Week 5 HW: Protein Design Part II
SOD1 Binder Peptide Design (From Pranam)
Generate Binders with PepMLM
Human SOD1 sequence:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutant SOD1 sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I inserted mutant sequence and set 12 as peptide length in PepMLM Google Colab code. To compare FLYRWLPSRRGG binding peptide pseudo perplexity, I added another code cell into Google Colab:

Here are the downloaded results rounded to hundredths:
| Binder | Pseudo Perplexity |
|---|---|
| KRYYVTGVRLKK | 30.16 |
| HRYPAVGVEHKX | 15.33 |
| WRYPAAAVAWWX | 10.03 |
| WRYPAAALAWKE | 11.92 |
| FLYRWLPSRRGG | 20.64 |
WRYPAAAVAWWX showed the lowest perplexity (10.03) upon this peptide-generation session, meaning highest plausibility. Interestingly, experimentaly approved FLYRWLPSRRGG showed the second lowest perplexity, whereas “the best” variant contains X (undefined) amino acid making it should be carefully interpreted. As the current LLMs’ outputs in general.
Evaluate Binders with AlphaFold3
Binders 2(HRYPAVGVEHKX) and 3 (WRYPAAAVAWWX, with the lowest pseudo perplexity) were excluded from the analyzis because of invalid “X” character.
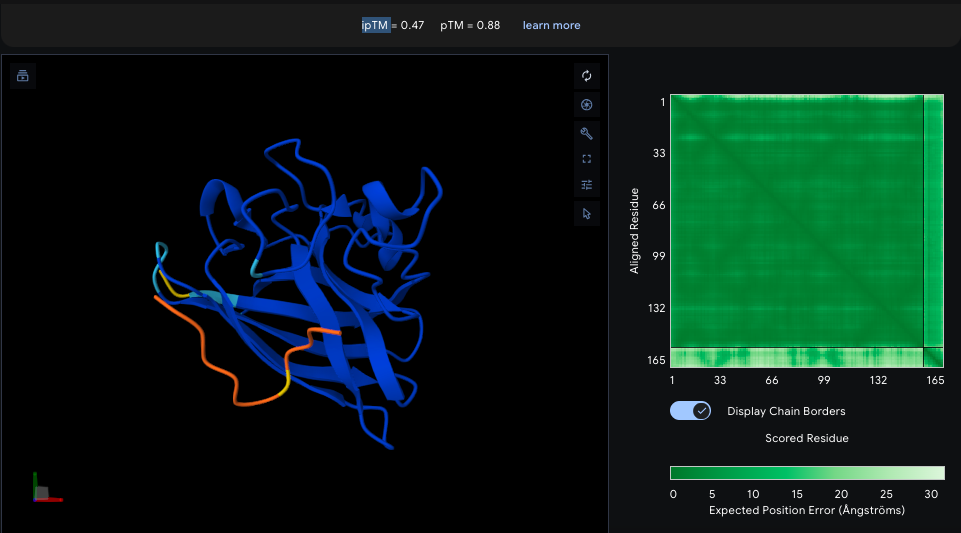
Binder 1 (KRYYVTGVRLKK), ipTM = 0.47:
Binder 4 (WRYPAAALAWKE), ipTM = 0.36:

Binder 5, experimentaly verified (FLYRWLPSRRGG), ipTM = 0.34:

Evaluate Properties of Generated Peptides in the PeptiVerse
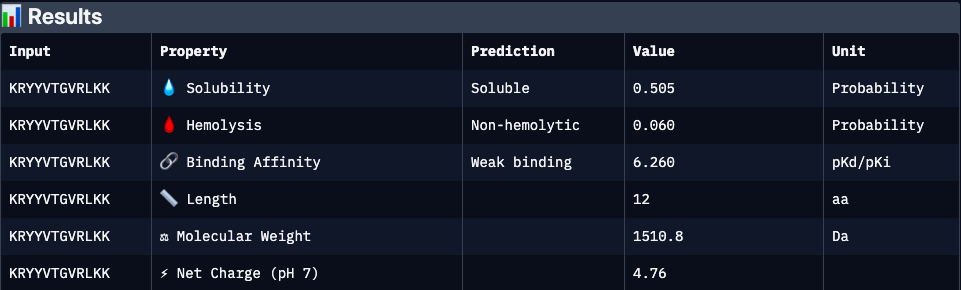
Binder 1 (KRYYVTGVRLKK):
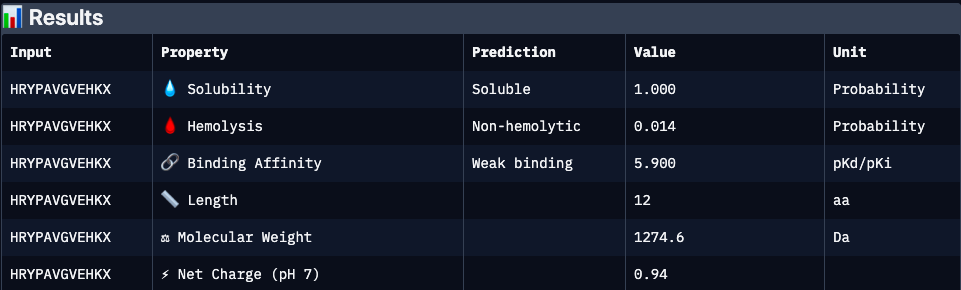
Binder 2 (KRYYVTGVRLKK):
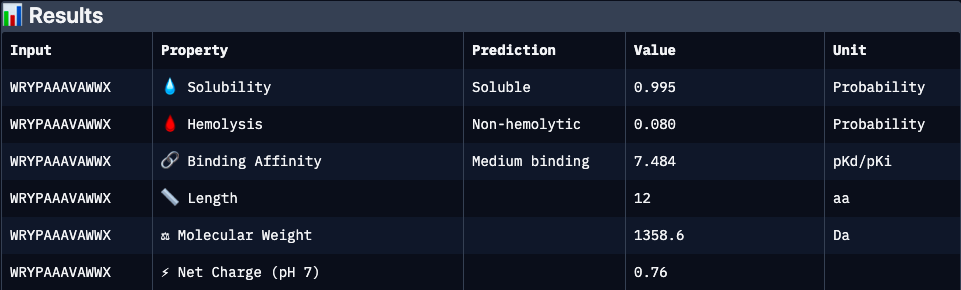
Binder 3 (WRYPAAALAWKE):
Binder 4 (WRYPAAALAWKE):
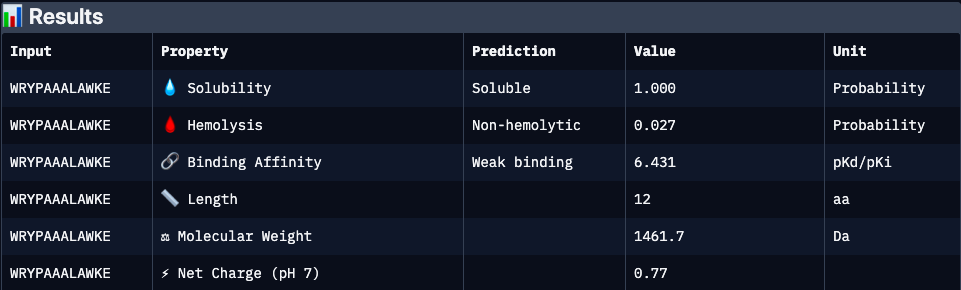
Binder 5, experimentaly verified (FLYRWLPSRRGG):
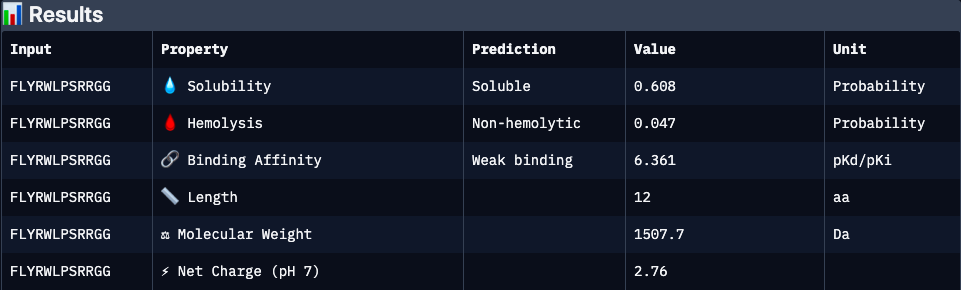
| Binder | ipTM | Binding Affinity | Solubility | Hemolysis | Net Charge (pH 7) | Molecular Weight | Interpretation |
|---|---|---|---|---|---|---|---|
| KRYYVTGVRLKK | 0.47 | 6.26 | 0.51 | 0.06 | 4.76 | 1510.8 | Moderate structural binding support; strongly cationic peptide may enhance target interaction but raises risk of nonspecific electrostatic binding and moderate cytotoxicity. Solubility is acceptable but not optimal |
| HRYPAVGVEHKX | N/A | 5.9 | 1.00 | 0.01 | 0.94 | 1274.6 | Excellent developability profile with very high solubility and minimal hemolysis; however, absence of structural validation (ipTM unavailable) and weaker predicted affinity make binding confidence uncertain |
| WRYPAAAVAWWX | N/A | 7.48 | 0.99 | 0.08 | 0.76 | 1358.6 | Strongest predicted affinity among candidates and highly soluble, but elevated hydrophobic/aromatic content may contribute to increased hemolytic potential and aggregation risk. Requires structural validation |
| WRYPAAALAWKE | 0.36 | 6.43 | 1.00 | 0.03 | 0.77 | 1461.7 | Balanced candidate with excellent solubility, low hemolysis, and moderate predicted affinity; relatively low ipTM suggests uncertain interface stability despite favorable developability properties |
| FLYRWLPSRRGG | 0.34 | 6.36 | 0.61 | 0.05 | 2.76 | 1507.7 | Experimentally validated binder with moderate predicted affinity and cationic character. Lower predicted solubility and interface confidence may reflect dynamic or partially disordered binding interactions rather than absence of activity |
Generate Optimized Peptides with moPPIt
moPPIt was run targeting hydrophobic motif positions 3–9 of A4V SOD1 (KVVCVLK). 5 peptide samples of 12 residues were generated considering Hemolysis, Solubility, Affinity, and Motif parameters optimization.
| Peptide | Hemolysis | Solubility | Binding Affinity | Motif | Interpretation |
|---|---|---|---|---|---|
| CTSGVNVGPGVP | 0.04 | 0.99 | 6.11 | 0.63 | Strong developability profile with very low predicted hemolysis and excellent solubility. Moderate motif alignment suggests partial but potentially indirect engagement of the hydrophobic KVVCVLK region; likely acts via peripheral or transient interaction rather than deep motif mimicry |
| ADSEIKAPSSGH | 0.08 | 1.00 | 5.52 | 0.67 | Highly favorable physicochemical properties with maximal solubility and low hemolysis risk. Motif similarity is moderate, suggesting a more electrostatically driven or scaffold-like interaction rather than direct hydrophobic motif mimicry |
| RSKYQWVPYHVT | 0.04 | 1.00 | 6.31 | 0.49 | Balanced candidate with strong predicted affinity and good solubility, but lower motif score indicates weaker structural motif mimicry. Likely binds through mixed hydrophobic–aromatic interactions rather than targeted motif recognition |
| SFAGICNVEQQT | 0.05 | 1.00 | 5.95 | 0.75 | Best overall motif alignment among the set, suggesting strongest intended targeting of the KVVCVLK hydrophobic patch. Favorable solubility and low hemolysis support developability, with potential for more direct motif-driven binding |
| QEPCEELQFNHF | 0.02 | 0.51 | 6.26 | 0.66 | Strong predicted affinity and acceptable motif similarity, but reduced solubility may limit practical usability. Likely engages target through distributed polar–aromatic interactions rather than strict hydrophobic motif complementarity |
Final Project: L-Protein Mutants
The goal of this project is to design mutant MS2 lysis protein (L-protein) to reduce its interaction with the bacterial chaperone DnaJ. Since DnaJ is important for proper folding and processing of the L-protein, weakening this interaction may help the phage remain functional even if bacteria modify DnaJ. To estimate this interaction, co-folding predictions were performed using AlphaFold2 Multimer with both proteins entered together. In AlphaFold2, both sequences are inserted into a single input field separated by a colon “:”. Since transmembrane domain affects the L-protein lysis activity, mutations were intoduced in N-terminal region.
L-Protein Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Otion 1: Mutagenesis
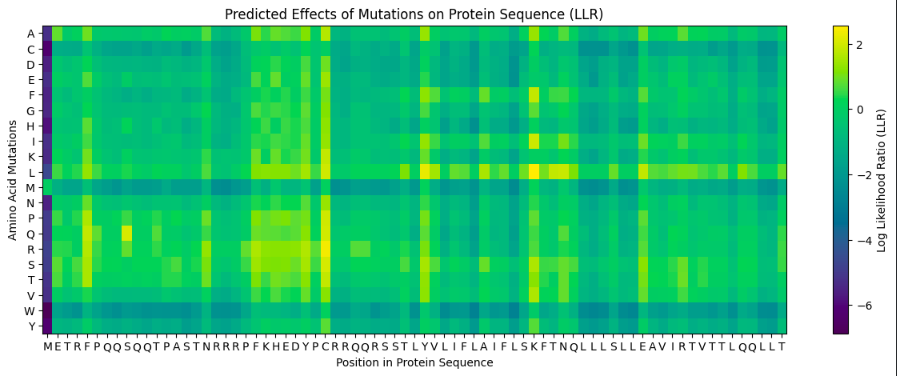
According to resulting heatmap and L-Protein Mutants Table data, I chose the following mutations:
- F5T
- S9Q
- C29S
- Y39L
- K50L
Mutant L-Protein Sequence:
METRTPQQQQQTPASTNRRRPFKHEDYPSRRQQRSSTLLVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
AlphaFold2-multimer-v3 querry:
METRTPQQQQQTPASTNRRRPFKHEDYPSRRQQRSSTLLVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR