Week 4 HW: Protein Design Part I
Conceptual Questions
How many amino acid molecules are in 500 g of meat?
Average amino acid ≈ 100 Da = 100 g/mol. 500 g meat (assuming mostly protein for estimation) ≈ 500 g protein ≈ 500 g / (100 g/mol) = 5 mol amino acids. Number of molecules = 5 × 6.02×10²³ ≈ 3.0 × 10²⁴ amino acid molecules (approximate).
Why humans do not become what they eat
Food proteins are broken down into amino acids during digestion. These amino acids are then reassembled into human-specific proteins according to human DNA instructions. The body does not preserve the original organism’s structure: instead, it uses the building blocks to synthesize its own molecules.
Why only 20 natural amino acids
The standard 20 amino acids represent an evolutionarily optimized set balancing chemical diversity, metabolic cost, and translational accuracy. This set provides sufficient structural and functional diversity for proteins while remaining compatible with ribosomal synthesis and tRNA charging systems. (That’s not a serious answer, but one of my professors joked thet there are only two laws in biology: i) it is the way it supposed to be, ii) it happened so. This is probably a second-law case)
Can we design non-natural amino acids?
Yes. Non-natural amino acids can be synthesized by modifying side chains or backbone structures. Examples include fluorinated amino acids (to increase stability), N-methyl amino acids (to restrict flexibility), and photo-reactive amino acids (e.g., azido groups for crosslinking). These are widely used in protein engineering and synthetic biology.
Origin of amino acids before life
Amino acids likely formed through prebiotic chemistry such as lightning-driven reactions (Miller–Urey experiment), hydrothermal vent chemistry, and extraterrestrial delivery via meteorites (e.g. Murchison meteorite). These processes produced simple organic molecules before enzymes existed.
Handedness of α-helix with D-amino acids
L-amino acids form right-handed α-helices, therefore, D-amino acids form left-handed α-helices.
Can we discover additional helices in proteins?
Yes. Beyond α-helices, proteins contain 3₁₀ helices, π-helices, and less common distorted helices. Computational modeling and structural biology continue to reveal new helical geometries, especially in membrane proteins and engineered peptides.
For instance, ε-Azido-lysine ((–N₃) on ε-amino group): azide interacts via click chemistry, such as strain-promoted azide-alkyne cycloaddition, enabling site-specific labeling, crosslinking, and drug conjugation. At the same time, it is inert to “natural” functional groups.
Why are most molecular helices right-handed?
Because life is built primarily from L-amino acids, and L-amino acids energetically favor right-handed α-helices due to steric constraints and optimal hydrogen bonding geometry. This chirality bias became fixed through evolutionary selection.
Why do β-sheets aggregate?
β-sheets expose alternating side chains that can form strong intermolecular hydrogen bonds. This allows extended stacking between sheets. Hydrophobic residues further drive aggregation by minimizing exposure to water, making aggregation energetically favorable.
Why do amyloid diseases form β-sheets? Can they be used as materials?
Under stress, mutation, aging, or chemical imbalance, some proteins fail to fold correctly. When misfolded proteins interact, they often rearrange into β-sheet-rich structures because β-sheets allow many strong hydrogen bonds between neighboring protein chains, maximize packing efficiency, cooperative stabilization between many molecules. So once aggregation begins, it becomes energetically favorable for more proteins to join the growing fibril
Misfolded protein strands → Alignment side-by-side → Intermolecular hydrogen bonding → Formation of extended β-sheets → Stacking into fibrils (amyloid fibers)
This stability is not always harmful. Amyloid β-sheets can be engineered as biomaterials, such as nanofibers, hydrogels, and scaffolds, though toxicity must be controlled. Their potential is promissing, since they are self-assembling nanoscale ordered structures.
β-sheet motif design
A stable β-sheet motif should include alternating hydrophobic and hydrophilic residues to promote proper side-chain packing.
Example sequence pattern: Val–Thr–Val–Thr–Val–Thr (repeating)
- Hydrophobic residues (Val) stabilize sheet core
- Polar residues (Thr) face solvent
- Multiple strands align antiparallel
- Interstrand hydrogen bonding forms stable sheet
- Edge residues capped to prevent uncontrolled aggregation
Protein Analysis and Visualization
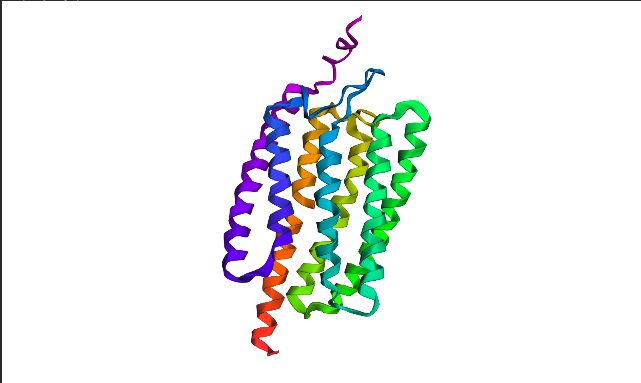
I chose Channelrhodopsin-2 (encoded by ChR2 gene) from Chlamydomonas reinhardtii. ChR2 is a light-gated ion channel and photoreceptor originally isolated from this green alga. When illuminated with blue light (maximally around 470 nm), ChR2 undergoes a conformational change that opens a pore, allowing positively charged ions (like sodium, calcium, and protons) to flow into the cell. Currently, I do a little research for my individual final project idea, and ChR2 might be relevant.
- ChR2 consists of 315 AA rsidues, 35 residues of which are leucine and 34 belong to glycine, making them the most frequent.
6EID_1|Chains A, B|Archaeal-type opsin 2|Chlamydomonas reinhardtii (3055) MDYGGALSAVGRELLFVTNPVVVNGSVLVPEDQCYCAGWIESRGTNGAQTASNVLQWLAAGFSILLLMFYAYQTWKSTCGWEEIYVCAIEMVKVILEFFFEFKNPSMLYLATGHRVQWLRYAEWLLTCPVILIHLSNLTGLSNDYSRRTMGLLVSDIGTIVWGATSAMATGYVKVIFFCLGLCYGANTFFHAAKAYIEGYHTVPKGRCRQVVTGMAWLFFVSWGMFPILFILGPEGFGVLSVYGSTVGHTIIDLMSKNCWGLLGHYLRVLIHEHILIHGDIRKTTKLNIGGTEIEVETLVEDEAEAGAVNKGTGK
This protein belongs to archaeal/bacterial/fungal opsin family. Uniprot BLAST finds 250 homologous sequences with identity range from 19.5% to 100%. 162 similar sequences belong to Archaea domain, 17—to Bacteria, 69 are found in distinct algae taxons, and just 2 belong to Fungi taxon.
Wild-type ChR2 3D structure at RCSB was released on 2017-12-06 and resolved at 2.39 Å scale.
Besides protein polymer, resolved structure includes 3 ligands: OLC ((2R)-2,3-dihydroxypropyl(9Z)-octadec-9-enoate), EDT [(BIS-CARBOXYMETHYL-AMINO)-ETHYL]-CARBOXYMETHYL-AMINO-ACETIC ACID), and phosphate.
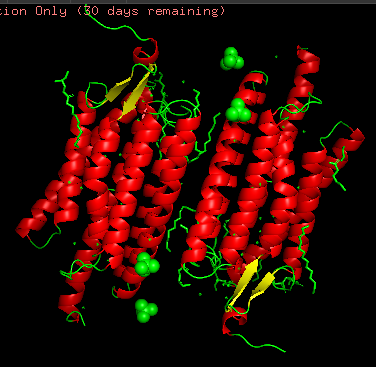
Structurally, this protein belongs to 7-transmembrane (7TM) α-helical membrane proteins.
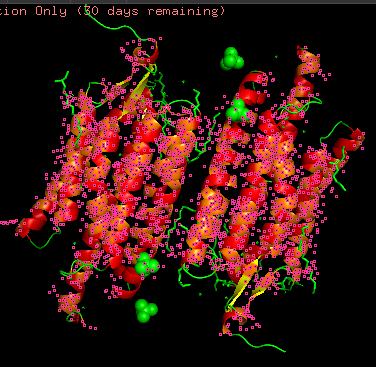
Protein structure visualized as “cartoon”, “ribbon”, and “balls & spheres (with different diameter)” respectively:
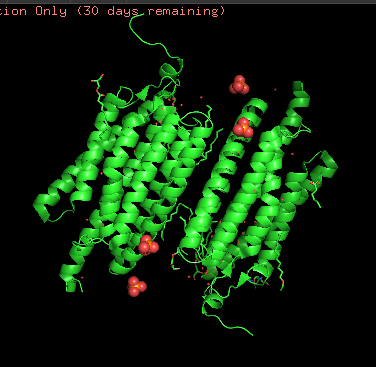
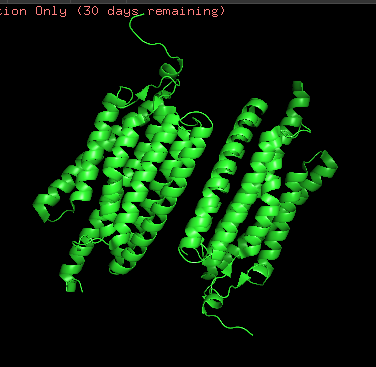
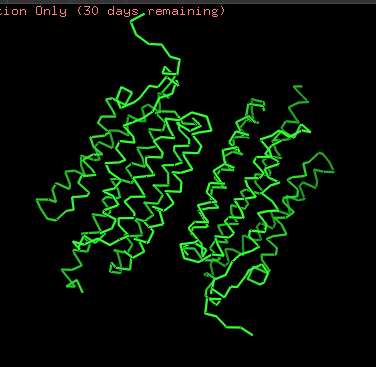
- Coloring the protein by secondary structure. As it follows from structure family name, it has many α-helices
- Coloring the protein by residue type. Being a transmembrane protein, Chr2 has hydrophobic residues. Hydrophobic residues are highly abundant and clustered in long continuous regions, consistent with transmembrane helices. Hydrophilic residues are more exposed on the surface and likely participate in solvent interactions. Hydrophobic vs. hydrophilic residues indication is shown below

- ChR2 surface visualization. It doesn’t actually show any obvious pockets, but contains narrow central channel and ligand binding sites
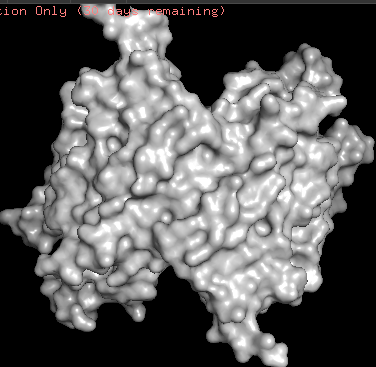
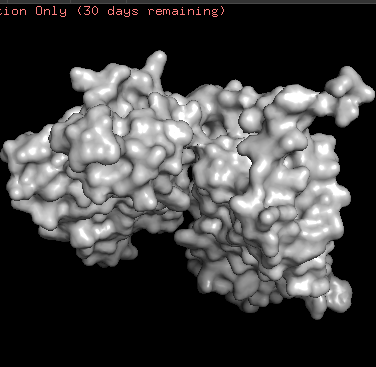
Using ML-Based Protein Design Tools
Protein Language Modeling
Deep Mutational Scans
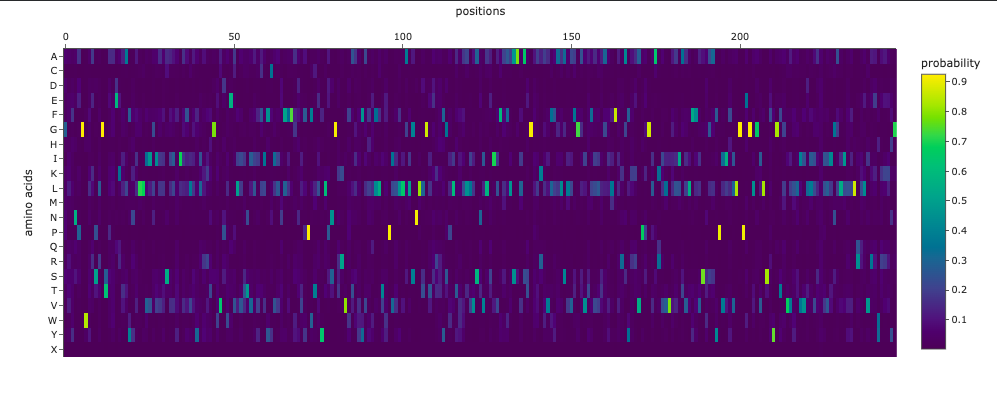
The resulting heatmap shows:
- Many bright yellow spots in leucine row at various positions. L is the most frequent residue
- Multiple bright yellow spots at position 27, predominantly regarding P, Q, R, S, T amino acids
- Almost all the W-row is blue or dark blue
- Mostly blue columns at 16-18, 53-58, 71-72 positions
- The heatmap ends on position 72, although there are 315 AA
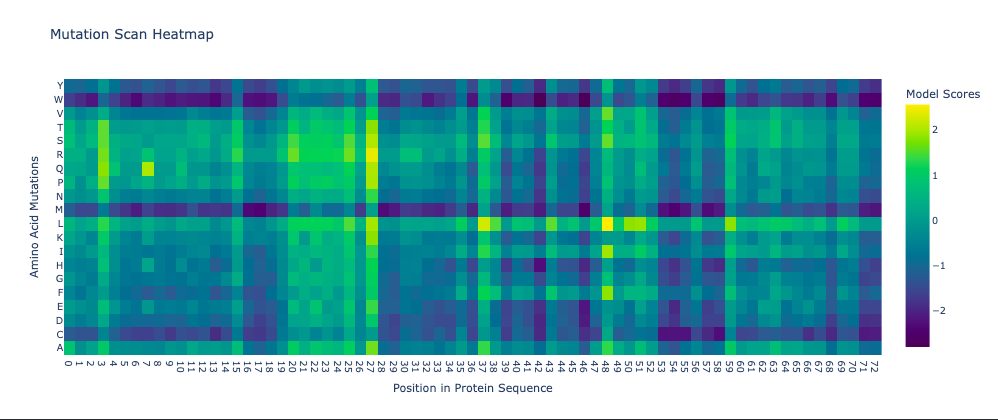
- Leucine (L) is strongly favored in hydrophobic environments, stabilizing protein cores and helices. It is very common in membrane proteins and buried regions, so it’s expected.
- Multiple tolerated substitutions at position 27. Probably, this position is structurally permissive or surface-exposed. Since bright yellow spots are concentrated in P, Q, R, S, T rows, it is likely on the protein surface or in a loop region. P (Proline) introduces kink suggests flexibility allowed. Q (Glutamine) is polar, surface-friendly. R (Arginine) is charged, usually solvent-exposed. S/T (Serine/Threonine) are small polar, often in loops.
- W (Tryptophan) is a very large bulky aromatic residue, sterically expensive, requiring space and hydrophobic packing. So, this is relevant for many proteins and not surprising.
- 16-18, 53-58, 71-72 columns represent likely structurally constrained positions. It could mean both protein core / folding nucleus and transmembrane helix core.
- It seems like ether an tokenizer is not aligned with amino-acid sequence length—which is doubtful—OR the algorythm reads only one of two polypeptide chain. Previous visualization show that ChR2 is a dimer.
Latent Space Analysis
Protein Folding
ESMFold visualized only a half of the functional protein, only one polypeptide chain. However, the chain itself looks pretty much like in PyMOL visualizations. The model is rather confident with ptm score of 0.751 and plddt of 75.483.
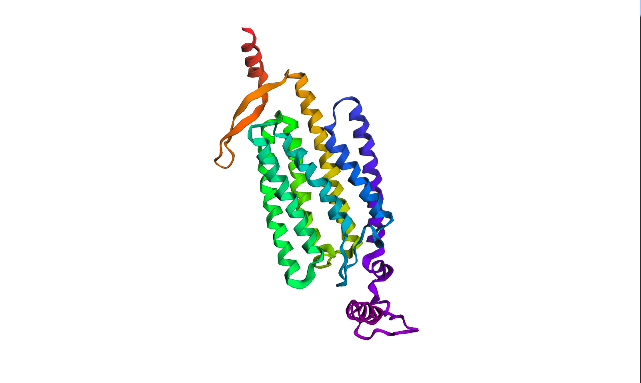
I inserted Lysine (K)—positively charged AA—into region that is supposed to be conservative according to Mutation Heatmap (near 50s positions). A51K point mutation sequence:
MDYGGALSAVGRELLFVTNPVVVNGSVLVPEDQCYCAGWIESRGTNGAQTKSNVLQWLAAGFSILLLMFYAYQTWKSTCGWEEIYVCAIEMVKVILEFFFEFKNPSMLYLATGHRVQWLRYAEWLLTCPVILIHLSNLTGLSNDYSRRTMGLLVSDIGTIVWGATSAMATGYVKVIFFCLGLCYGANTFFHAAKAYIEGYHTVPKGRCRQVVTGMAWLFFVSWGMFPILFILGPEGFGVLSVYGSTVGHTIIDLMSKNCWGLLGHYLRVLIHEHILIHGDIRKTTKLNIGGTEIEVETLVEDEAEAGAVNKGTGK
Not being very high initialy, the folding prediction confidence didn’t decrease much: to ptm: 0.740, plddt: 74.659.
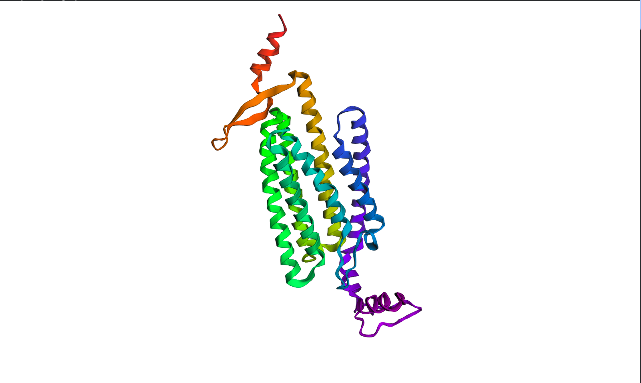
Larger segment mutation—insert of of KKKKKKKKKKK instead of A51—lead to even higher plddt of 75.643 and slightely decreased ptm: 0.737.
MDYGGALSAVGRELLFVTNPVVVNGSVLVPEDQCYCAGWIESRGTNGAQTKKKKKKKKKKKSNVLQWLAAGFSILLLMFYAYQTWKSTCGWEEIYVCAIEMVKVILEFFFEFKNPSMLYLATGHRVQWLRYAEWLLTCPVILIHLSNLTGLSNDYSRRTMGLLVSDIGTIVWGATSAMATGYVKVIFFCLGLCYGANTFFHAAKAYIEGYHTVPKGRCRQVVTGMAWLFFVSWGMFPILFILGPEGFGVLSVYGSTVGHTIIDLMSKNCWGLLGHYLRVLIHEHILIHGDIRKTTKLNIGGTEIEVETLVEDEAEAGAVNKGTGK
Probably, initially moderate confidence of the model does not allow to catch specific mutation induced folding destabilization. Alternatively, even this mutation did not affect critical region.
Protein Generation
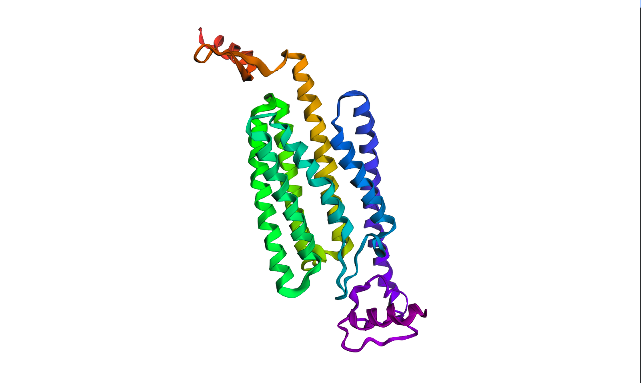
Using ProteinMPNN (model v_48_020, sampling temperature 0.1), I performed ChR2 (PDB ID: 6EID) inverse folding. The resulting sequence recovery score is 0.4024. meaning 40% of the original amino acids were reproduced by the model. That could mean many ChR2 positions are not strictly sequence-specific, its structure can tolerate multiple amino acids, and the backbone constraints are more important than exact residues.
The new sequence consists of only 247 AA in contrast to original ChR2. Visualization looks pretty much similar, althoug one of the terminal regions was noticeably dropped. However, new ptm is 0.866 and plddt is 84.774 vs. 0.751 and 75.483 of original sequence. ProteinMPNN has probably replaced “hard-to-fit” residues with those that are more structurally compatible to “general vision” of this protein.