How does this compare to the length of the human genome?
Human genome consist on 3.2 Giga base pairs, so at least more than 3 000 errors could be made in the synthesis of an entire human genome by one polymerase. But, since human methabolic pathways are at most 10kbp long, its negligible in the majority of context.
How does biology deal with that discrepancy?
There are a lot of polymerase and DNA genome is separated in chromosomes, so chances of committing errors in replication are low, also thera are DNA mismatch reparation systems (MutS Repair System).
How many different ways are there to code for an average human protein?
Considering that the average human protein has 1036 bp, and the genetic code is degenerate, you can make the same protein with different codons that in the end encode the same aminoacid, so there should be a lot of different ways.
In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
This is primarily because, when using different base conjugations, some bases will be compatible with others, and in the case of biological synthesis, we could risk the formation of hairpin-like structures between them (as in the case of RNA proteins), which would force the termination of the translation when we want to obtain the proteins.
There is also the error rate.
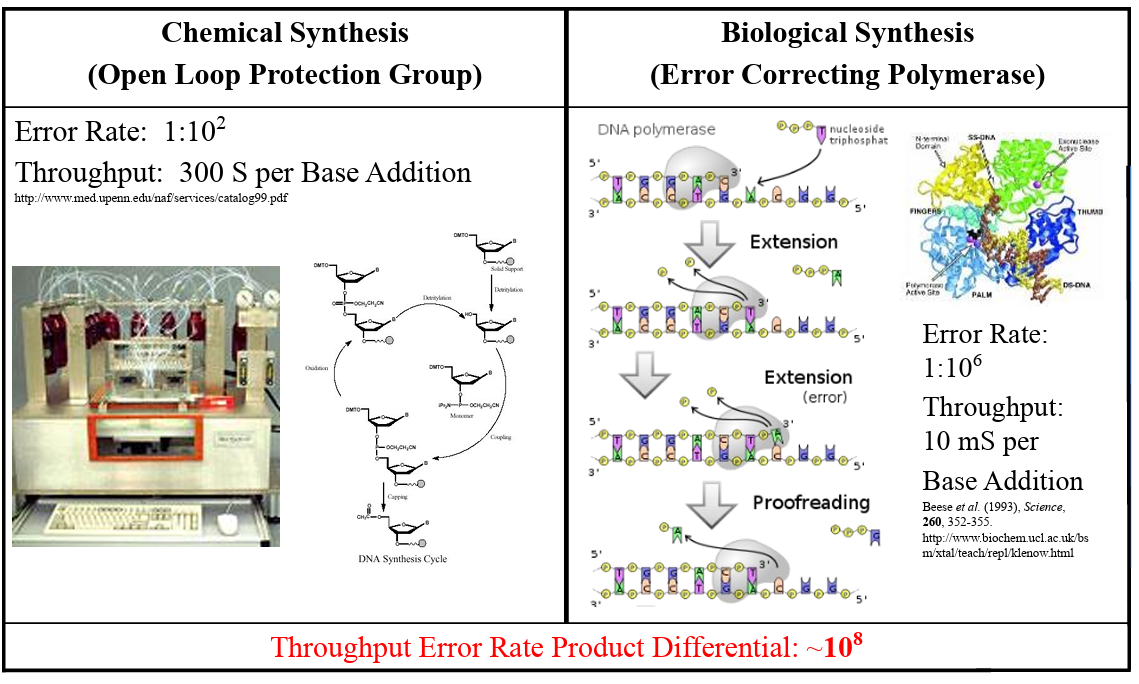
From DNA Synthesis Development and Application:
What’s the most commonly used method for oligo synthesis currently? Phosphoramidite method
Why is it difficult to make oligos longer than 200nt via direct synthesis? An accumulation of errors due to the error rate of chemical synthesis
Why can’t you make a 2000bp gene via direct oligo synthesis? Because the error rate makes it, probabilistically speaking, almost impossible to achieve. So instead gene assembly is used.
From reading and writing life:
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
10 essentials: Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, Arginine
The lysine contingency It would work well if dinosaurs were as easy to isolate from external resources as bacteria in a petri dish. After all, essential amino acids are those we can’t produce ourselves, but obtain by eating other organisms that do possess them. It would be very difficult to cut off any other available source of lysine from the enviroment, so I would recommend another aproach.
Google Search: “Lysine Contigency”
Week 2 assingments:
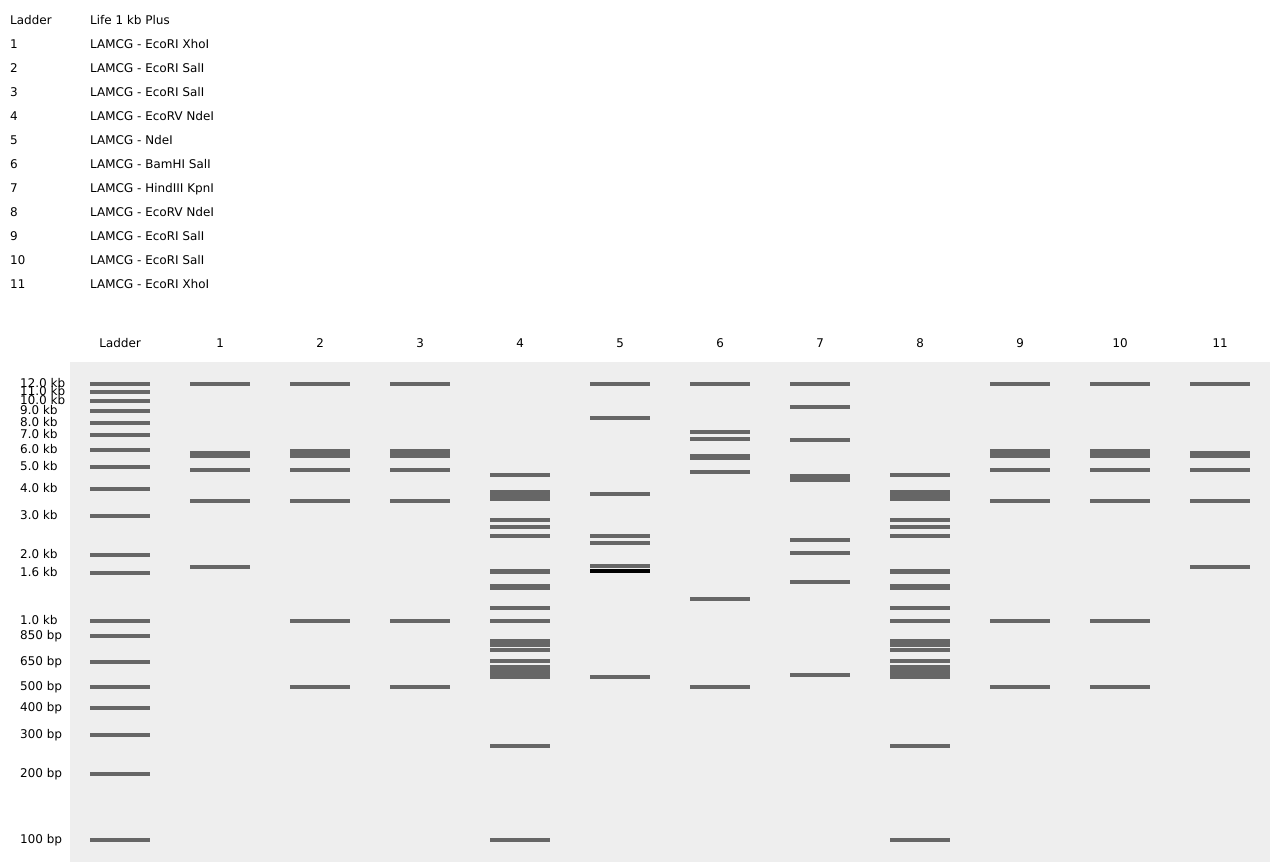
(Part 2 consisted of “Gel Art - Restriction of Digests and Gel Electrophoresis”, intended to be carried out in the laboratory, since I do not have access to one, I did not complete it)
Using a ladder Life 1 kb Plus and different restriction enzymes (EcoRI, XhoI, SalI, EcoRV, NdeI, BamHI, HindIII and KpnI) on the Lambda fage genome, I tried to draw a Teletubbie.
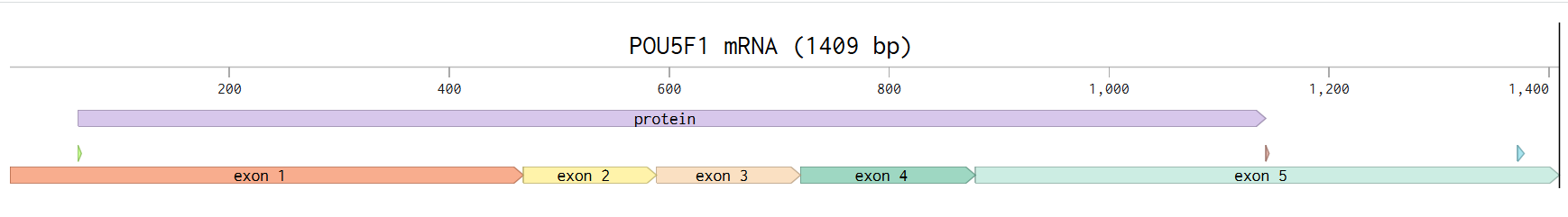
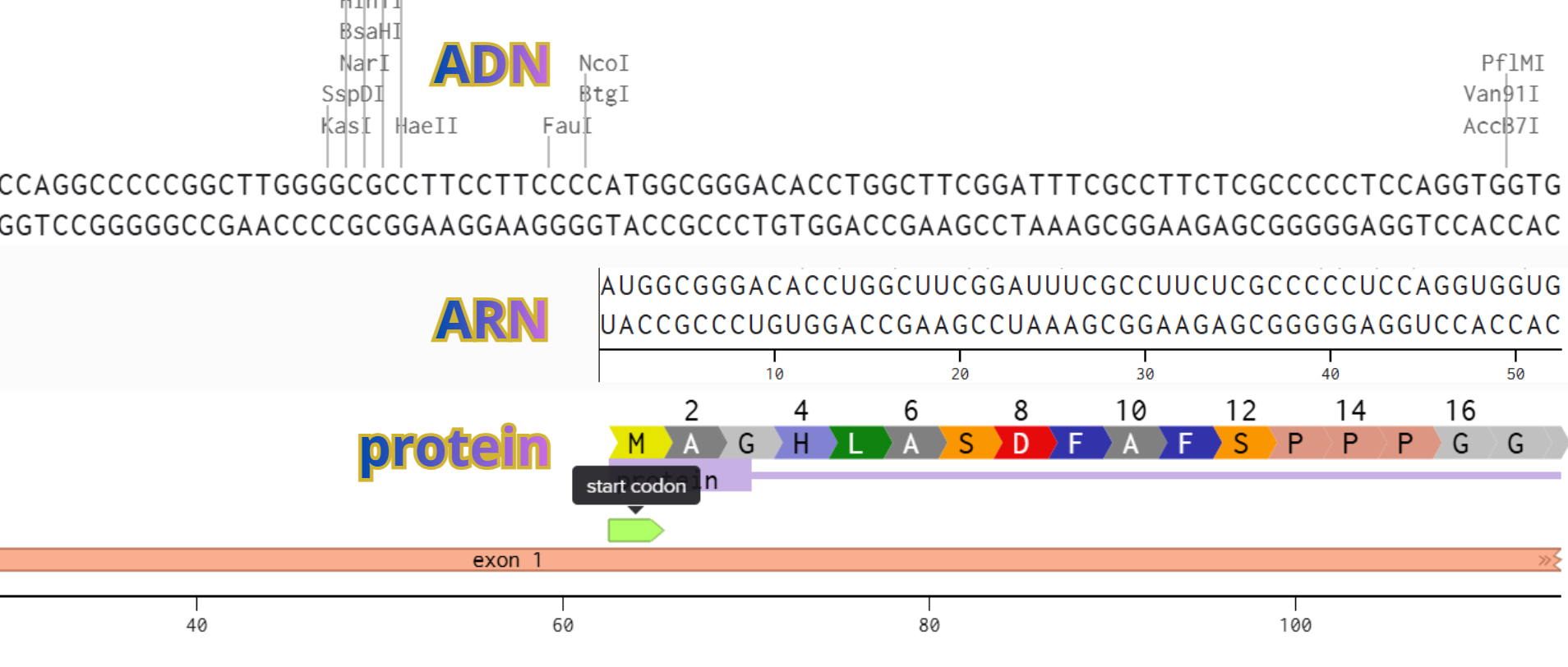
PO5F1 protein DNA sequence
ATGGCGGGACACCTGGCTTCGGATTTCGCCTTCTCGCCCCCTCCAGGTGGTGGAGGTGATGGGCCAGGGGGGCCGGAGCCGGGCTGGGTTGATCCTCGGACCTGGCTAAGCTTCCAAGGCCCTCCTGGAGGGCCAGGAATCGGGCCGGGGGTTGGGCCAGGCTCTGAGGTGTGGGGGATTCCCCCATGCCCCCCGCCGTATGAGTTCTGTGGGGGGATGGCGTACTGTGGGCCCCAGGTTGGAGTGGGGCTAGTGCCCCAAGGCGGCTTGGAGACCTCTCAGCCTGAGGGCGAAGCAGGAGTCGGGGTGGAGAGCAACTCCGATGGGGCCTCCCCGGAGCCCTGCACCGTCACCCCTGGTGCCGTGAAGCTGGAGAAGGAGAAGCTGGAGCAAAACCCGGAGGAGTCCCAGGACATCAAAGCTCTGCAGAAAGAACTCGAGCAATTTGCCAAGCTCCTGAAGCAGAAGAGGATCACCCTGGGATATACACAGGCCGATGTGGGGCTCACCCTGGGGGTTCTATTTGGGAAGGTATTCAGCCAAACGACCATCTGCCGCTTTGAGGCTCTGCAGCTTAGCTTCAAGAACATGTGTAAGCTGCGGCCCTTGCTGCAGAAGTGGGTGGAGGAAGCTGACAACAATGAAAATCTTCAGGAGATATGCAAAGCAGAAACCCTCGTGCAGGCCCGAAAGAGAAAGCGAACCAGTATCGAGAACCGAGTGAGAGGCAACCTGGAGAATTTGTTCCTGCAGTGCCCGAAACCCACACTGCAGCAGATCAGCCACATCGCCCAGCAGCTTGGGCTCGAGAAGGATGTGGTCCGAGTGTGGTTCTGTAACCGGCGCCAGAAGGGCAAGCGATCAAGCAGCGACTATGCACAACGAGAGGATTTTGAGGCTGCTGGGTCTCCTTTCTCAGGGGGACCAGTGTCCTTTCCTCTGGCCCCAGGGCCCCATTTTGGTACCCCAGGCTATGGGAGCCCTCACTTCACTGCACTGTACTCCTCGGTCCCTTTCCCTGAGGGGGAAGCCTTTCCCCCTGTCTCCGTCACCACTCTGGGCTCTCCCATGCATTCAAAC
I went to NCBI genbank and retrieved POU5F1 mARN transcript variant, 1409bp long
I learned that there are spliced variants of OCT4 gene, that includes some introns and can have a more pivotal role in the induction of stemness properties (Yazd et al., 2011)
4. Central Dogma of biology
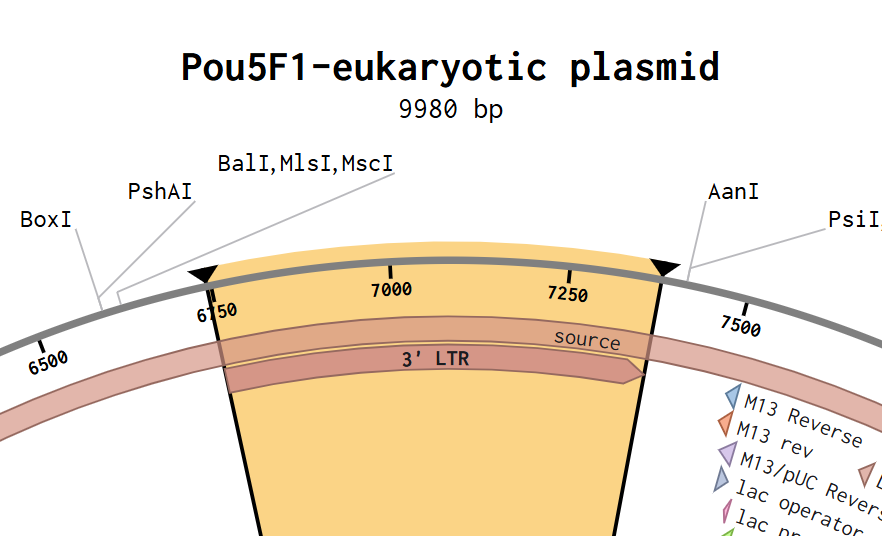
Part 4: DNA Synthesis order, building my first plasmid
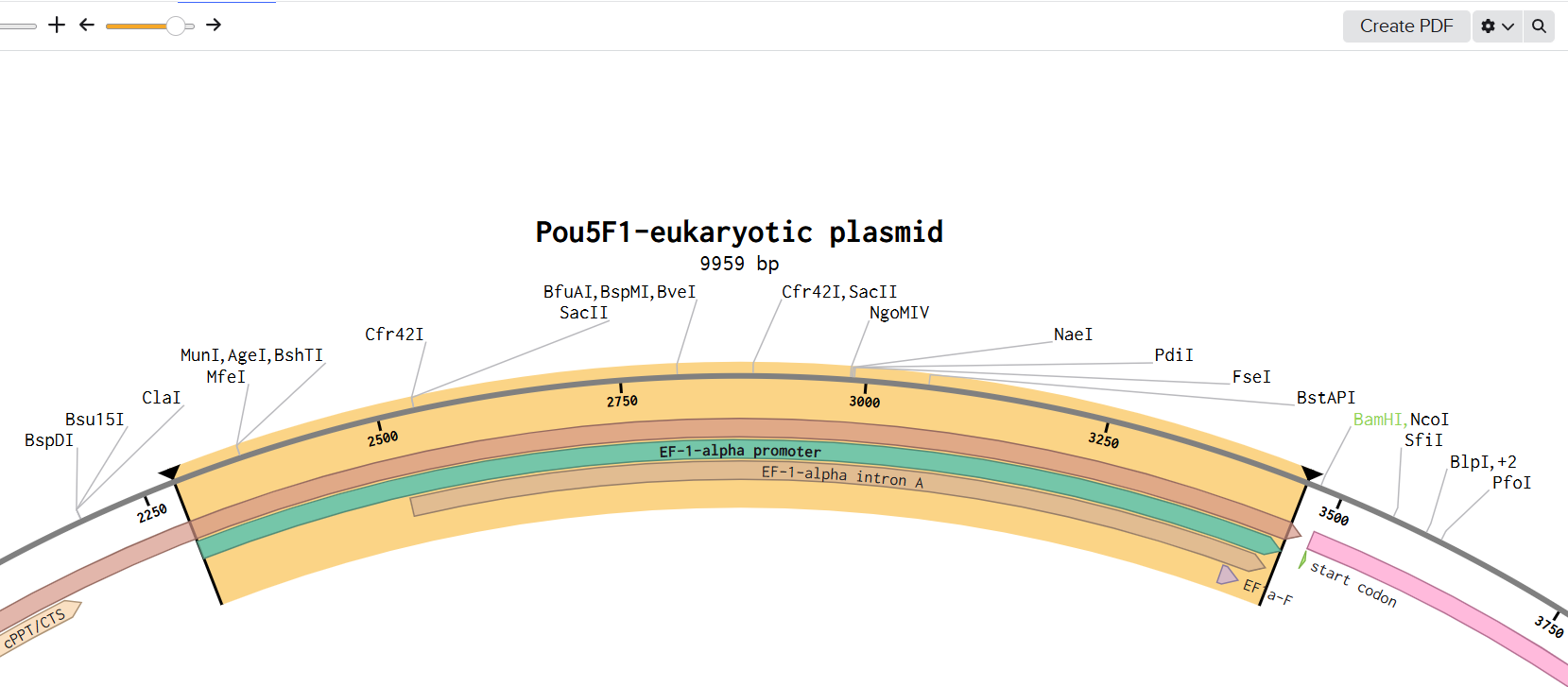
1. Finding an appropriate plasmid backbone:
Reading the Yang et al. (2023) article, I saw in their “Materials and Methods” section, that they transfected pLVX-EF1alpha
2xGFP:NES-IRES-2xRFP:NLS to generate NCC-stable cells, so I thought it would be a good backbone for the assembly. This “shuttle vector” was originally described by Mertens et al. (2015) (AddGene ID: 71396)
Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Mertens J, Paquola AC, Ku M, Hatch E, Bohnke L, Ladjevardi S, McGrath S, Campbell B, Lee H, Herdy JR, Goncalves JT, Toda T, Kim Y, Winkler J, Yao J, Hetzer MW, Gage FH. Cell Stem Cell. 2015 Oct 6. pii: S1934-5909(15)00408-7. doi: 10.1016/j.stem.2015.09.001. 10.1016/j.stem.2015.09.001 PubMed 26456686
2. Assembly
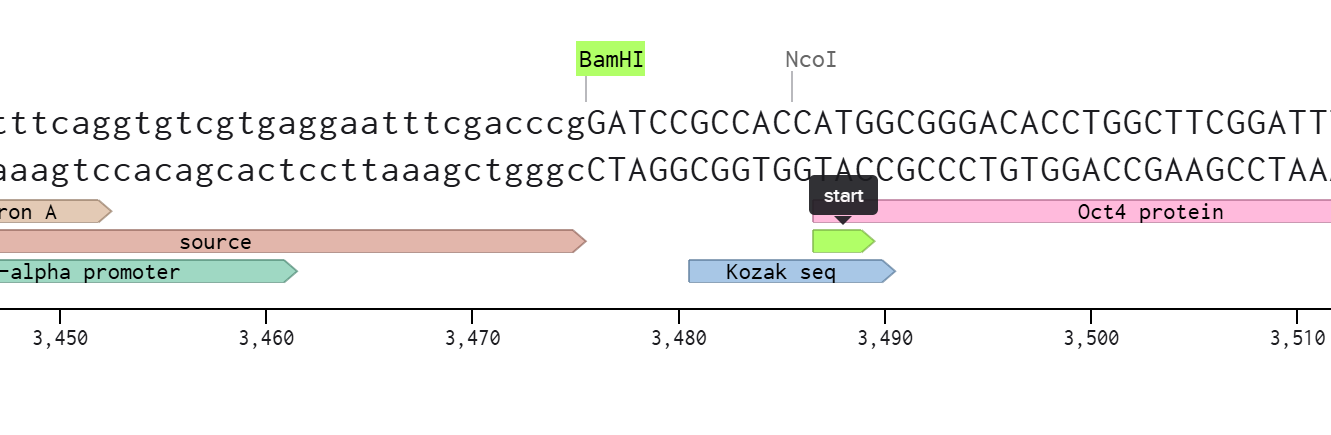
I selected restriction enzymes BamHI and EcoRI to cut the GFP reporter out, and then I built my DNA insert sequence (POU5F1 gene for Oct4) with sticky ends ideal to connect with the backbone.
I I incorporated a Kozak consensus sequence (GCCACC) upstream of the ATG instead of an RBS
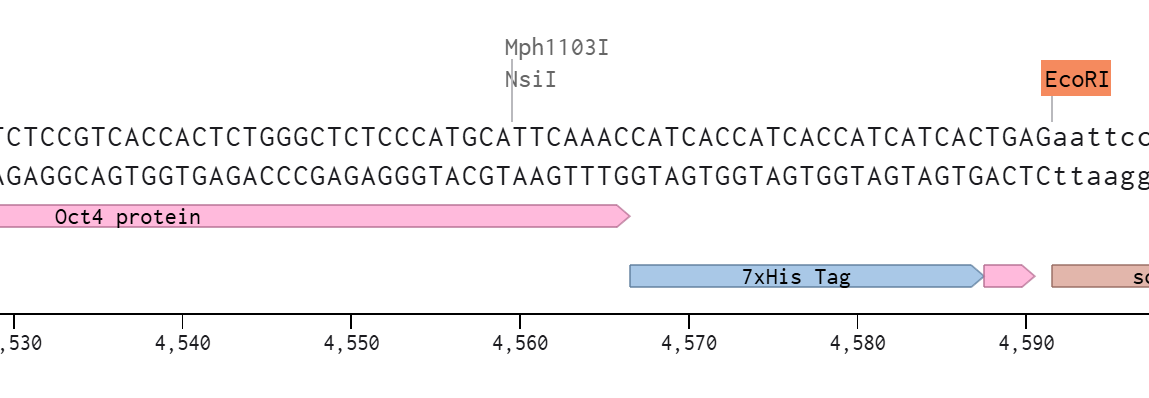
And to add the 7xHis at the C-terminus of the protein, before the stop codon
The design leverages the backbone’s endogenous promoter and polyadenylation signal, so I didn’t incorporate those to the insert
What DNA would you want to sequence (e.g., read) and why?
I would like to sequence the plasmid I modified (pLVX-EF1alpha-POU5F1-7xHis-IRES-mCherry) as a way of experimentally verifying that there were no mutations and that the junctions integrated properly.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use the “Oxford Nanopore” sequencing method because it would allow me to find accidental recombinations and deletions by being able to sequence the entire plasmid.
DNA Write
What DNA would you want to synthesize (e.g., write) and why?
I would like to synthesize de novo the entire modified POU5F1 (Oct4) cassett, because that way I can perform codon optimization and add the necessary elements (histidine tag and Kozak sequence) in the most efficient way.
What technology or technologies would you use to perform this DNA synthesis and why?
I would use silicon-based DNA synthesis on microchips. Given that the Oct4 cDNA is approximately 1 kb, I really appreciate the ability to synthesize thousands of oligonucleotides in parallel with extremely high accuracy and at a much lower cost than traditional column synthesis.
DNA Edit
What DNA would you want to edit and why?
I would like to edit the genome of the plasmid recipient cells so that they possess a specific locus where the POU5F1 cassette can be inserted without activating oncogenes or disrupting vital genes.
What technology or technologies would you use to perform these DNA edits and why?
I would use Prime Editing because it would allow me to edit a small portion of the genome with considerable precision and turn it into a specific recognition site. Since it uses a Reverse Transcriptase (fused to Cas9 nickase) to write the new sequence directly into the DNA, it works perfectly in neurons or resting fibroblasts.