Energy depletion
Improve ATP regeneration, add fresh energy substrate, optimize Mg/P balance, use glucose-based regeneration systems
Protein misfolding or aggregation
Reduce reaction temperature, add chaperones, include detergents/liposomes, optimize redox conditions
RNase or protease contamination
Use inhibitors, prepare fresh lysate
Codon bias
codon-optimize the gene, supplement rare tRNAs
HW question from Kate Adamala: Example Synthetic Minimal Cell Design
a) Function: Smart inflammation-sensing therapeutic synthetic cell
This synthetic minimal cell is designed to detect inflammatory signals associated with intestinal disease (such as inflammatory bowel disease, IBD) and respond by producing an anti-inflammatory therapeutic protein.
It would:
Sense inflammatory biomarkers in the gut
Process the signal internally
Produce and release an anti-inflammatory molecule only when inflammation is detected
Input
nitric oxide (NO)
reactive oxygen species (ROS)
inflammatory cytokines (e.g. TNF-α)
Output
interleukin-10 (IL-10), an anti-inflammatory cytokine
or
a fluorescent reporter (GFP) for testing purposes
Inflammation (NO) → activation of promoter → IL-10 production → therapeutic effect
b) Encapsulation over CFPS
A pure bulk cell-free system could detect NO and express IL-10 transiently. However, encapsulation is highly advantageous because it provides:
compartmentalization
protection from degradation
localized delivery
controlled diffusion
sustained operation
c) Could this function be realized by genetically modified natural cells?
Yes.
But, synthetic minimal cells offer advantages:
no replication
reduced biosafety risks
no mutation/evolution
easier containment
programmable lifespan
lower immune complexity
d) Desired outcome
Minimal synthetic cell for proposed purpose should:
remain inactive under healthy conditions
detect inflammation-associated NO
produce IL-10 locally
reduce inflammation without systemic immunosuppression
degrade safely after use
a) Membrane composition
POPC (phosphatidylcholine) + cholesterol. Also, DOPE and cardiolipin if there would be resources for these details.
Phosphoenolpyruvate (PEP), pyruvate kinase, or glucose metabolism enzymes
GroEL/ES or DnaK, glutathione, DTT
c) Choice of Tx/Tl system
Prokaryotic as proof-of-concept
Mammalian or hybrid systems may be preferable for therapeutic-grade IL-10: it could be syntheside without mammalian post-translational modifications but it is probably unavoidable for immune acceptance.
d) Communication with the environment
no membrane channel is strictly necessary for sensing NO
NO dose-response curve (RT-qPCR and Western blot assays for srecific genes/proteins)
Functional inflammation assay (cytokine profiles)
HW question from Peter Nguyen
1. One sentence pitch
A “living fragrance textile” that uses freeze-dried cell-free systems embedded in fashion fabrics to sense body chemistry and dynamically produce personalized perfume molecules throughout the day.
How the idea works
The concept is a smart fashion fabric containing microencapsulated freeze-dried cell-free transcription/translation (Tx/Tl) systems integrated into clothing, scarves, jewelry textiles, or wearable accessories. These systems remain dormant while dry but become activated when exposed to moisture from sweat, humidity, or skin contact. The activated cell-free biosensors detect biochemical signals such as skin pH, stress metabolites, temperature changes, or sweat composition and respond by enzymatically generating fragrance compounds directly within the textile.
For example:
Increased sweat/stress biomarkers
↓
Rehydration of freeze-dried Tx/Tl system
↓
Expression of fragrance-producing enzymes
↓
Localized release of perfume molecules
Customization would likely become one of the most attractive features of the system. The textile could create various scents during stress, exercise, or related with outer temperature.
Trigger
Customized scent response
Stress detected
Lavender / chamomile calming scent
Exercise
Citrus / mint freshness
Evening temperature drop
Warm amber / sandalwood
Different scent molecules are produced by different enzymatic pathways:
Fragrance note
Example molecule
Floral
Linalool
Citrus
Limonene
Pine/fresh
Pinene
Vanilla/warm
Vanillin
Rose
Geraniol
This is conceptually realistic because:
cell-free systems are modular
DNA templates are easily replaceable
fragrance synthesis pathways already exist in plants and microbes
freeze-dried reactions can be pre-programmed
The major challenge is not sensing itself, but:
stability
precise dosage control
long-term scent consistency
scalable manufacturing
Societal challenge or market need
1. Personalized fragrance experiences
Traditional perfumes are static and identical for all users, despite body chemistry strongly affecting scent perception. A responsive biosynthetic textile could create individualized fragrances that adapt in real time to:
body chemistry
stress
environment
activity level
This enables hyper-personalized fashion experiences.
2. Sustainable perfumery
Conventional perfume manufacturing often involves:
petrochemical synthesis
excessive packaging
solvent-heavy production
environmentally intensive ingredient sourcing
Cell-free fragrance synthesis could reduce overproduction, waste, and transportation of volatile compounds. The textile would produce scent only when needed.
3. Long-lasting wearable fragrance
Perfumes typically evaporate rapidly and require reapplication. A cell-free fragrance textile could continuously regenerate scent molecules over time, extending fragrance longevity without carrying additional products.
4. Emotional wellness and sensory fashion
Fashion is increasingly integrating wellness and multisensory design. Adaptive scent-producing textiles could:
reduce stress
improve mood
enhance confidence
create immersive sensory experiences
Thus, the idea escapes narrow fashion context with luxury wearables, and performance costumes, being theoretically expandable onto therapeutic fashion and wellness accessories.
Addressing limitations of cell-free systems
1. Moisture-triggered activation
Sweat and humidity naturally serve as activation mechanisms:
2. Protective encapsulation for stability
Heat, oxidation, and UV exposure are primary sources of Tx/Tl components destabilization of wearable freeze-dried systems.
They could be encapsulated in:
silk fibroin microcapsules
hydrogel nanofibers
trehalose sugar matrices
These materials are also compatible with textiles and fashion materials.
3. Replaceable fragrance patches
Rather than washing the biological system repeatedly, garments could contain:
detachable fragrance inserts
replaceable biosensing textile modules
refillable collar or cuff patches
This makes the fashion piece reusable while replacing only the active biological component.
4. Controlled one-time or slow-release activation
The system could be designed for single-event luxury scent release or gradual activation over hours. Hydrogels and layered polymer barriers could regulate:
water diffusion
enzyme activation
fragrance release rate
5. Stabilizing the energy supply
The freeze-dried system would include:
ATP regeneration enzymes
glucose or maltodextrin energy reservoirs
slow-release substrates.
This could extend fragrance production during wear.
6. One-time use as a luxury feature
In haute couture or perfumery, ephemerality can become part of the artistic concept and experience. For example:
a dress that releases a unique fragrance only during a runway performance
a perfume scarf activated once during an event
personalized scent “moments” linked to environmental conditions
The transient nature of cell-free reactions could therefore enhance exclusivity rather than limit functionality.
HW question from Ally Huang
Background
In space, astronauts face increased exposure to cosmic radiation and microgravity, which can damage DNA and disrupt cellular function. This raises risks such as cancer, immune dysfunction, and accelerated aging. Monitoring DNA damage in real time is challenging due to limited lab infrastructure aboard spacecraft. Freeze-dried cell-free systems like BioBits® provide a safe, stable, and portable platform for biological sensing without using living cells. These systems can be deployed on-demand and rehydrated when needed. Developing reliable DNA damage sensors is important for astronaut safety and for understanding how biological systems respond to extreme environments beyond Earth.
The recA promoter is part of the bacterial SOS response, which activates when DNA damage occurs due to radiation stress. In the BioBits® cell-free system, a plasmid containing the recA promoter linked to GFP is added after rehydration. Importantly, the system does not interact with human DNA; instead, it detects environmental stress signals by responding only to the engineered DNA template. When DNA damage conditions are simulated, activation of the promoter leads to GFP production. This provides a controlled, indirect biosensing method for estimating radiation-related stress in space-relevant conditions.
Hypothesis
The hypothesis is that a freeze-dried BioBits® cell-free system containing a recA promoter–GFP reporter will show significantly higher fluorescence when exposed to DNA-damaging conditions compared to non-damaged controls. This is based on the principle that the recA promoter is activated during the SOS response to DNA damage, leading to increased downstream gene expression. In our system, GFP production acts as a direct, measurable output of promoter activation. When exposed to simulated space radiation stress, such as UV light, experimental samples should exhibit increased fluorescence intensity relative to controls. This would demonstrate that cell-free biosensors can reliably detect genotoxic stress. The results would support the use of BioBits®-based systems for monitoring astronaut exposure to radiation and for developing autonomous, low-resource diagnostic tools for long-duration space exploration missions.
Experiment design
Freeze-dried BioBits® reactions will be prepared containing a recA promoter–GFP plasmid. Samples will be rehydrated and divided into experimental and control groups. Experimental groups will be exposed to UV light as a radiation proxy, while controls remain unexposed. The miniPCR® thermal cycler will be used to amplify DNA constructs if required before reaction setup. After incubation, GFP fluorescence will be measured using the P51 Molecular Fluorescence Viewer. Negative controls will include reactions without plasmid DNA. Fluorescence intensity will be compared across conditions to quantify activation of the DNA damage response biosensor.
Several aspects of the nanoparticle-mediated plant transfection system will be measured to evaluate delivery efficiency, functional gene expression, and electrophysiological response.
The primary measurable parameters include:
efficiency of nanoparticle-mediated DNA delivery into plant tissues
expression of fluorescent reporter proteins
functional activity of Channelrhodopsin-2
electrophysiological responses after light stimulation
physicochemical properties of peptide-based nanoparticles (size and charge)
Additional measurements may include comparison between magnetic nanoparticles and peptide-based nanoparticles in terms of transfection efficiency and signal intensity.
Elements to be measured and methods of measurement
Nanoparticle formation and stability
The size and surface charge of peptide–DNA nanoparticles will be evaluated to confirm successful self-assembly and stability.
Measurements:
nanoparticle diameter
zeta potential
aggregation behavior
Methods:
dynamic light scattering (DLS)
zeta potential analysis (under consideration)
microscopy-based visualization
Presence of plasmid DNA
Successful incorporation of plasmid DNA into nanoparticles will be verified.
Measurements:
presence and integrity of plasmid DNA
DNA binding efficiency.
Methods:
agarose gel electrophoresis
PCR
gel retardation assay
Gene expression in plant tissues
Successful transfection will be assessed through expression of GFP reporter.
Measurements:
fluorescence intensity
localization of expression
Methods:
fluorescence microscopy
confocal microscopy (if available)
image analysis software
Functional activity of Channelrhodopsin-2
The activity of the optogenetic ion channel will be evaluated after blue-light stimulation.
Measurements:
changes in membrane potential
electrophysiological response amplitude
signal timing after stimulation
Methods:
extracellular electrode recording
optical stimulation using blue LED light
Technologies used
Fluorescence Microscopy
Fluorescence microscopy will be used to detect expression of reporter proteins such as GFP or mCherry in plant tissues. This provides visual confirmation of successful transfection and allows comparison of delivery efficiency between nanoparticle systems.
Dynamic Light Scattering (DLS)
DLS will be used to characterize nanoparticle size distribution and stability in solution. Nanoparticle size is an important parameter influencing plant tissue penetration and delivery efficiency.
Electrophysiological Recording
Extracellular electrophysiological measurements will be used to detect electrical responses generated after activation of Channelrhodopsin-2 by blue light stimulation. These measurements will provide functional validation of successful gene expression.
Agarose Gel Electrophoresis
Agarose gel electrophoresis will be used to verify plasmid DNA integrity and evaluate peptide–DNA complex formation. Gel retardation assays can demonstrate successful nanoparticle assembly by reduced migration of DNA complexes through the gel matrix.
Computational Design Tools
Benchling will be used for plasmid design, sequence annotation, and in silico cloning simulations. Automation workflows for nanoparticle preparation may additionally be developed using Python scripting and Opentrons OT-2 protocols.
Waters Part I — Molecular Weight
His-tagged and linker-containing eGFP MWth = 28,006.60Da, according to ExPASy portal calculator
I chose the following peaks:
m/z_n = 1000.4302
m/z_n+1 = 965.9684
Using the first formula: z = (m/z_n+1) / (m/z_n - m/z_n+1)
z = 965.9684 / (1000.4302 - 965.9684)
z = 28.03
meaning the higher m/z peak is 28 and the lower is 29
Calculating molecular weight: MW = z × (m/z - proton mass)
for the higher peak, MW = 28 × (1000.4302 - 1.0073)
MW = 27,983.84 Da
for the lower peak, MW = 29 × (965.9684 - 1.0073)
MW = 27,983.87 Da
Thus, average experimental MW MW_exp = (27,983.84 + 27,983.87) / 2
MW_exp = 27,983.86 Da
Calculating mass accuracy against the mature theoretical MW:
MW_theory = 27,986.57 Da
MW_exp = 27,983.86 Da
In the zoomed-in peak around m/z ≈ 1473. If moving toward higher m/z and decreasing the charge by one for each peak, charge state can be estimated from isotope spacing.
Δm/z ≈ 0.053
hence, z = 1 / 0.053
z ≈ 19
Thus, we can observe the charge state for the zoomed-in peak in the intact eGFP mass spectrum.
Waters Part III — Peptide Mapping - primary structure
ExPASy PeptideMass tool with the parameters given in Figure 4 outputs 19 peptides.
The eGFP peptide map (Figure 5a) contains 21 chromatographic peaks between 0.5 and 6 minutes with >10% relative abundance. These are the labeled peaks at retention times (min):
0.61
0.79
1.20
1.43
1.80
1.85
1.93
2.17
2.26
2.54
2.78
3.27
3.53
3.59
3.70
4.30
4.48
4.64
4.87
5.06
5.43
All of them exceed 10% relative abundance.
No, they differ. There are slightly more observed chromatographic peaks than predicted peptides (21 vs. 19).
Some peptides may co-elute at the same retention time
Some peptides may be too low in abundance to detect
Some peptides may ionize poorly
Some peaks may include modified peptides, missed-cleavage products, or non-peptide signals
The chromatogram peak count is not always equal to the theoretical peptide count
The most abundant peak in the spectrum is at m/z 525.76712.
The isotope spacing is approximately 0.49206 ≈ 0.5 m/z
isotope spacing is approximately: 1 / z => 2
Thus, the most abundant peptide is 2+
By comparing determined [M+H]+ ≈ 1050.53 Da with the theoretical peptide masses from the ExPASy PeptideMass tool, the peptide eluting at 2.78 min most likely corresponds to: FEGDTLVNR. The theoretical monoisotopic mass ([M+H]+) is: 1050.5214 Da
ppm = (Δ / theoretical mass) × 1,000,000
ppm ≈ 5.25 ppm
From Figure 6, 88% of the eGFP sequence is confirmed by peptide mapping
Fragment masses (122.07, 214.09, 388.22, 501.31, 602.35, 537.25, 774.41, 903.44, 1050.52) match the b- and y-ion series for FEGDTLVNR. Key y-ions: y1 (R) = 175.12, y2 (NR) = 289.16, y7 (GDTLVNR), and the immonium ion at 122.07 corresponds to F (phenylalanine immonium). Confirms FEGDTLVNR
Waters Part IV — Oligomers
Oligomer
Expected Mass (MDa)
Peak in Fig.7 (MDa)
7FU Decamer
3.4
3.4
8FU Didecamer
8.0
~8.33
8FU 3-Decamer
12.0
12.67
8FU 4-Decamer
16.0
16.0
The 4.013 MDa peak is likely an 8FU decamer (10 × 400 = 4.0 MDa). The 0.1982, 0.79, 1.52 peaks are sub-decameric assemblies/free subunits.
Waters Part V — Did I Make GFP?
Theoretical
Observed / Measured on Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28.007
~28.097
~3,203 ppm
Theoretical mature eGFP-His₆ MW = 28,007 Da
Observed intact LC-MS MW = 28,097 Da
ppm error = |28,007 - 28,097| / 28,097 × 1,000,000
ppm error ≈ 3,203 ppm
Combined with 88% peptide map coverage and confirmed FEGDTLVNR fragmentation → yes, this is eGFP