Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When you eat beef or fish, your body does not keep the meat intact and turn it into “cow tissue” or “fish tissue.” Instead, your digestive system breaks everything down into basic molecules, like proteins into amino acids, fats into fatty acids + glycerol, carbohydrates into simple sugars and DNA into nucleotides.
Why are there only 20 natural amino acids?
Life could have used more (and sometimes does), but 20 appears to be a near-optimal balance. But why is that so? The Genetic Code has limits. Proteins are built using codons — 3-letter sequences in DNA/RNA. ➜ 4 bases (A, U/T, C, G), 3 positions per codon, 4³ = 64 possible codons. But 3 are stop signals and many codons are redundant (multiple codons for the same amino acids). Therefore, The code settled on 20 standard amino acids early in evolution and became highly conserved. Changing the code would break nearly all exiting proteins and would be catastrophically disruptive!
Can you make other non-natural amino acids? Design some new amino acids.
Synthetic biology and medicinal chemistry routinely create non-natural amino acids, and some are even genetically encoded in engineered organisms. Some are chemically synthesized and incorporated during peptide synthesis, while others are genetically encoded using engineered tRNA/synthetase systems.
One amino acid would be Alkyne-Lysine (Bioorthogonal Handle). The lysine’s side chain is modified to include a terminal alkyne. Alkynes allow click chemistry (azide–alkyne cycloaddition), site-specific labeling and fluorescent tagging. This amino acid could be used in protein imagin, drug conjugationg and synthetic protein networks.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids are within all living things on Earth, being the building blocks of proteins. Proteins are essential for many processes within living organisms, including catalysing reactions (enzymes), replicating genetic material (ribosomes), transporting molecules (transport proteins) and providing a structure to cells and organisms (e.g. collagen). Therefore, amino acids would have been needed in significant amounts within the region where life began on Earth. The Miller–Urey Experiment (1953) showed, that organic molecules can spontaneously form under plausible early-Earth conditions. Chemists simulated early Earth’s atmosphere and within days, the flask contained amino acids like Glycine, Alanine and Aspartate. Without enzymes or cells, just chemistry. That means, enzymes didn´t invent amino acids. Instead, Geochemistry made amino acids, those amino acids accumulated, some began forming short peptides, eventually, self-replicating systems emerged and only later did enzyme-based metabolism evolve.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If you build an α-helix entirely from D-amino acids, it will form a left-handed helix. The reason why is that natural proteins use L-amino acids. In Biology, almost all amino acids are L and standard α-helices in proteins are right-handed. D-amino acids are mirror images of L-amino acids. So if you build A peptide from L residues ➜ right-handed α-helix and the exact mirror molecule (all D residues) must adopt the mirror conformation. Therefore, the entire structure inverts and the mirror image of a right-handed helix is a left-handed helix.
Can you discover additional helices in proteins?
Yes, and in fact, there already has been discovered additional helices beyond the standard α-helix. But whether new helices can exist is a deeper structural question. Other helices are: 3₁₀ Helix and π-Helix. There can be more, but it is very constrained.
Why are most molecular helices right-handed?
Most molecular helices in biology are right-handed because life uses L-amino acids, and L stereochemistry makes the right-handed α-helix energetically favored.
Why do β-sheets tend to aggregate?
β-sheets aggregate because their backbone hydrogen bonding is unsatisfied at the edges, and the easiest way to satisfy it is by binding to another β-sheet.
8.1 What is the driving force for β-sheet aggregation?
The driving force for β-sheet aggregation is driven by a combination of backbone hydrogen bonding, hydrophobic interactions, and water-mediated entropy effects, with cooperativity making it autocatalytic.
Why do many amyloid diseases form β-sheets?
Amyloid diseases form β-sheets because β-sheets have exposed hydrogen-bonding edges at misfolded regions, β-strands are geometrically compatible with stacking and fibril formation, hydrophobic and polar side chains stabilize sheet stacking, cross-β fibrils represent a low-energy, highly stable state and misfolding exposes β-prone sequences that nucleate aggregation.
9.1 Can you use amyloid β-sheets as materials?
Yes, amyloid β-sheets are not just pathological; their structural properties make them ideal building blocks for engineered materials. Some amyloid-based materials are:
- Hidrogels ➜ Short amyloidogenic peptides form cross-β networks in water and creates soft, viscoelastic gels that can be used in tissue engineering scaffolds, drug delivery systems and 3D cell culture matrices.
- Nanofibers and Films ➜ Amyloid fibrils can be aligned to make strong, thin fibers and they can be embedded in composites for e.g. biocompatible electronics
- Functionalized Materials ➜ Side chains can be chemically modified to bind metals, fluorophores, or enzymes and enables catalytic amyloid materials, light-responsive materials, and sensing platforms
Part B: Protein Analysis and Visualization
I selected the human hemoglobin because it is a crucial and very well-known protein that transports oxygen from the lungs to tissues and carbon dioxide back to the lungs.
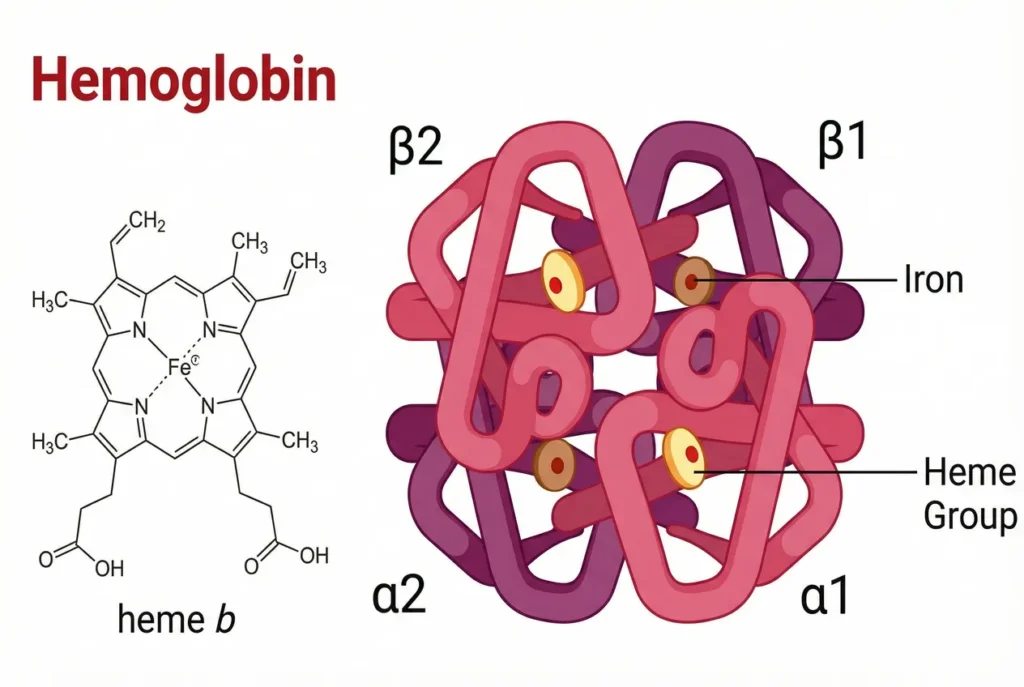
The structure of hemoglobin. Source: https://chemistwizards.com/wp-content/uploads/2026/01/hemoglobin-structure-1024x687.webp
- 🩸Sequence (from FASTA):
sp|P69905|HBA_HUMAN Hemoglobin subunit alpha OS=Homo sapiens OX=9606 GN=HBA1 PE=1 SV=2 MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHG KKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTP AVHASLDKFLASVSTVLTSKYR
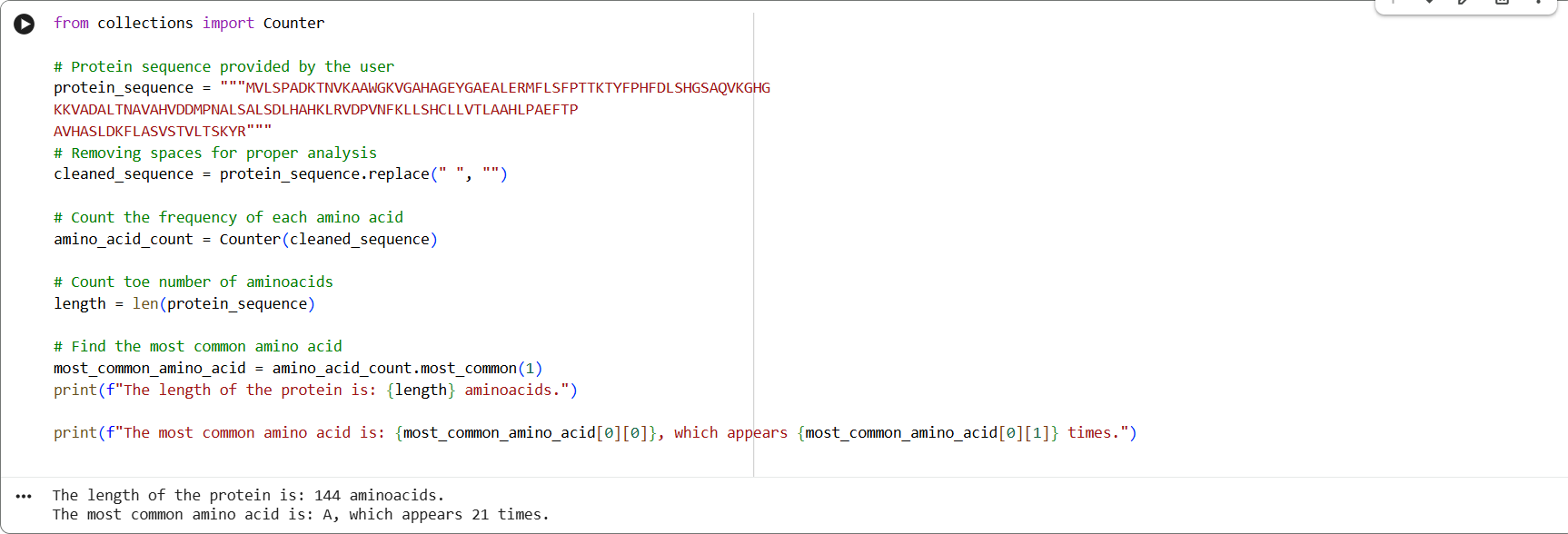
🩸This is the frequency of amino acids from Google Colab:
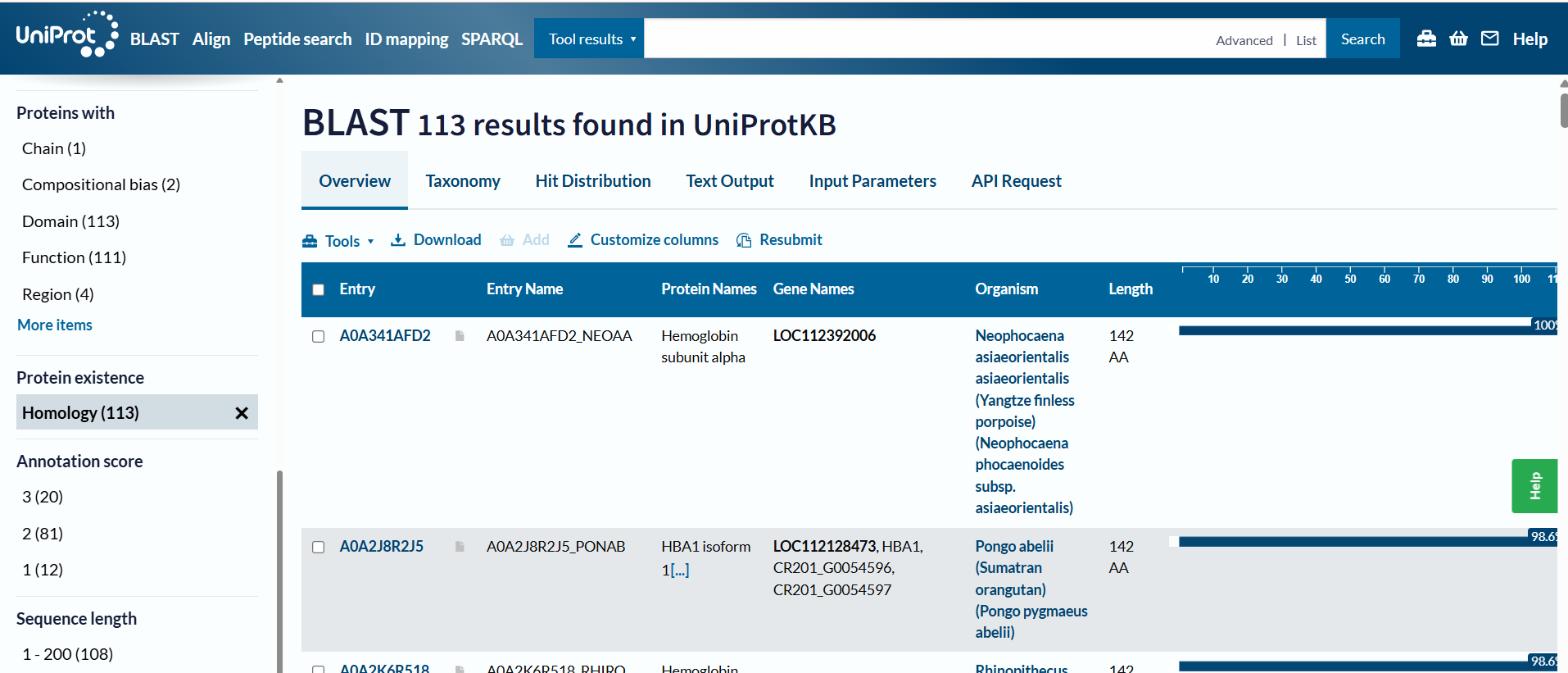
🩸On UniProt´s Blast tool, it showed that there are 113 protein sequence homologs.
🩸Hemoglobin belongs to the globin superfamily, which is a large group of proteins that bind heme and transport or store oxygen. Some common features of globins are globin fold, heme-binding pocket and conserved residues. Also, within the globin superfamily, hemoglobin has a subfamily ➜ Alpha-globin and Beta-globin.
- 🩸The structure from RCSB was released in 1998-04-29. The resolution is 1.80 Å
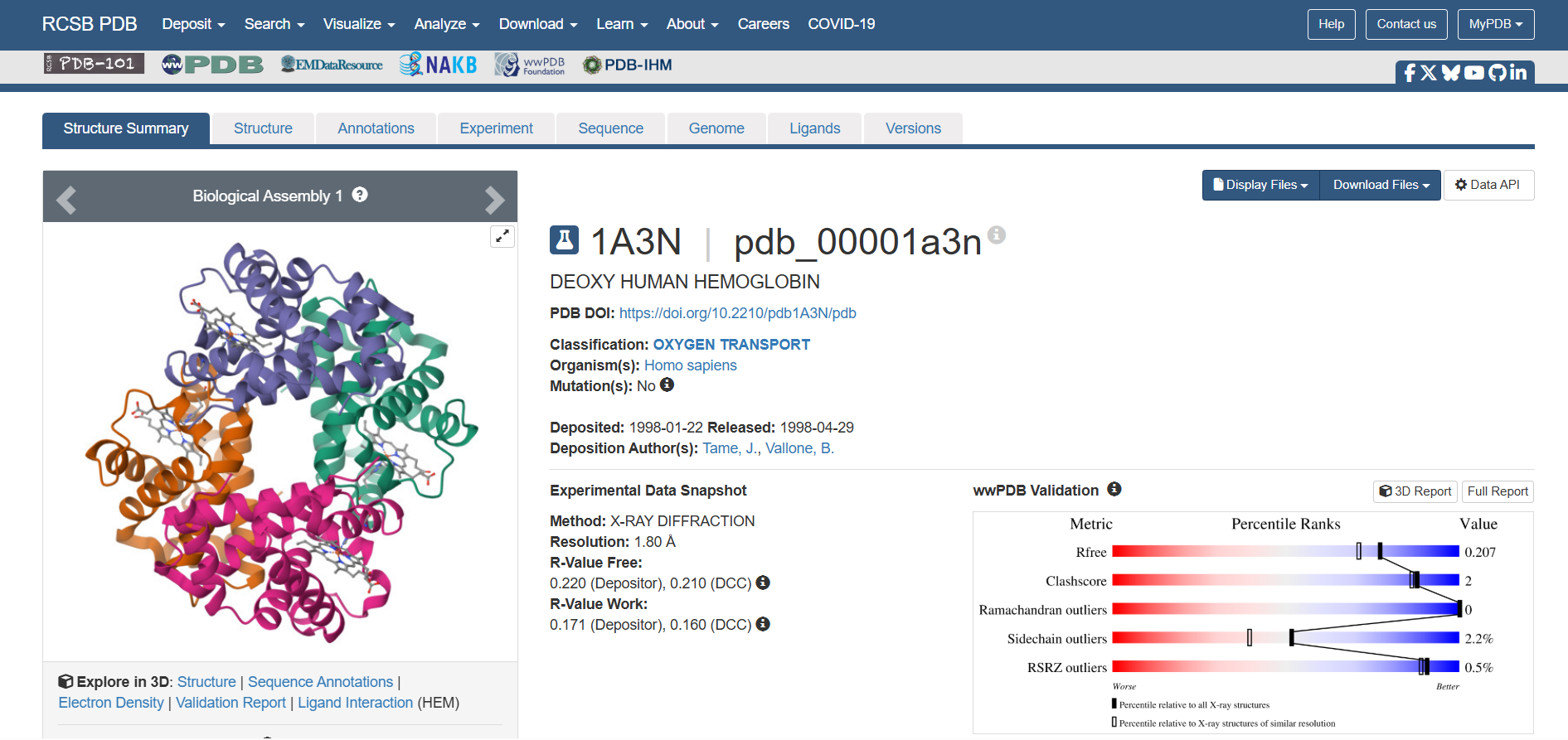
🩸There is a molecule in the structure. A ligand called “PROTOPORPHYRIN IX CONTAINING FE”
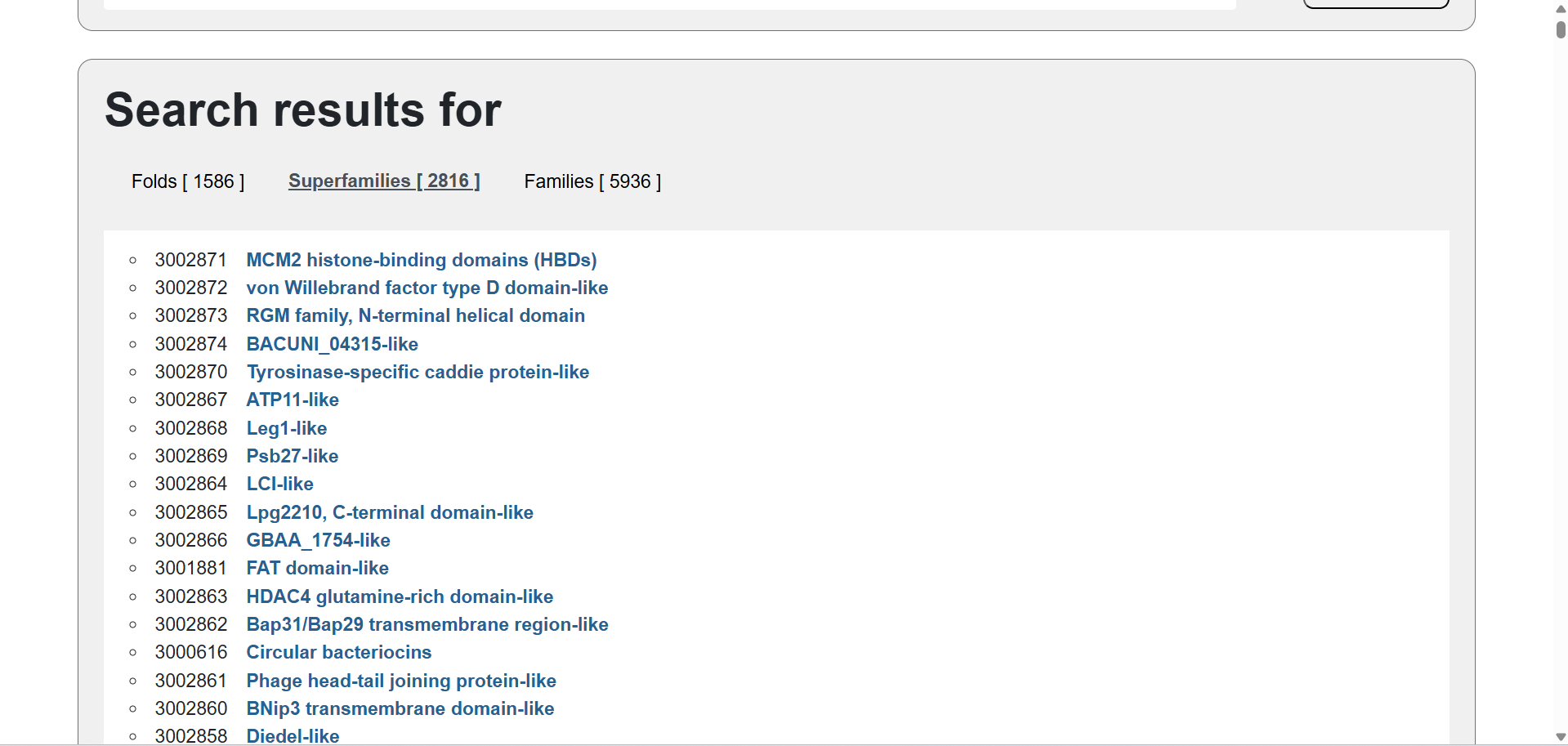
🩸SCOP showed me the following structure classification families:
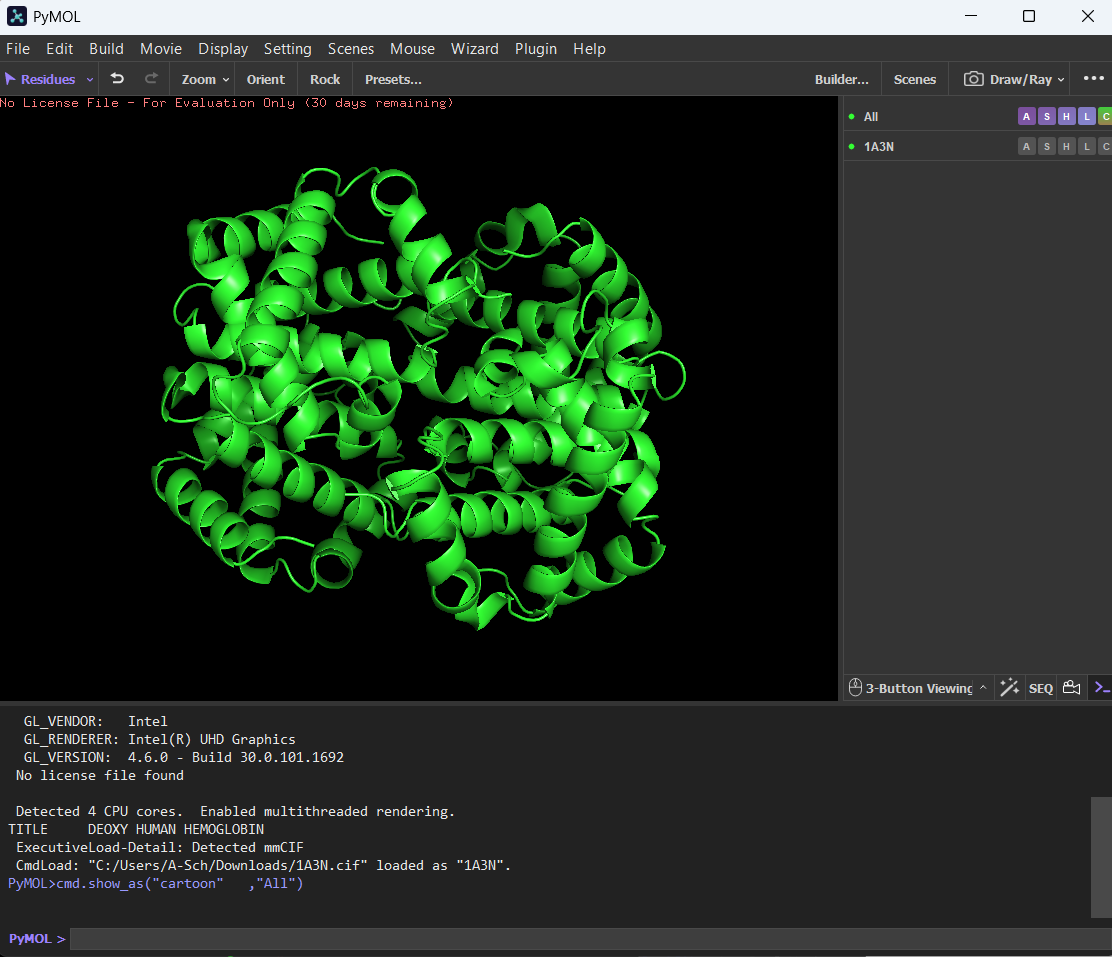
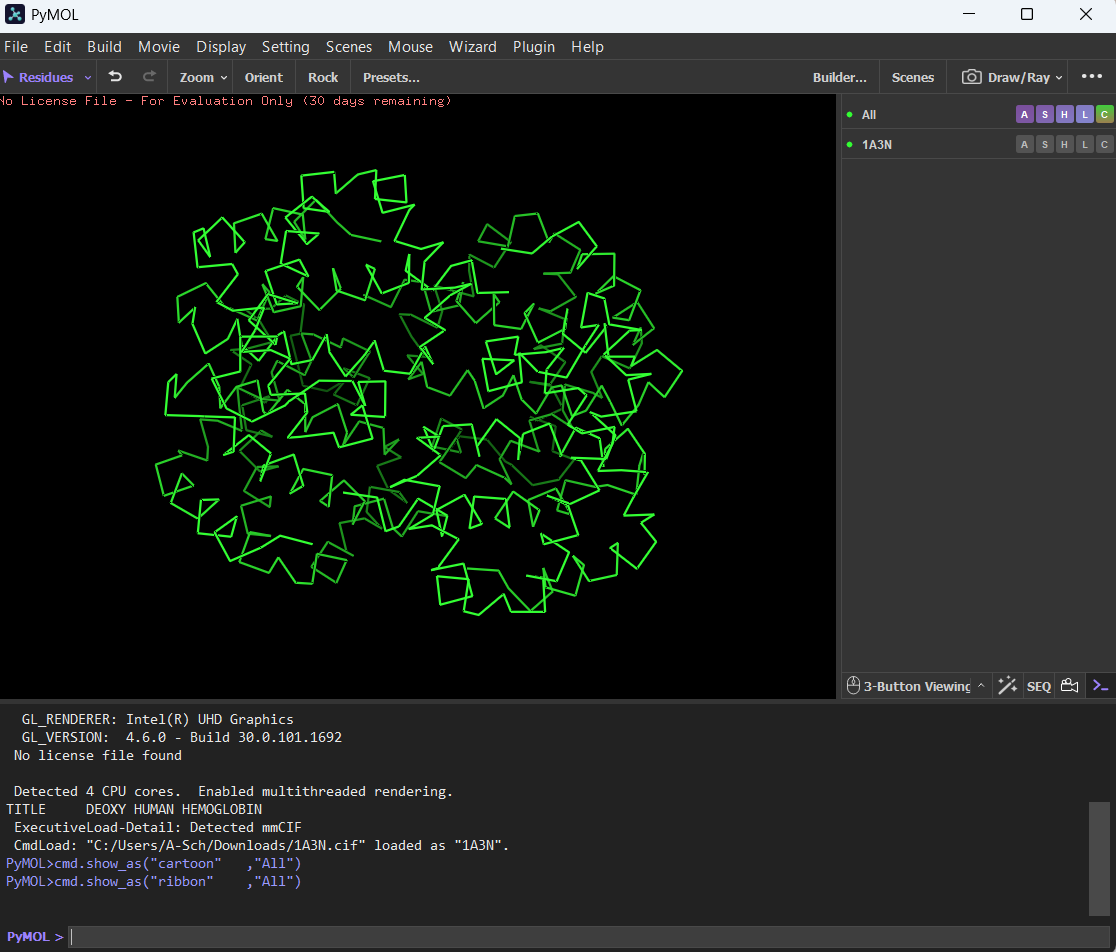
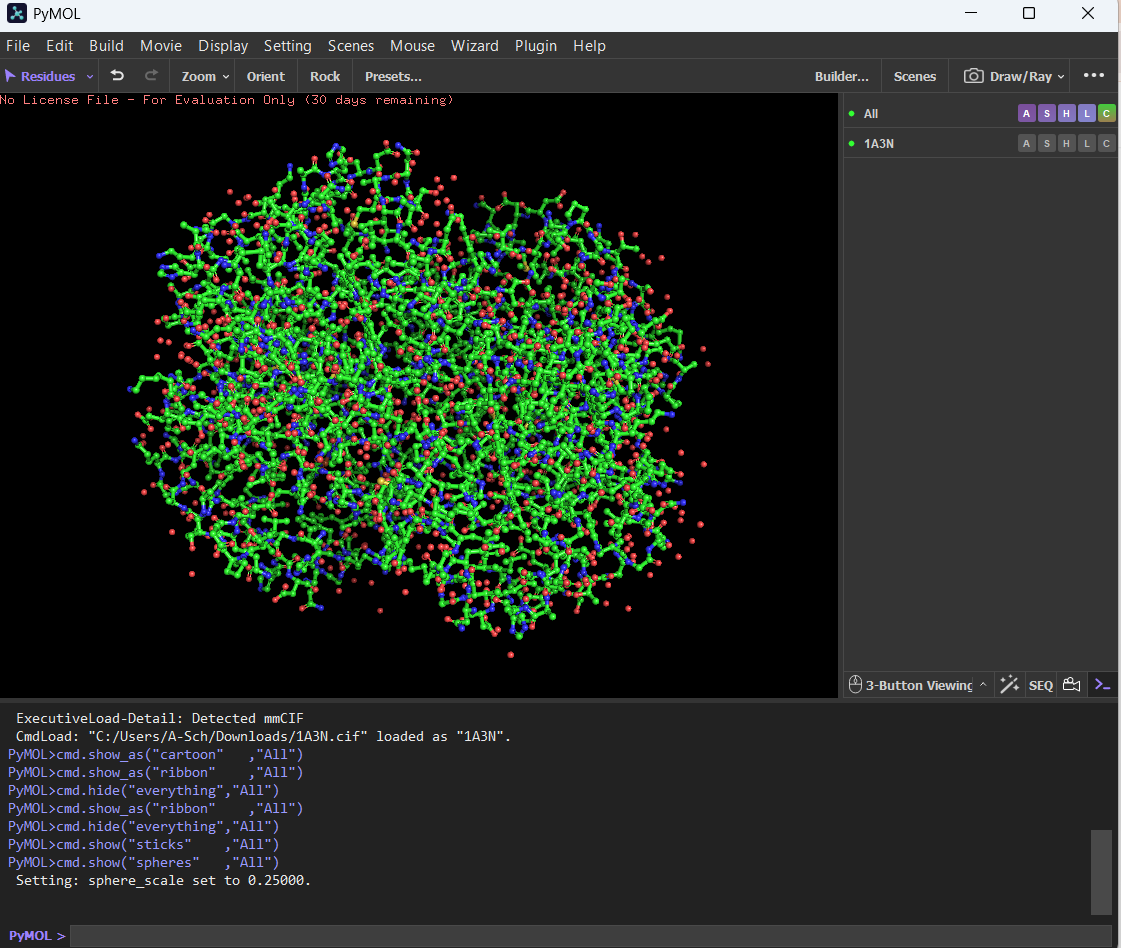
🩸Visualizing the protein on PyMol as:
Cartoon:
Ribbon:
Ball and Stick:
🩸By coloring the protein by secondary structure, it showed more helices than sheets.
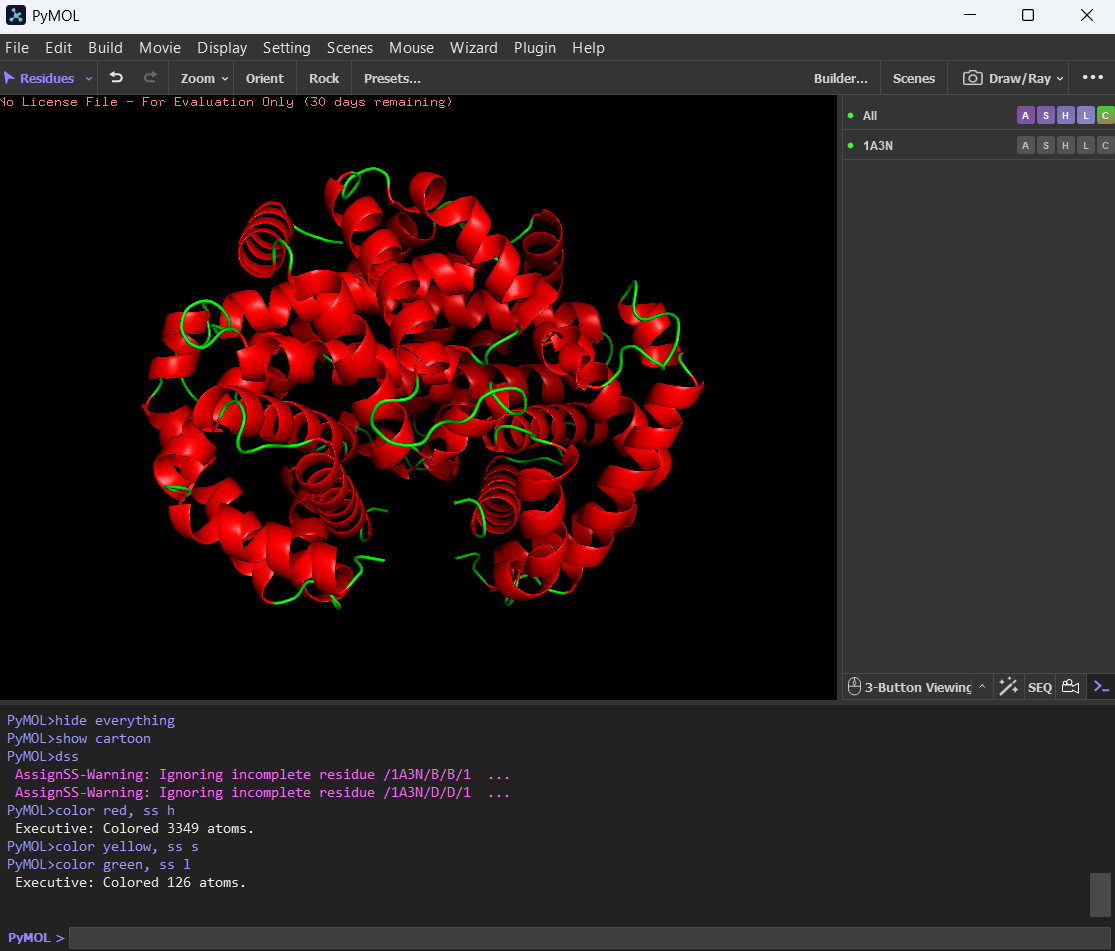
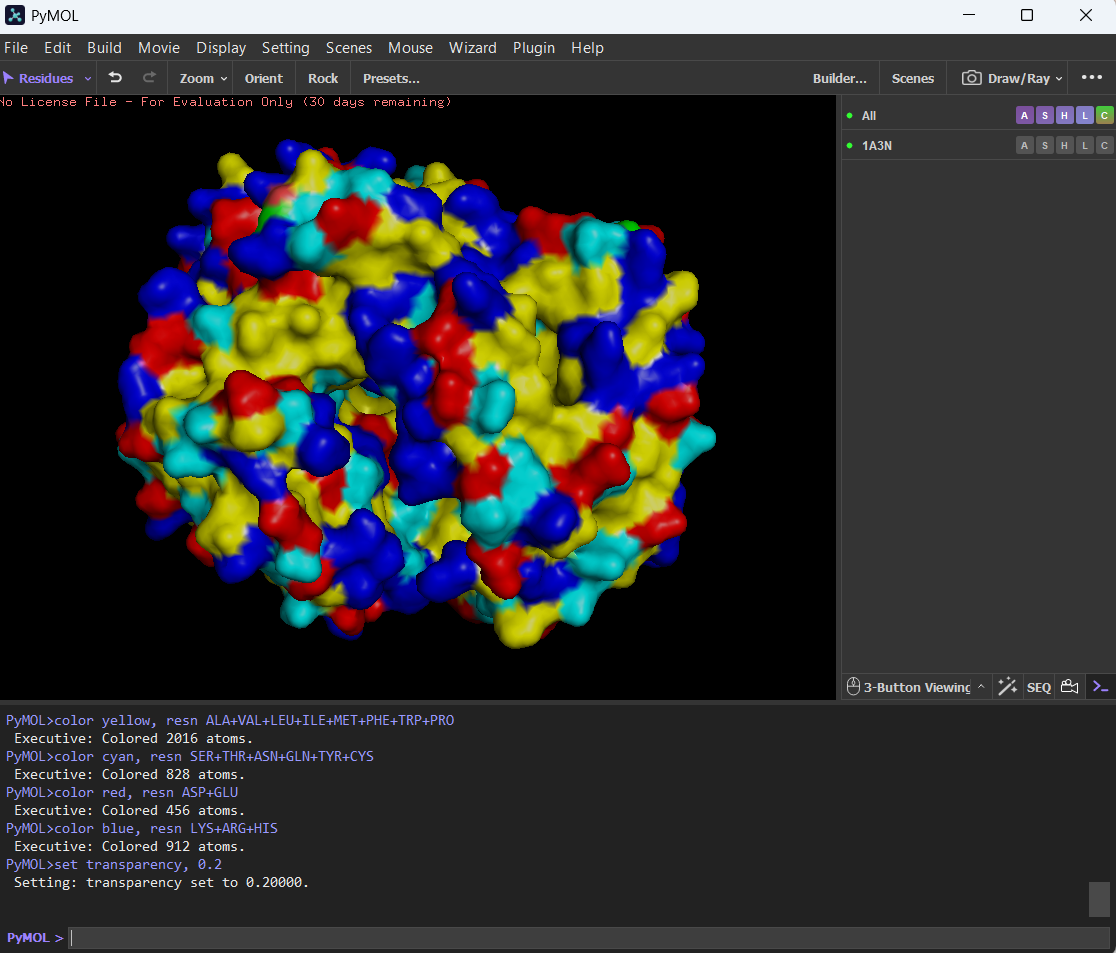
🩸The hydrophobic residues are (color yellow in image):
ALA, VAL, LEU, ILE, MET, PHE, TRP, PRO
The hydrophilic residues are (color cyan in image):SER, THR, ASN, GLN, TYR, CYS
And the charged residues (also hydrophilic):ASP, GLU (negative, color red in image), LYS, ARG, HIS (positive, color blue in image)
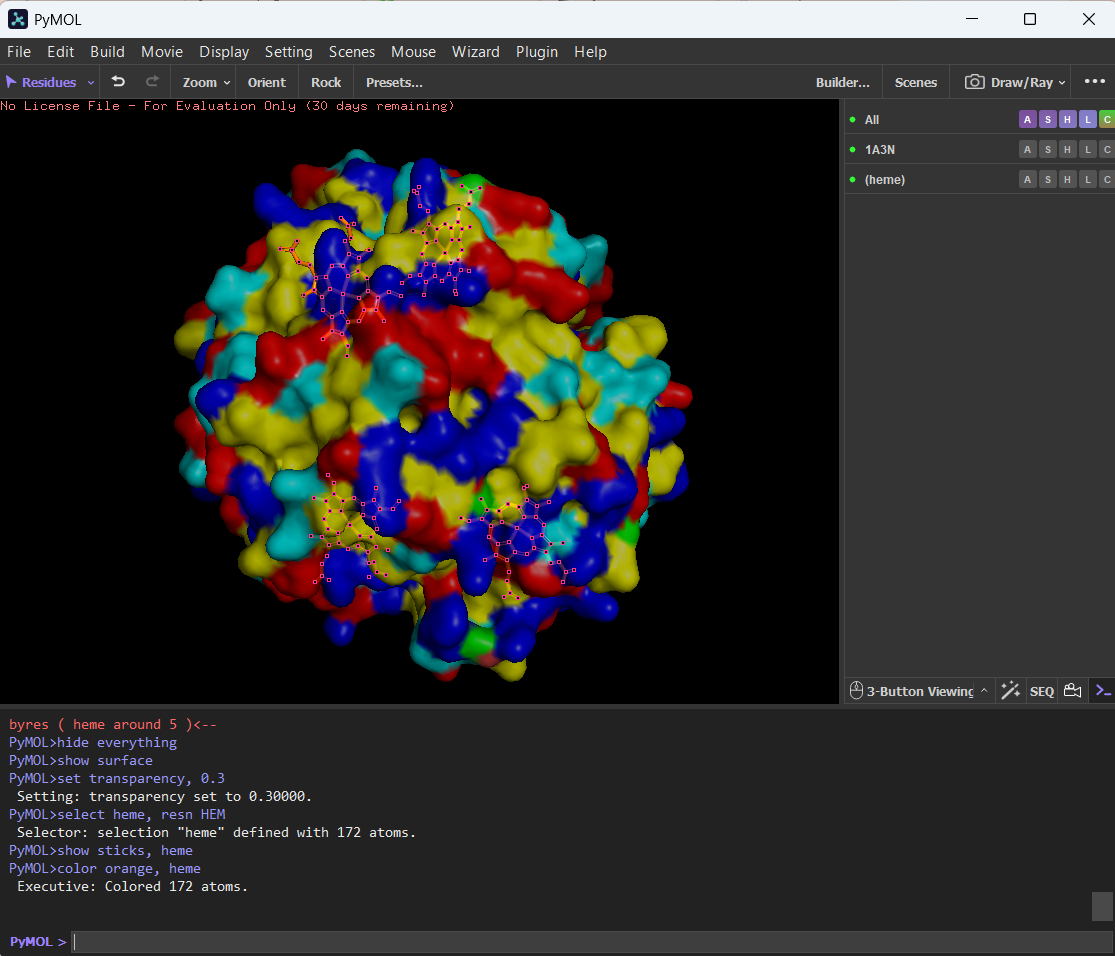
🩸Here you can see the surface of hemoglobin and the cavity (binding pocket) where the heme sits.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
For this part, I chose the lysozyme protein from PDB. Sequence:
168L_1|Chains A, B, C, D, E|T4 LYSOZYME|Enterobacteria phage T4 (10665) MNIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAKSELDKAIGRNCNGVITKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRCALINMVFQMGETGVAGFTNSLRMLQQKRWDAAAAALAAAAWYNQTPNRAKRVITTFRTGTWDAYKNL
1. Deep Mutational Scans
a.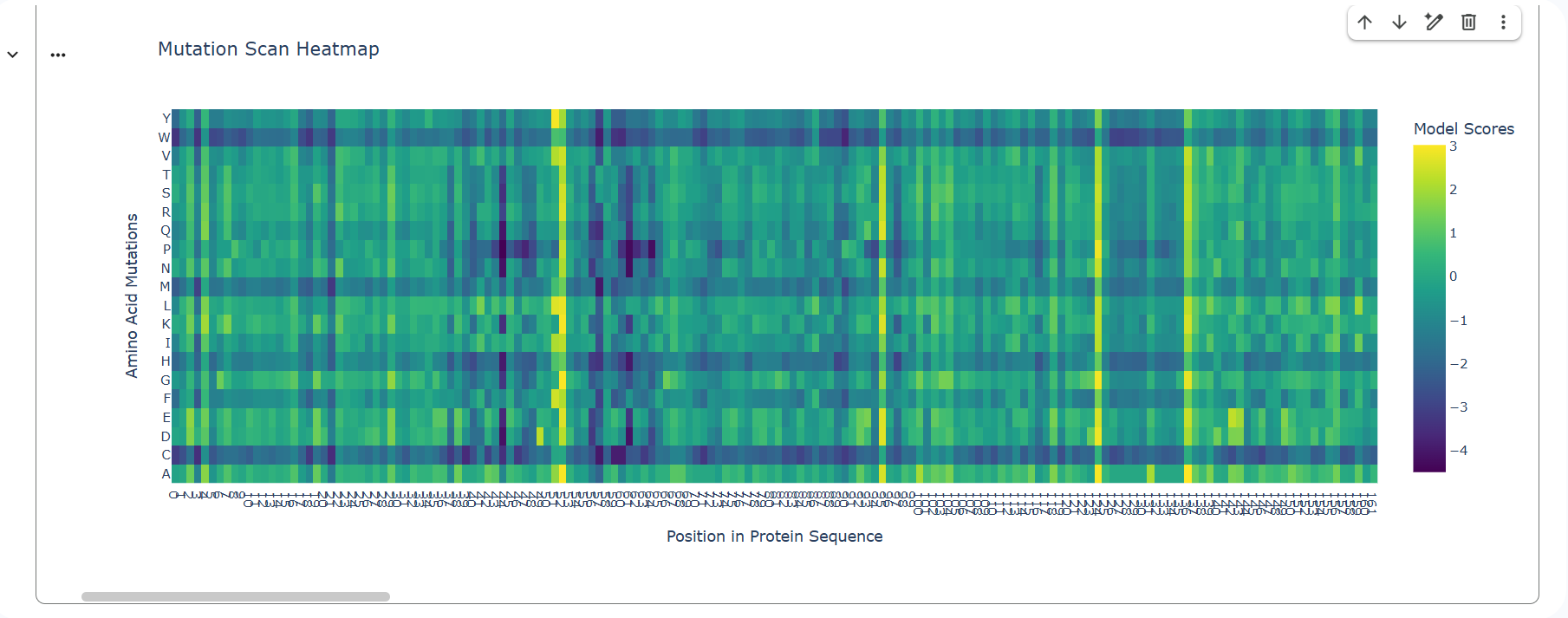
b. Two patterns stand out immediately: A strong horizontal dark band for C (cysteine) across many positions and a few bright vertical columns where almost every substitution is beneficial. The C row is strongly negative because it has a deep purple color and it is darker than surrounding columns. This is very likely one of lysozyme’s disulfide-forming cysteines. Plus, there is a column where many substitutions are bright yellow/green across many amino acids. That usually means that the wild-type residue is suboptimal, the position is surface-exposed, many substitutions improve stability or packing or the model predicts energetic relief.
A mutation that stands out is Cys → Ser. Even though serine is chemically similar (small, polar), the heatmap shows it as strongly negative at those positions. That is because Ser cannot form a disulfide bond, even subtle size changes can disrupt precise geometry and disulfides in lysozyme are deeply integrated into folding topology. That means these cysteines are structurally essential, not just chemically similar residues.
2. Latent Space Analysis
This is the resulting map: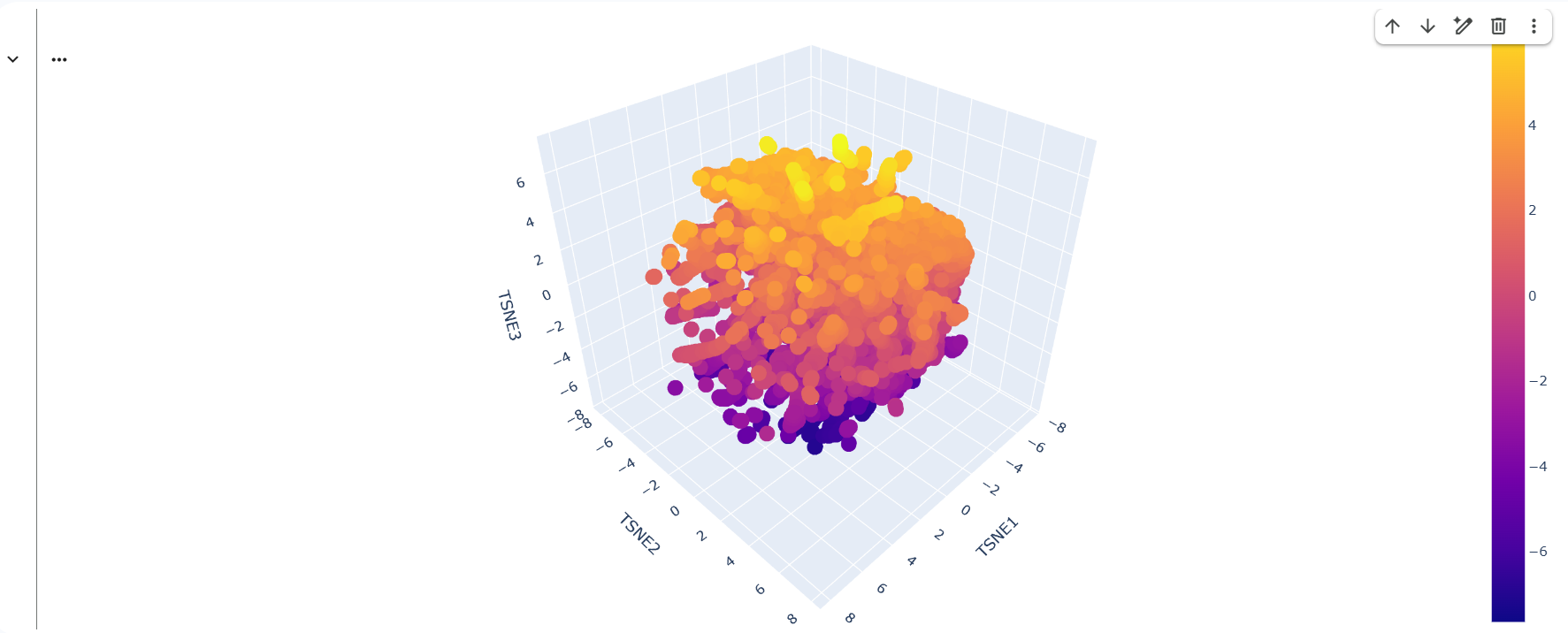
C2. Protein Folding
1. Folded protein with ESMFold:
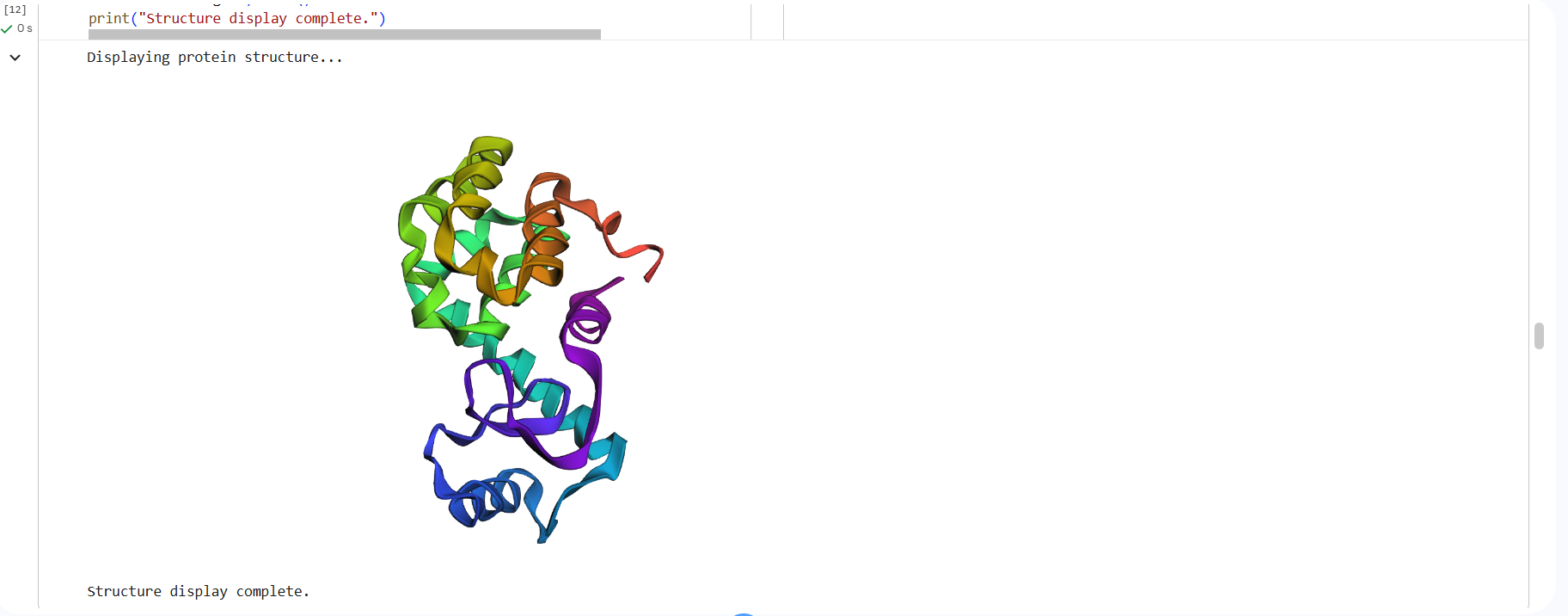
This is the 3D structure in PDB:
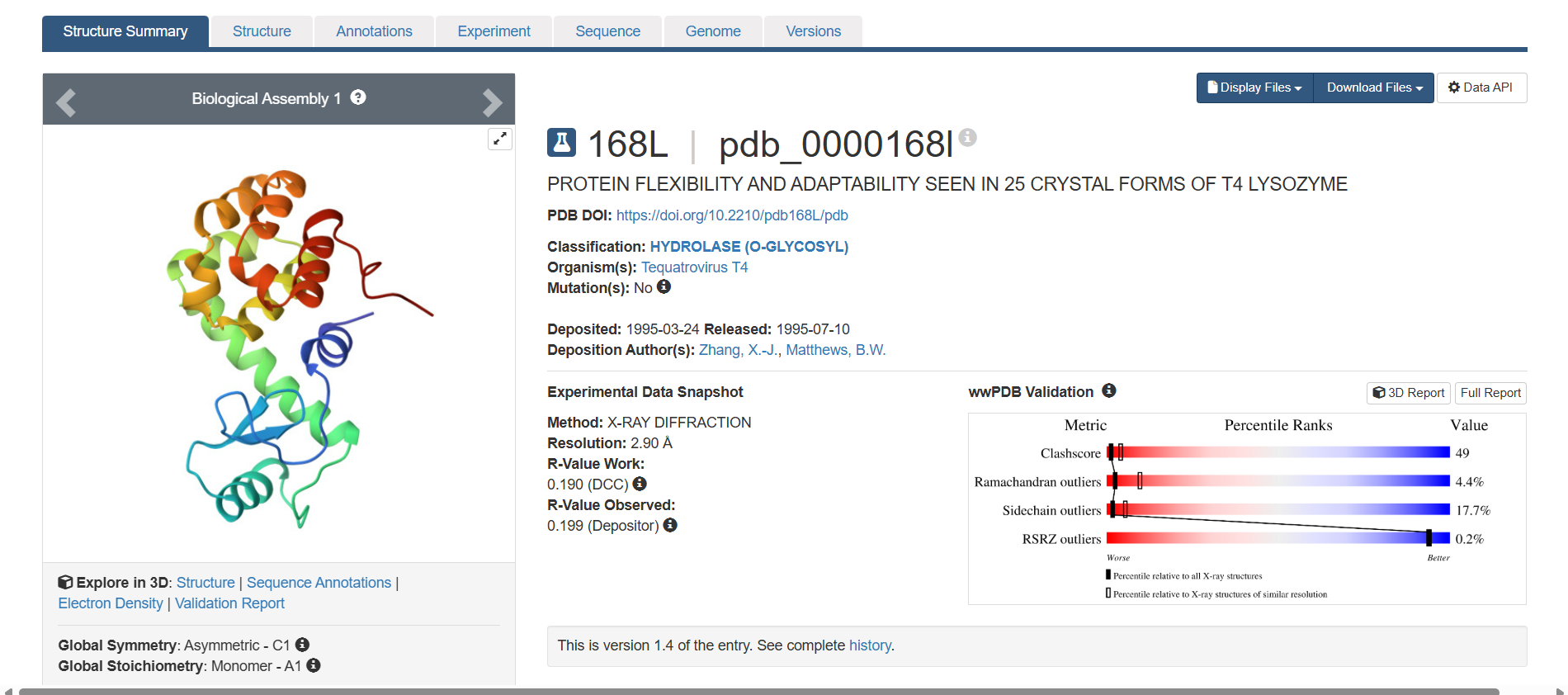
Part D. Group Brainstorm on Bacteriophage Engineering
The main goal would be to computationally design mutations that increase the structural stability of the bacteriophage L lysis protein using structure prediction and stability analysis tools. It is the easiest and most common computational protein-engineering task, but very useful as well.
The second goal would be computationally analyzing conserved residues in L-like lysis proteins to identify mutations that may increase the toxicity of the protein. This method is more difficult, but quite interesting because this goal tries to make the protein kill bacteria more efficiently. The challenge is that toxicity often depends on complex cellular interactions and membrane effects, which are harder to model.
➜ Increased Stability of the Lysis (L) ProteinTools/approaches 🧬
- Protein language models to perform in-silico mutagenesis and identify mutations that are evolutionarily compatible with the L protein sequence. For exapmle, ESM-2
- Approach: Input of the L-protein sequence into a protein language model, performing in-silico mutagenesis (substituting amino acids at different positions), scoring mutations based on their likelihood or predicted fitness and selecting mutations predicted to be tolerated or beneficial.
- Structure Prediction with AlphaFold to evaluate structural effects with 3D structures
- Approach: Predicting the structure of the wild-type L protein, modeling mutant variants suggested by language models or other methods and comparing structural confidence scores (pLDDT) and structural changes.
- Protein language models allow large-scale in-silico mutagenesis, filter out mutations likely to destabilize the protein and suggest mutations that resemble evolutionarily acceptable variants.
- Structure predictions help locate buried vs. exposed residues, identify functional or interaction sites and see whether mutations affect structural packing.
- With energy calculations, you can quantitatively compare mutations and select variants predicted to produce a more stable protein fold.
- Bacteriophage lysis proteins are relatively poorly characterized compared with many other proteins. This means that there may be few experimentally validated structures and limited functional data for mutations. Because of this, models trained on general protein datasets (e.g., ESM-2) may not fully capture the specific biology of phage lysis mechanisms in Escherichia coli.
- The activity of lysis proteins depends on the complex environment of the bacterial cell, including membranes and host proteins like DnaJ. A mutation predicted to improve stability could accidentally reduce interaction with the membrane, alter timing of lysis or disrupt important host interactions.
➜ Higher Toxicity of the Lysis Protein
Tools/approaches 🧬- Structure Prediction and Structural Analysis with AlphaFold and PyMol
- Approach: Predicting the 3D structure of the L protein with AlphaFold, visualizing the structure in PyMOL and identifying structural features, such as exposed residues, potential interaction sites and membrane-facing regions.
- Analysis of Lysis Proteins with BLAST and Clustal Omega
- Approach: Using BLAST to find homologous lysis proteins from other bacteriophages, aligning the sequences using Clustal Omega and identifying highly conserved residues or motifs.
- The Structure Prediction and Analysis helps identify regions that could be modified to strengthen the interactions with the bacterial membrane or other proteins.
- Analysing the Lysis Protein with the given tools help identifying the position of conserved residues. By identifying these positions, it is possible to locate functional regions responsible for lysis and design mutations near these sites to potentially enhance activity
- Protein toxicity often depends on complex interactions inside the bacterial cell, such as membrane disruption or interactions with host proteins in Escherichia coli. Computational tools like AlphaFold can predict protein structures and interactions, but they cannot fully model the cellular environment.
- Sometimes mutations that improve function can reduce protein stability or folding efficiency. Even if a mutation increases interaction with bacterial targets, it might cause misfolding, make the protein degrade faster or reduce expression levels.
References
- https://pmc.ncbi.nlm.nih.gov/articles/PMC10105836/
- https://www.chemistryworld.com/features/why-are-there-20-amino-acids/3009378.article
- https://www.pittwire.pitt.edu/pittwire/features-articles/liu-chemistry-proteins-synthesis#:~:text=to%20Pittwire%20Today-,A%20new%20chemical%20process%20makes%20it%20easier%20to%20craft%20amino,proteins%20or%20their%20smaller%20cousins.
- https://astrobiology.com/2023/04/how-were-amino-acids-formed-before-the-origin-of-life-on-earth.html#:~:text=After%20several%20millions%20of%20years,other%2C%20similar%20to%20human%20hands.
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8508955/#:~:text=Abstract,conductive%20materials%2C%20and%20catalytic%20materials.