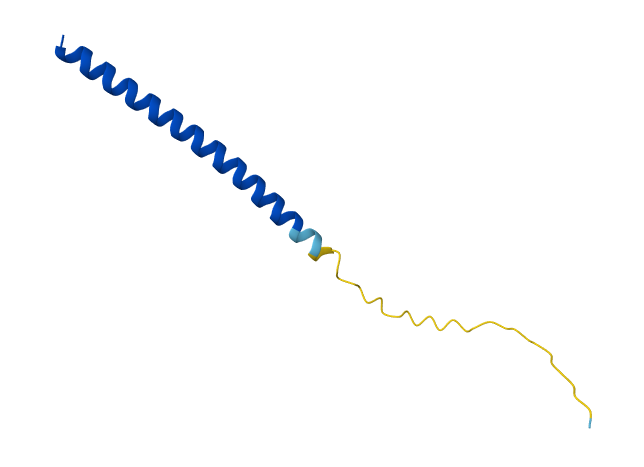
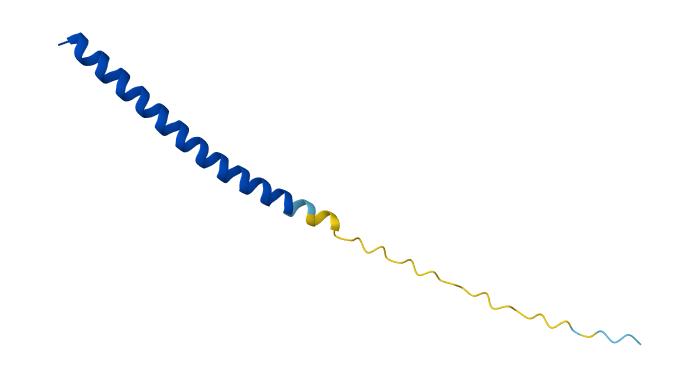
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
The human SOD1 sequence without the mutation:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
The human SOD1 sequence with the A4V mutation:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
These are the 4 generated peptides and the added peptide "FLYRWLPSRRGG" I obtained from Google Colab:
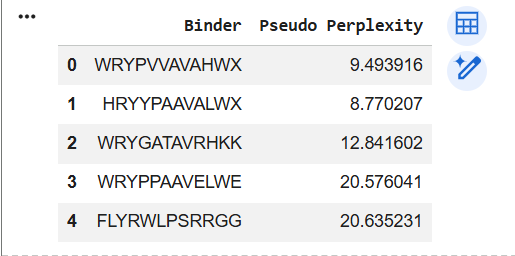
Perplexity measures how surprised the model is by a peptide sequence — lower values mean the sequence looks more natural or compatible with the protein context. The higher the score, the stranger, more unlikely the sequence. Therefore, according to PepMLM, peptide no. 1 has the best perplexity score with 8.770207. That means PepMLM thinks the generated peptides are more likely / better fitting sequences for the mutant SOD1 context than the known peptide.
Part 2: Evaluate Binders with AlphaFold3
Because peptides 0 and 1 have the variable "X", which means unknown amino acid, I used the other peptides, which don´t contain any unknown amino acids. These are the following results:
🟢(1) 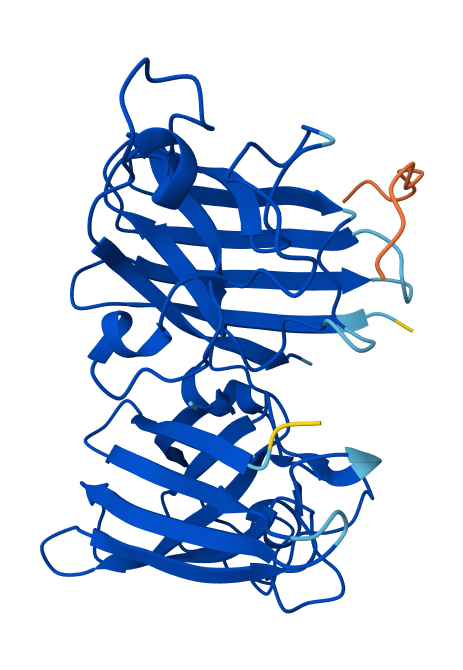
🟠(2)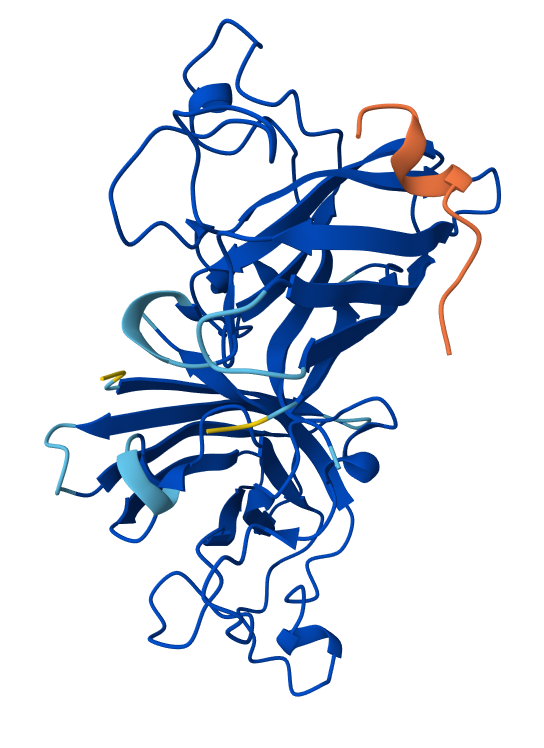
🟣(3) 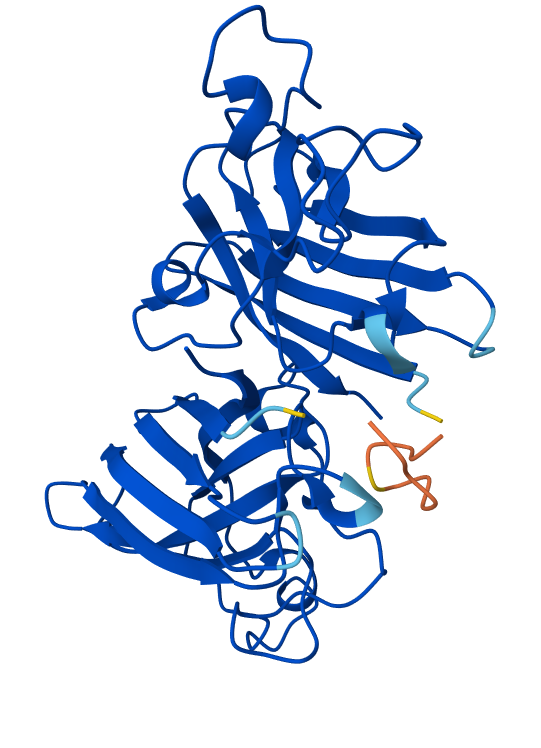
All of the peptides seem to "float" over the SOD1 protein structure. That means that these peptides are not buried within the structure, but rather surfaced-bound.
🟢(1) contains peptide no. 2 from the table above. Its ipTM is 0.86 and pTM is 0.9
🟠(2) contains peptide no. 3 from the table above. Its ipTM is 0.81 and pTM is 0.86
🟣(3) contains the known SOD1-binding peptide "FLYRWLPSRRGG". Its ipTM is 0.9 and pTM is 0.92
By comparing all three of the binding peptides, the one with the best results is the known peptide 🟣(3). pTM measures how confident AlphaFold is in the overall 3D structure of the protein and ipTM measures confidence in the interaction between the protein and peptide chains. 🟣(3) scores the highest, which means that it is predicted by AlphaFold to bind most stably to the mutant SOD1.
In the structure from 🟣(3), the peptide (orange) binds on the surface of SOD1, near loops on one end of the β-barrel. It is close to the N-terminal region where the A4V mutation sits but does not penetrate the β-barrel core. The peptide does not appear to directly engage the dimer interface; rather, it interacts with one monomer of the SOD1 dimer.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
These are the following predictions for each peptide on PeptiVerse:
🟢(1) 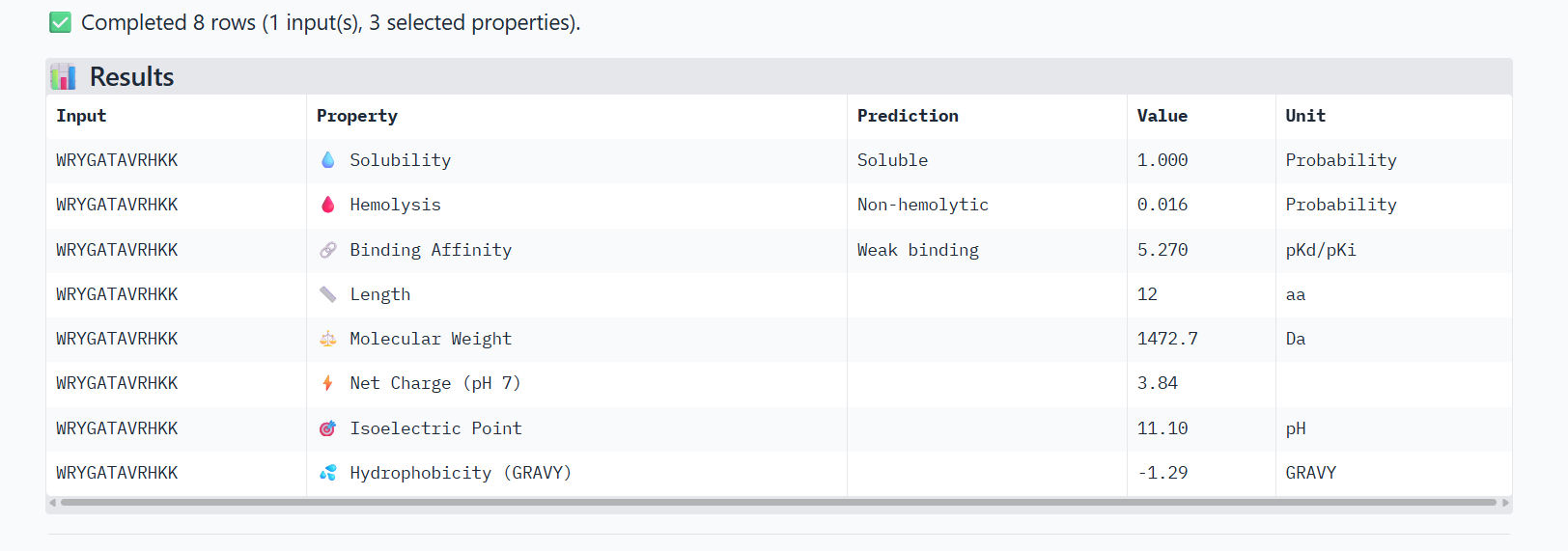
🟠(2)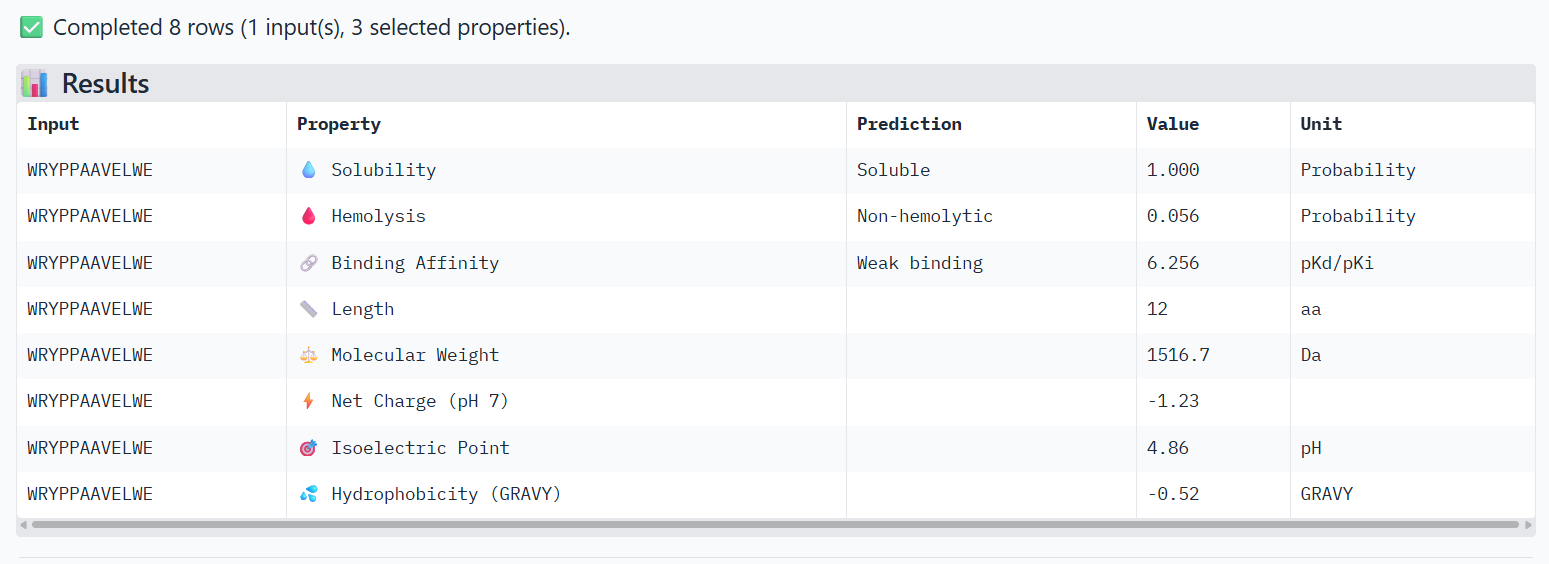
🟣(3) 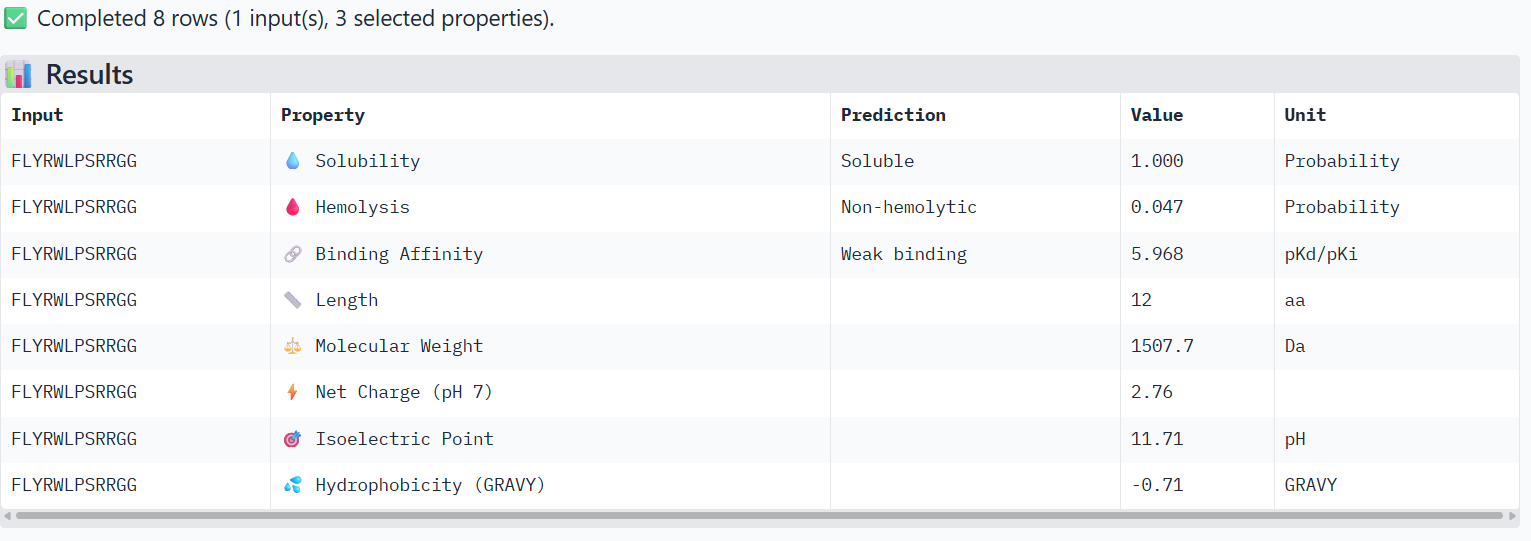
➜ Solubility: All of the 3 peptides are soluble.
➜ Hemolysis: The lower, the safer. The safest one is Peptide 1
➜ Binding Affinity: The lower the value, the better/stronger predicted binding. All 3 of them show weak binding, but Peptide 1 has the best score. Although AlphaFold3 showed Peptide 3 with the best ipTM score, this measurement difference is normal, because the tools measure different things. AlphaFold3 measures the structural confidence of the interaction and PeptiVerse measures the predicted binding energy.
➜ Net Charge: Positive charge helps interaction with proteins and membranes. Peptide 1 scores the best, followed by Peptide 3.
➜ Hydrophobicity: Moderate values are usually ideal, so Peptide 3 has the best value, followed by Peptide 1.
To conclude, Peptide 🟢(1) has the best overall balance. It has the strongest predicted binding, the lowest hemolysis (safest), good positive charge and good hydrophilicity and solubility.
Part 4: Generate Optimized Peptides with moPPIt
moPPIt generated the following binding peptides:
KTFAQFKKIFLQ
PQKEITRCQFFE
VTYCAYYWVTCV
Part C: Final Project: L-Protein Mutants
I chose Option 3: Random Mutagenesis.
I used ChatGPT for helping me create a python function to generate random mutation combinations. It generated the following mutations:
Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Mutations:
E → A at position 2
L → F at position 45
T → G at position 64
This is the new sequence with the mutations:
MATRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFFAIFLSKFTNQLLLSLLEAVIRTVTGLQQLLT
This is the 3D structure of the L-protein: