Evolutionary Scale Model (ESM): A model that generates likelihood values for the existence of an amino acid at a specific position within a protein sequence. It is essentially a Masked Language Modeling (MLM) architecture.
Deep Mutational Scanning (DMS): A 2D map representing the probability values generated by the ESM for every possible mutation in a sequence.
Latent Space: Upon inputting a FASTA sequence, the language model adds start and end tokens, generating a high-dimensional vector (320 dimensions in the case of the recitation model). This vector classifies the protein; when projected into a 3D space, it creates a map that clusters the protein alongside others from a provided database based on functional or family similarities.
Steps for 3D Protein Structure Prediction with ESM-2:
Likelihood Generation: We utilize the fundamental property of the ESM (Masked Language Modeling) to calculate amino acid probabilities based on the sequence context.
Attention Mechanism: These data points are processed as Queries (Q), Keys (K), and Values (V). This generates an Attention Matrix, where vectors for each amino acid indicate the relevance and position of other residues in the chain.
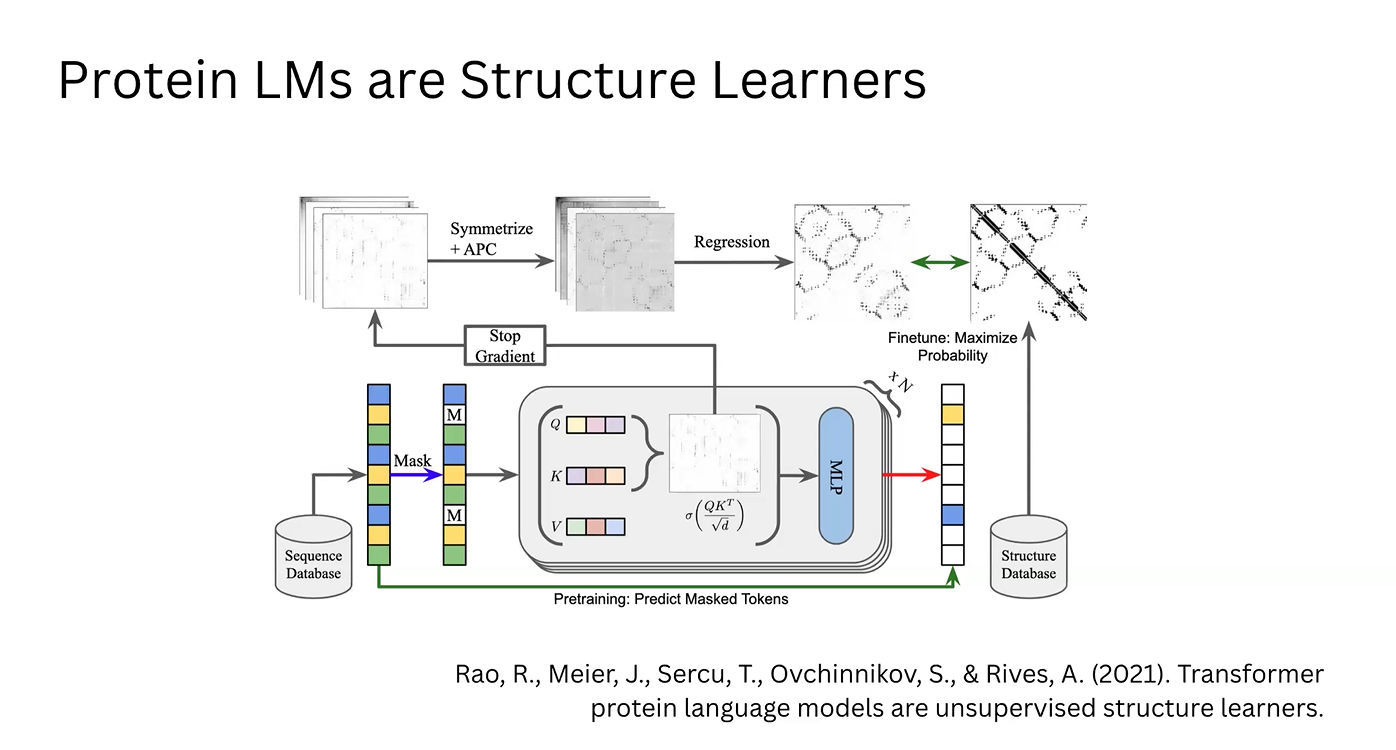
Structural Extraction (Simple ESM): The raw attention matrix is symmetrized to ensure physically plausible distances and refined using Average Product Correction (APC) to eliminate correlation noise. Through regression, a 2D contact map is obtained.
Model Optimization: This map is compared against structural databases (like the PDB) to adjust the model. This feedback loop updates the model’s weights, maximizing the log-likelihood of real-world protein structures found in nature.
Enrichment via MLP: In parallel, the correlation vectors from the attention matrix are processed by a Multi-Layer Perceptron (MLP). This generates enriched vectors that describe the physicochemical implications of the attention data.
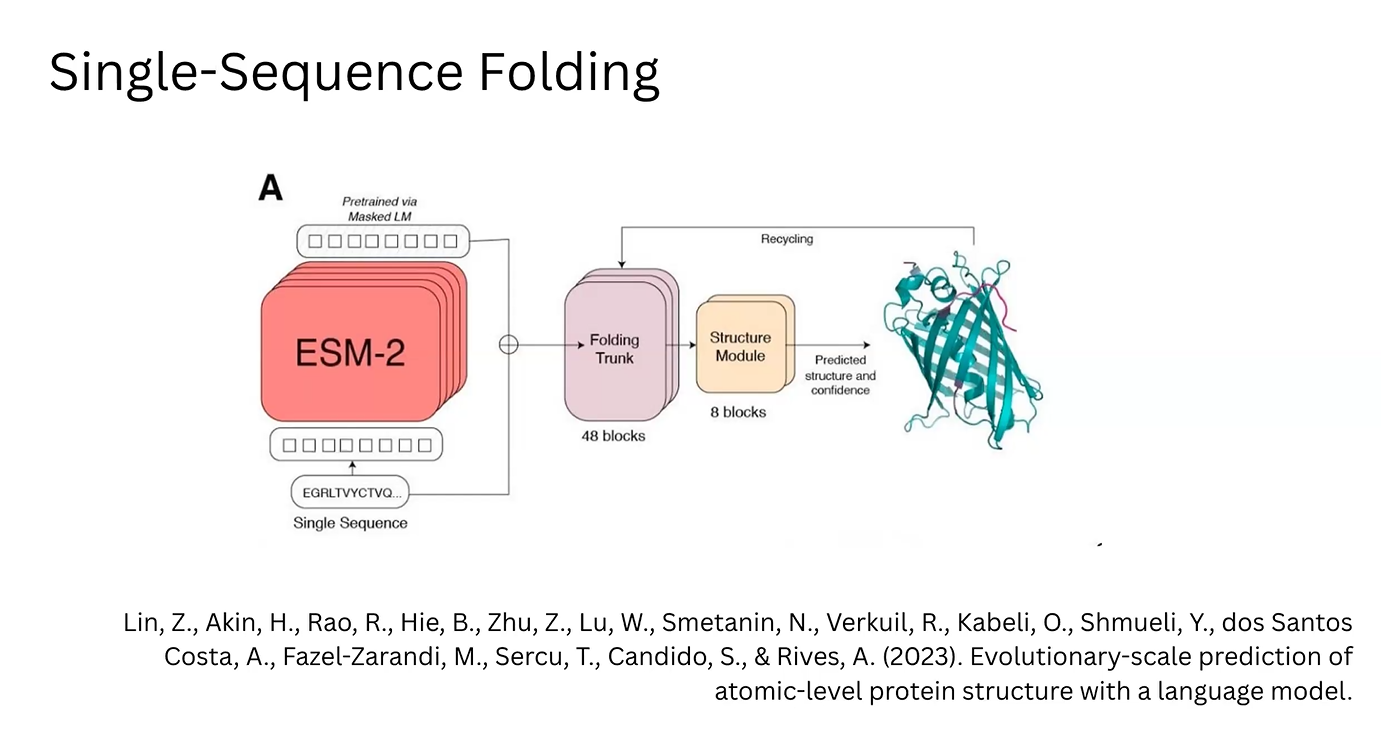
3D Prediction (ESM-2/ESMFold): In the ESM-2 architecture, these MLP-enriched vectors are fed into a “Folding Trunk” module. This module applies learned rules to predict the 3D structure, including bond angles and atomic coordinates.
About AlphaFold ans MSA
Key Concepts to Know:
Multiple Sequence Alignments (MSA): The different dialects of the tree of life for the same protein
For this part I used transcription factor Klf4 and realized a Deep mutational Scan, Latent Space Analysis, folding protein exercise, and inverse-folding protein exercise.
Subsections of Weeek 4 HW: Protein design - Part 1
Conceptual questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
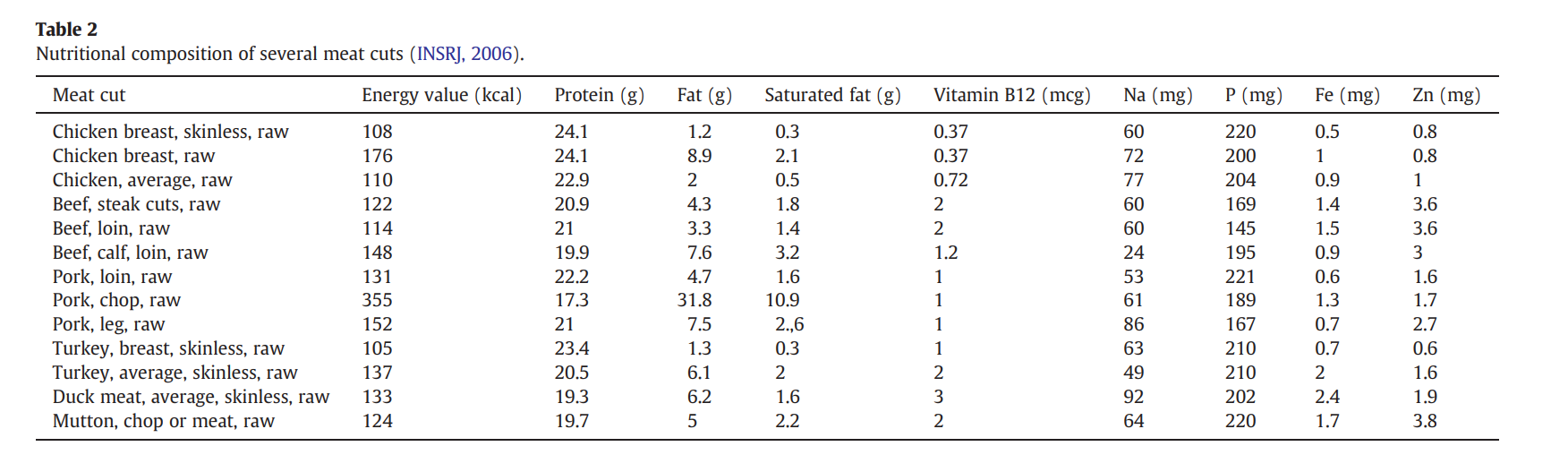
A: Averaging out the nutritional composition of common meats (like the data from Pereira & Vicente, 2013), meat is about 21.25% protein by weight.If you eat a 500-gram steak, you are consuming roughly 106.25 grams of pure protein. Since 1 Da is exactly 1 g/mol, an average amino acid weighing 100 Da has a molar mass of 100 g/mol.By dividing your total protein (106.25 g) by the average amino acid mass (100 g/mol), we get 1.0625 moles of amino acids. Multiply that by Avogadro’s number 6.022e+23, and you get approximately 6.399e+23 molecules of amino acids
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
A: Because exogenous DNA is either degraded before consumption or degraded by our own enzymes during the digestion process, it cannot be processed to alter our cells. So, the only materials our body takes are the disassembled amino acids; then, it rebuilds them into human proteins, following our human genome.
Why are there only 20 natural amino acids?
A: Certainly, there are only 20 amino acids in Choanoflagellatea, and this set of amino acids provides just enough chemical diversity (acidic, basic, hydrophobic, and hydrophilic shapes) to build complex and versatile proteins. However, I believe it’s a little bit biased to say they are the only “natural” ones, because there are also species of methanogenic archaea that use other amino acids, like selenocysteine and pyrrolysine.
Can you make other non-natural amino acids? Design some new amino acids.
A: Yes, it’s possible to synthesize new amino acids with synthetic biology or organic chemistry. You could also find them as non-canonical amino acids, like 2-azatyrosine, 3-azatyrosine, or azetidine-2-carboxylic acid.
Where did amino acids come from before enzymes that make them, and before life started?
A: It’s believed that they formed completely abiotically, as was proven in the famous Miller-Urey experiment, where they showed that if you mix simple gases present on early Earth (like methane, ammonia, and water) and strike them with electricity (simulating lightning), amino acids form naturally and spontaneously.
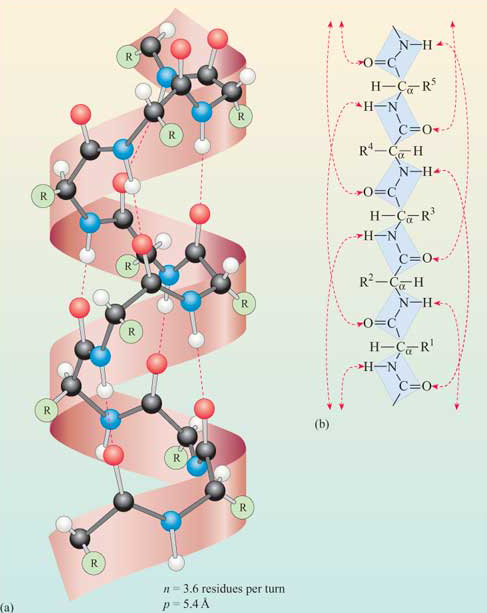
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A: Left. Because D-amino acids are enantiomers of our natural L-amino acids, the most energetically stable way for them to fold is also completely mirrored.
Why are most molecular helices right-handed?
A: This conformation is much more energetically favorable because it minimizes atomic collisions (steric hindrance) between side chains and the backbone of the L-amino acids.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
A: Because the unpaired hydrogen bond donors and acceptors of a beta strand attain a highly stable conformation by bonding with another strand, reaching a lower energy state. The driving forces are hydrogen bonds and hydrophobic interactions.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
A: Because amyloid diseases are characterized by protein denaturation or misfolding, these proteins expose hydrofobic regions in this process so the end up crashing ther strands together and “zippering up” into tightly packed cross-β-sheet structures. Yes, because of its stability and self-assembly capacity, we could use it as diverse materials: nanowires, hydrogel, scaffolds, etc.
References:
Pereira, P. M. de C. C., & Vicente, A. F. dos R. B. (2013). Meat nutritional composition and nutritive role in the human diet. Meat Science, 93(3), 586–592. https://doi.org/10.1016/j.meatsci.2012.09.018
This is primarily because is the next protein in the yamanaka factors and participates in a SOX2/OCT4-bound, being OCT-4 the protein I selected for week’s two homework.
In relation to Sox2 aminoacid sequence
Sequence length and composition: This protein is 317 amino acids long. Based on the frequency analysis, Serine (S) is the most frequent amino acid, appearing 36 times (11.36% of the sequence), followed closely by Glycine (G) with 35 occurrences (11.04%).
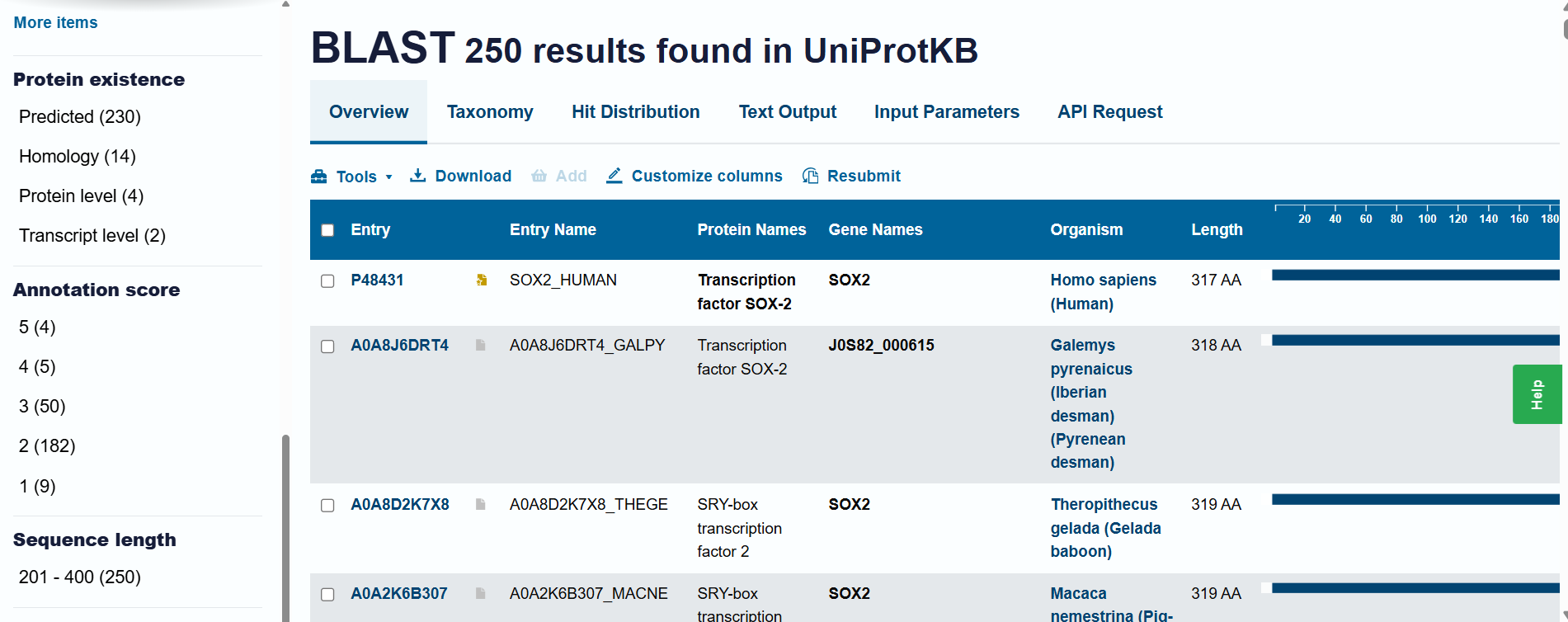
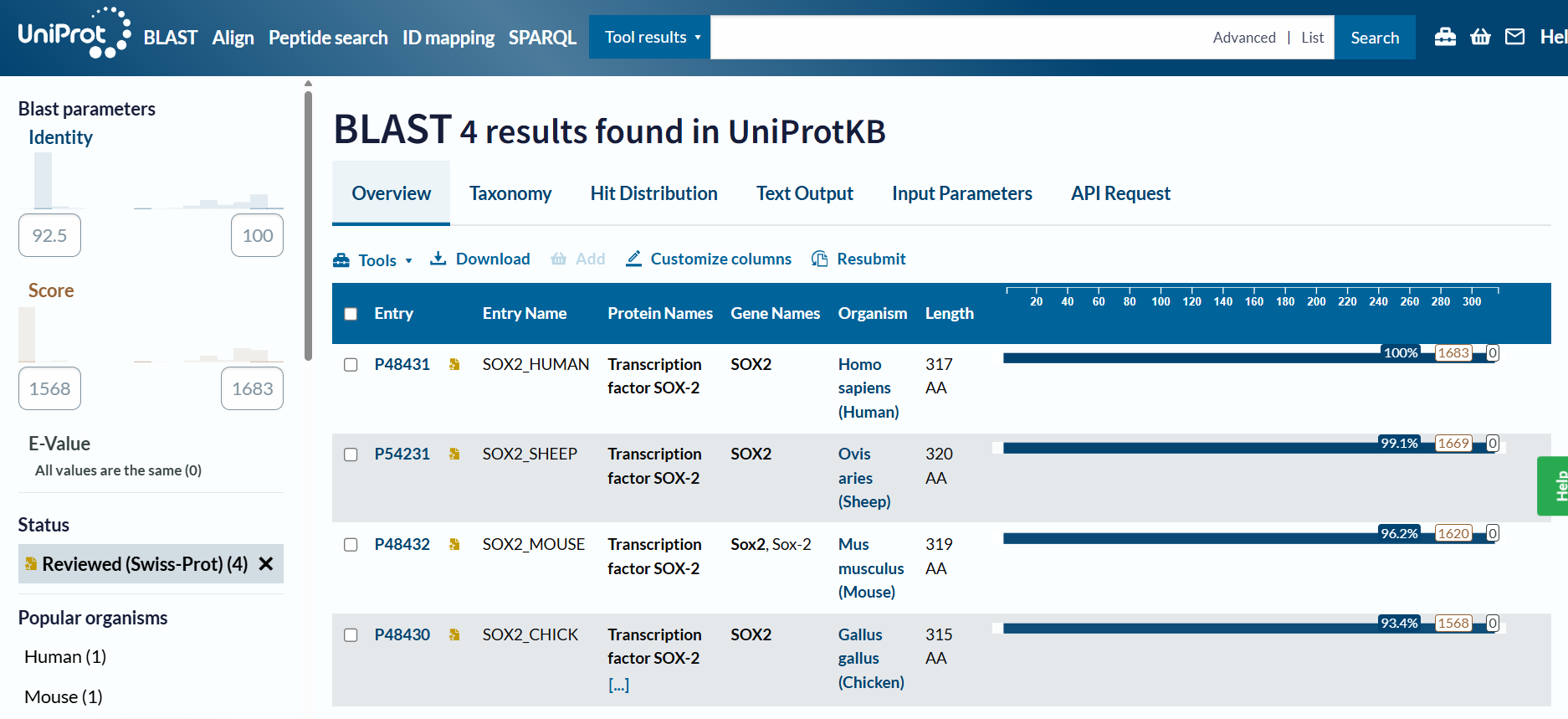
Homology and BLAST results: When including “predicted” and “homology” evidence levels, the search reached the limit of 250 results in the UniProt BLAST tool. This high number of hits reflects that Sox-2 is an extremely conserved protein across species. However, for high-confidence analysis, I primarily considered the results from the reviewed Swiss-Prot database.
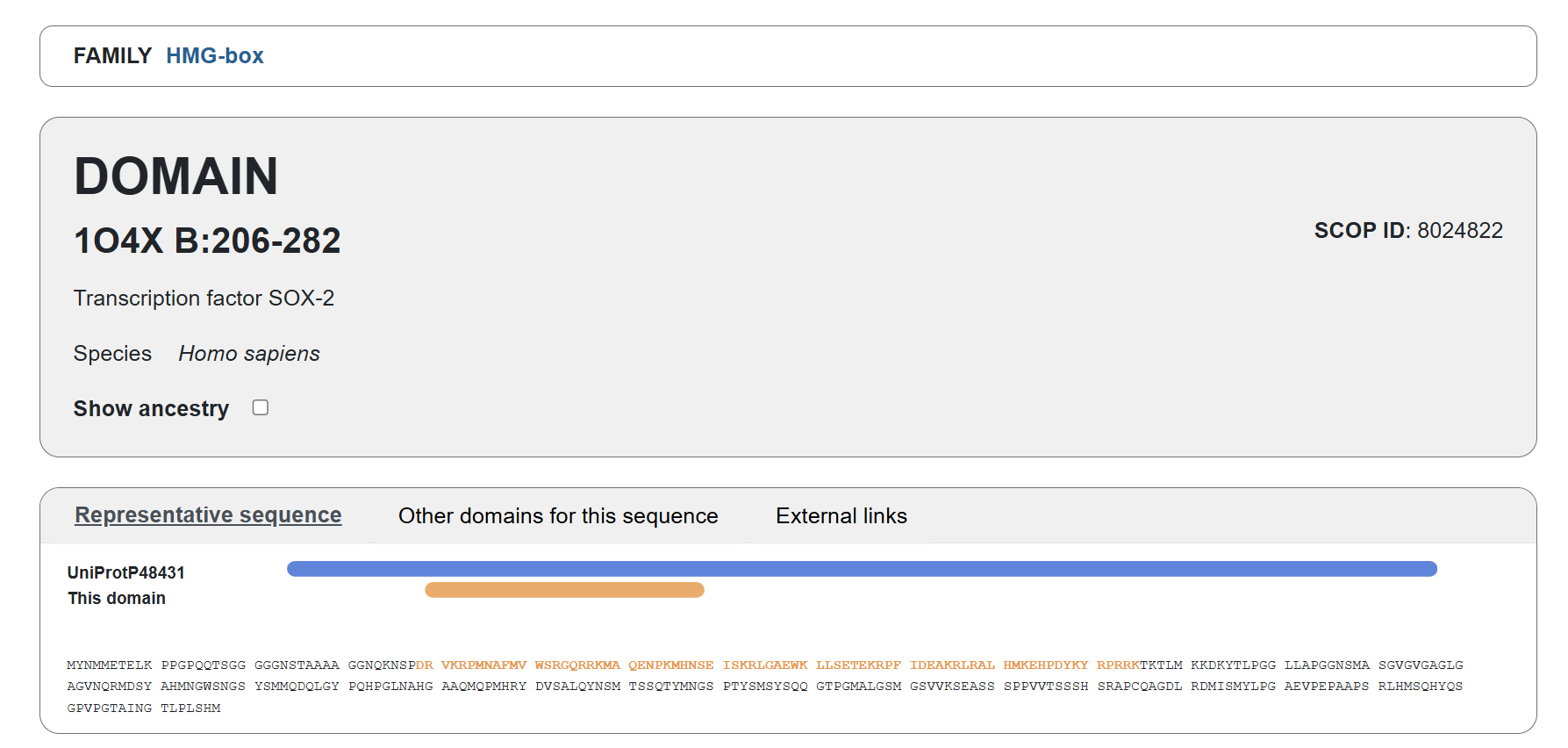
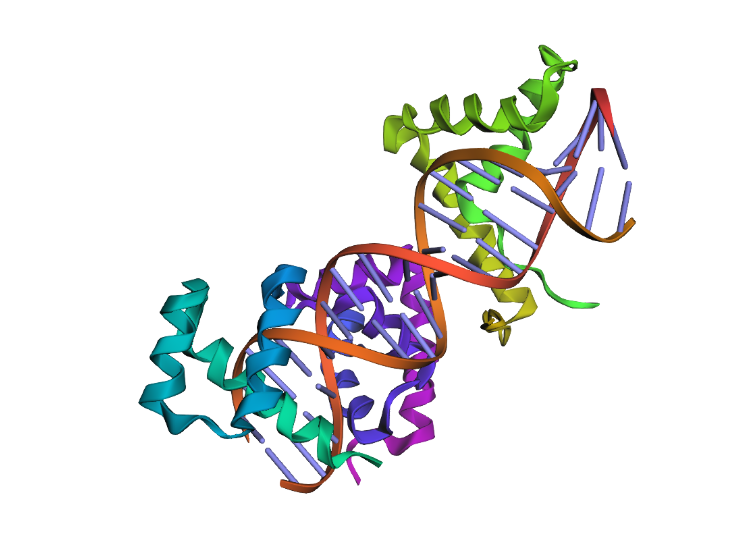
Familiy Classification: SCOP results showed that Sox-2 contains a High Mobility Group (HMG) box structural domain located between residues 206-282. This domain is characterized by its L-shaped triple α-helix fold that binds to the minor groove of DNA. While it shares this structural “HMG-box” fold with other architectural proteins, Sox-2 functionally belongs to the SOX transcription factor family. Despite recognizing the same DNA consensus motif ( 5-(A/T)(A/T)CAA(A/T)G-3 ) different Sox factors achieve target gene selectivity through differential affinity for particular flanking sequences next to consensus Sox sites, homo- or heterodimerization among Sox proteins, posttranslational modifications of Sox factors, or interaction with other co-factors (Wegner, 2010)
The structure was solved and published in 2003 by Reményi et al. in the journal Genes & Development (https://doi.org/10.1101/gad.269303).
Since the article its about the crystal structure of a POU/HMG/DNA ternary complex, the resolution is not below 2.70 Å, wich is not ideal for high-detail drug design, but I think the 2.70 Å resolution obtained is considered a good quality structure for analyzing protein-DNA ternary complexes.
As I mentioned before, the solved structure is often a ternary complex. Apart from the SOX2 protein, it includes the POU domain of the OCT4 protein and a specific DNA oligonucleotide (the FGF4 enhancer)
Acording to SCOP, it belongs to the HMG-box family within the structural class all alpha protein
In relation to the molecule visualization:
Since I ran into some strange errors while trying to use Conda, I used pip to import the PyMOL libraries instead
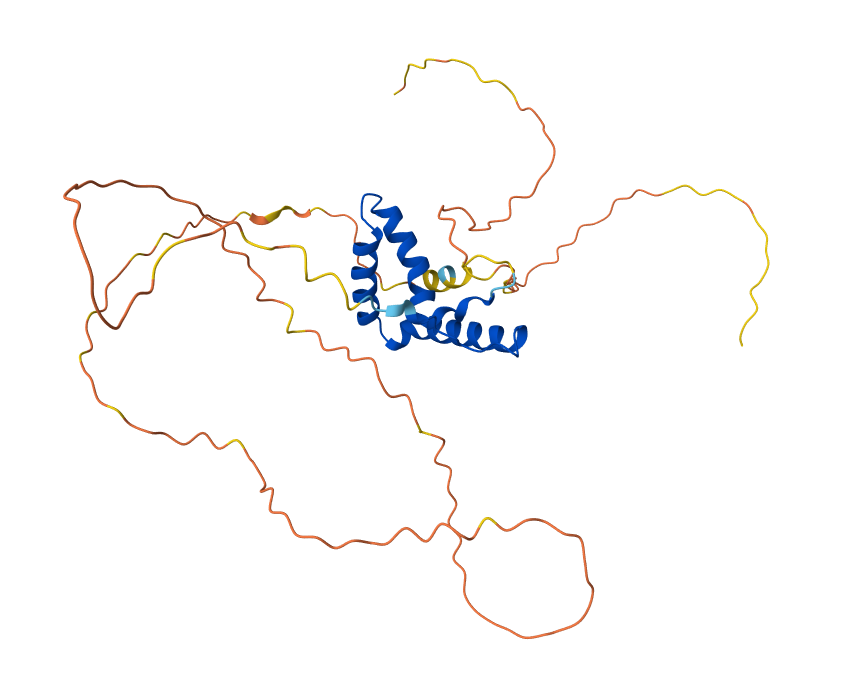
I used py3Dmol to visualize Sox2 in “cartoon” and “ball and stick” representations, and PyMOL for the “ribbon” view
To highlight the secondary structures, I added colors: red for alpha helices, blue for beta sheets, and grey for loops
I then used the following code to count the atoms and determine the predominant structure:
By coloring the hydrophilic residues blue, the hydrophobic residues orange, and others yellow, it becomes clear how they are distributed. You can notice that the hydrophilic residues tend to interact with the phosphates in the DNA chain, while the hydrophobic ones are positioned to fit within the DNA grooves.
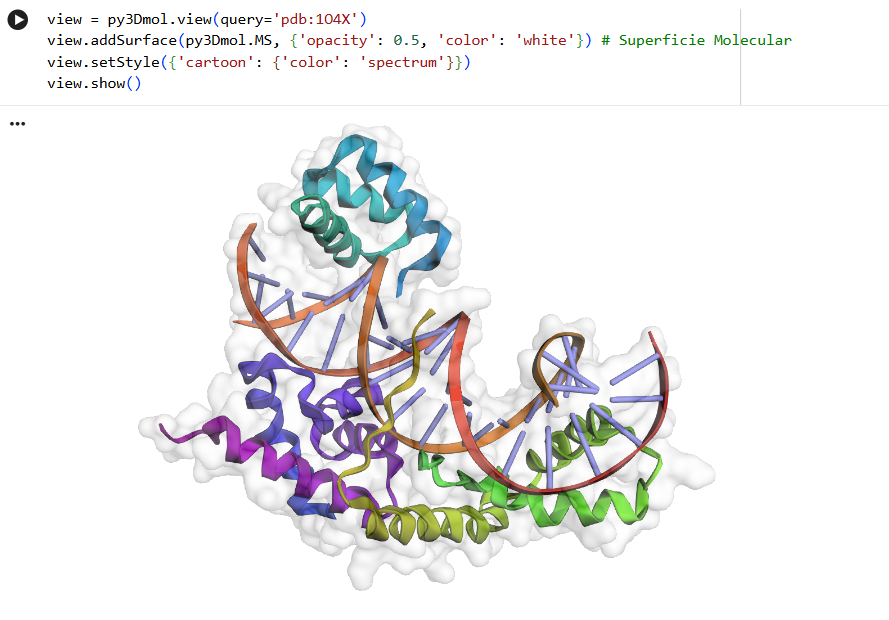
As seen in the surface visualization, the “binding pockets” of transcription factors aren’t exactly “holes.” Instead, they act more like a contoured surface that forms “hooks” around the DNA backbone.
Using ML-Based Protein Design Tools
Protein Language Modeling
Deep Mutational Scans
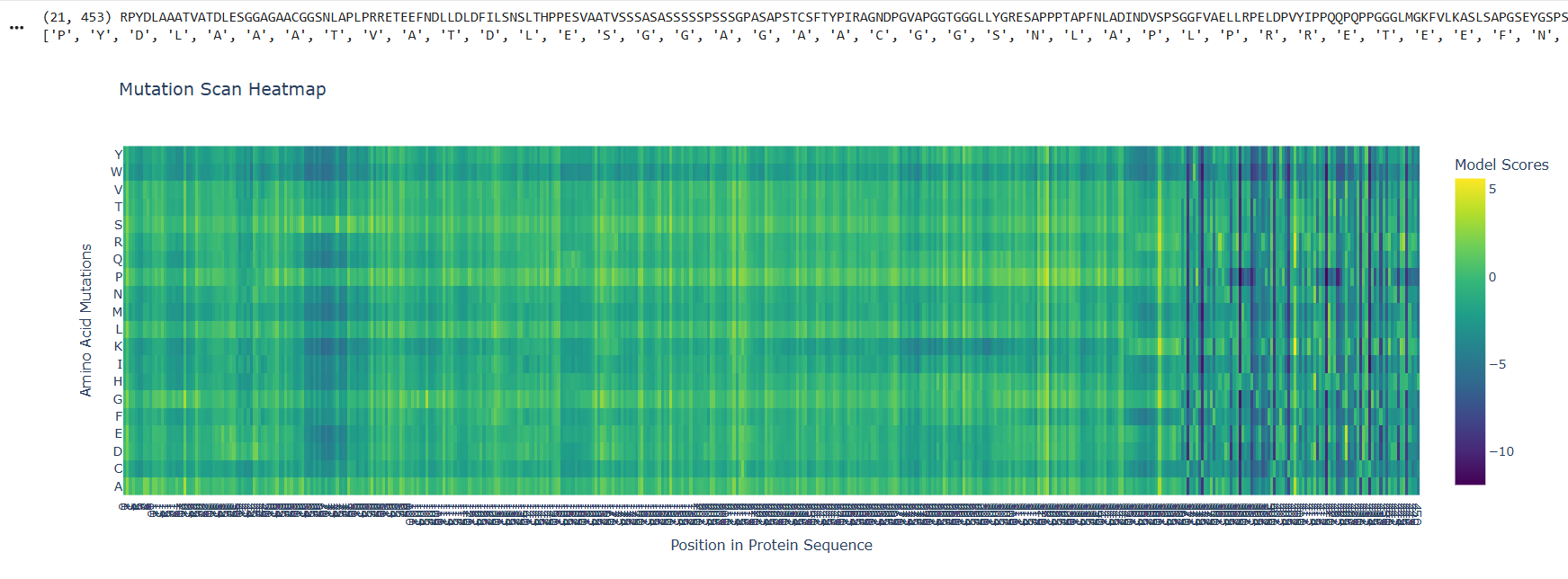
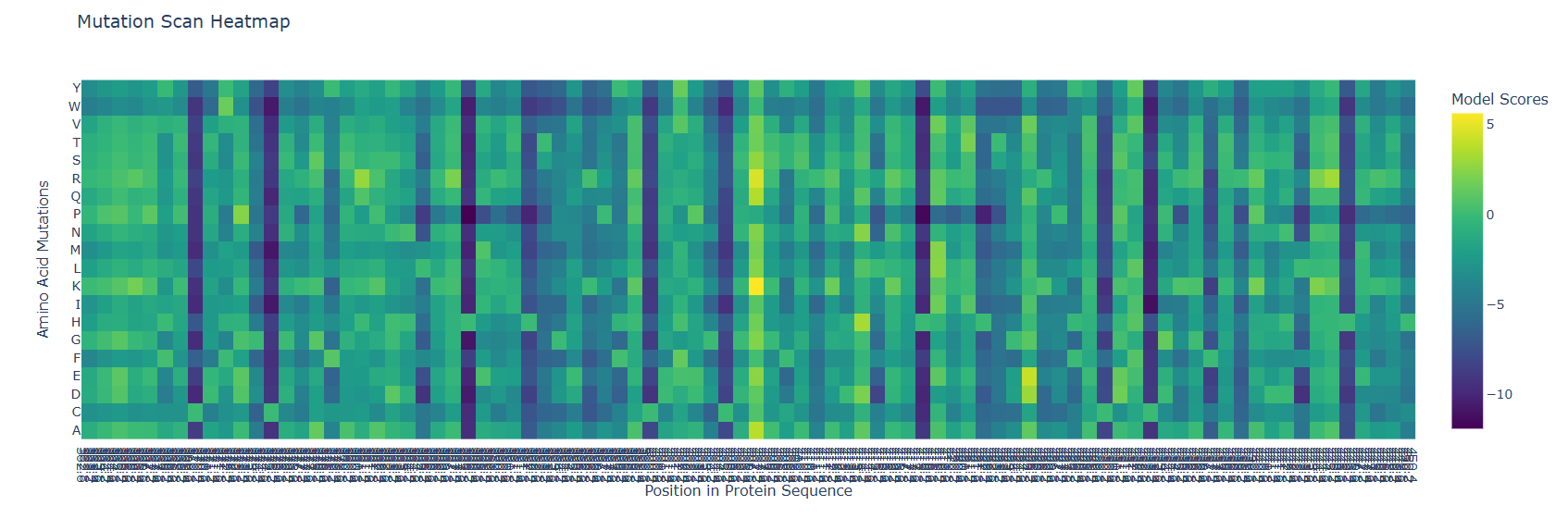
At the beggining and the mid part of the protein there are visible and abundant green/yellow zones, indicating that the protein function its not likely to be compromised by a mutation on this parts, but there is also a lot of dark violet zones at the end of the sequences, indicating that the probability of a critical mutation increases in that zone
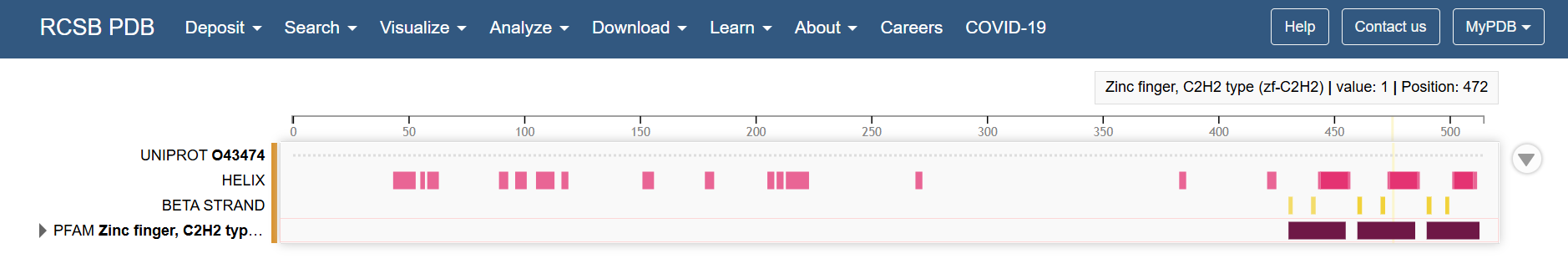
Looking at the Klf4 in the RCSB PDB page, the structural features of the last part of the sequence corresponds to the zinc fingers
So it makes sense that in a zoom in of the last part, the mutational scan looks like this
Latent Space Analysis
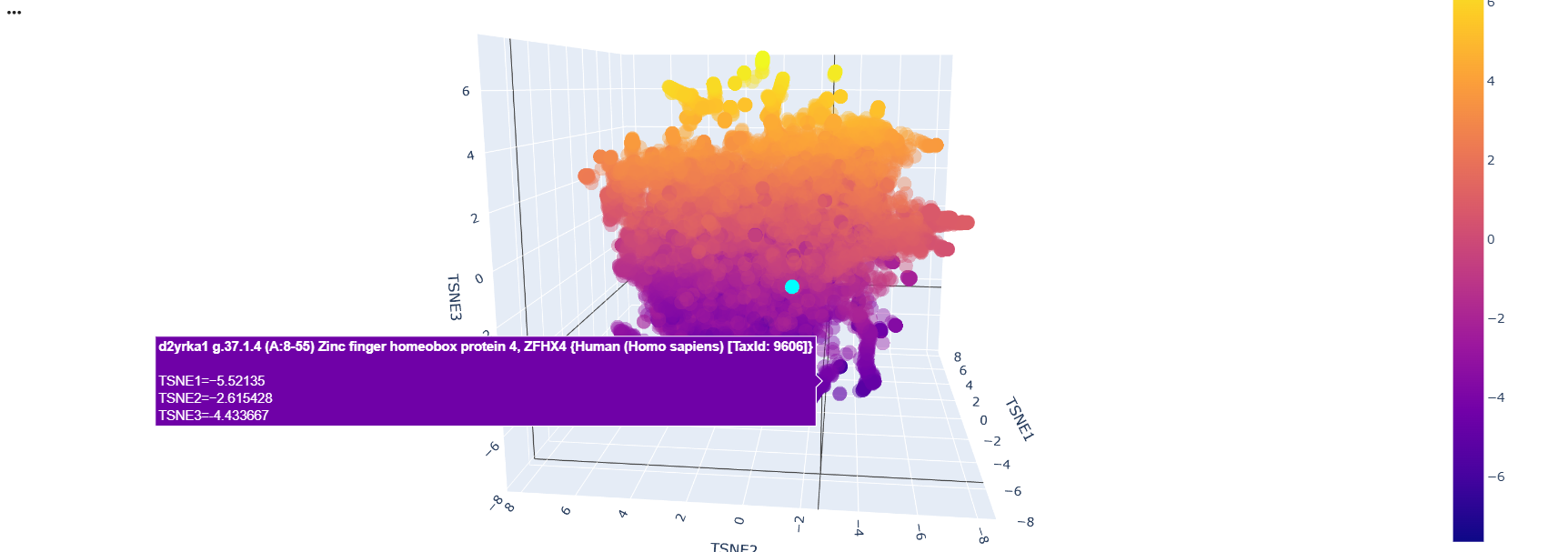
After running the code and analyzing the positions of tokens of sequences, I could find some clusters of similar proteins within the 3D depiction. just like these transcription factors whose position variability was almost constant on the TSNE2 axis
Then I added the Klf4 protein sequence to the “sequences” variable in the colab
And modified some visualization components to highlight the Klf4
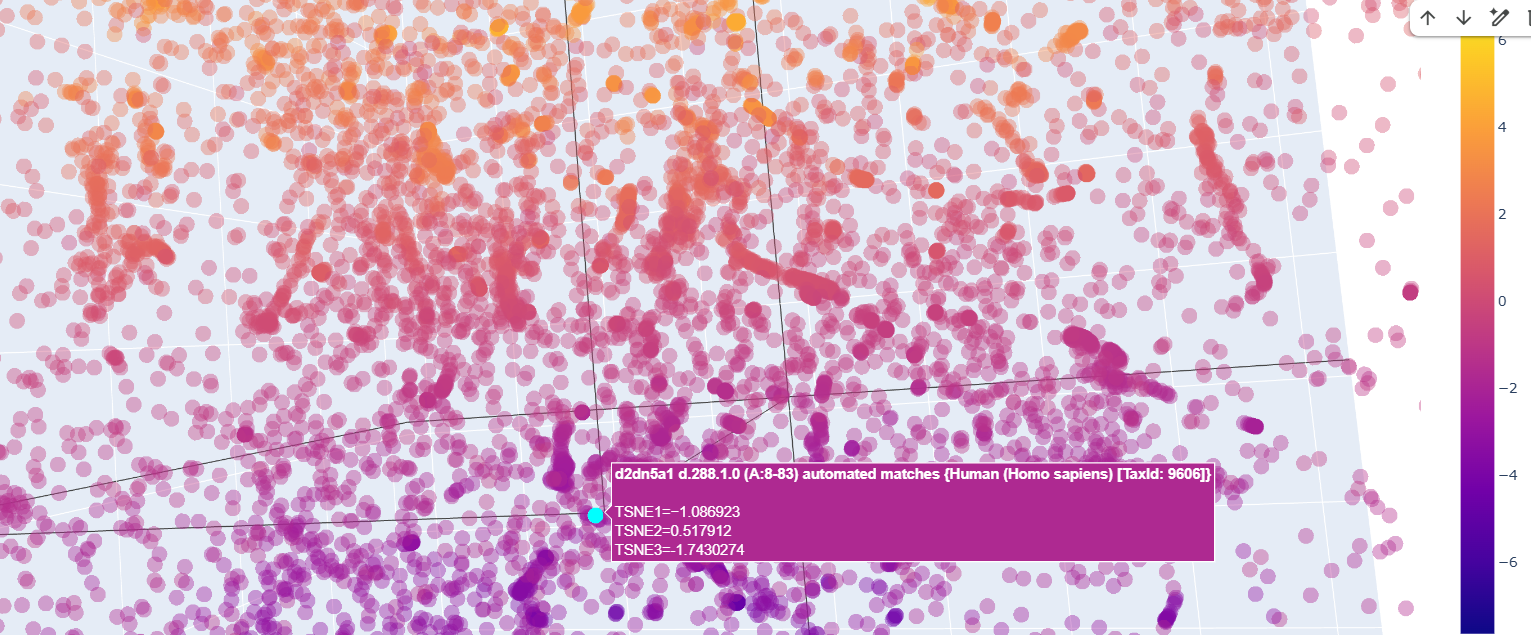
The neighbors of the protein were commongly also transcription factors.
There was a lot of Homo sapiens general transcription factors, as the d2dn5a1 seen in the image below
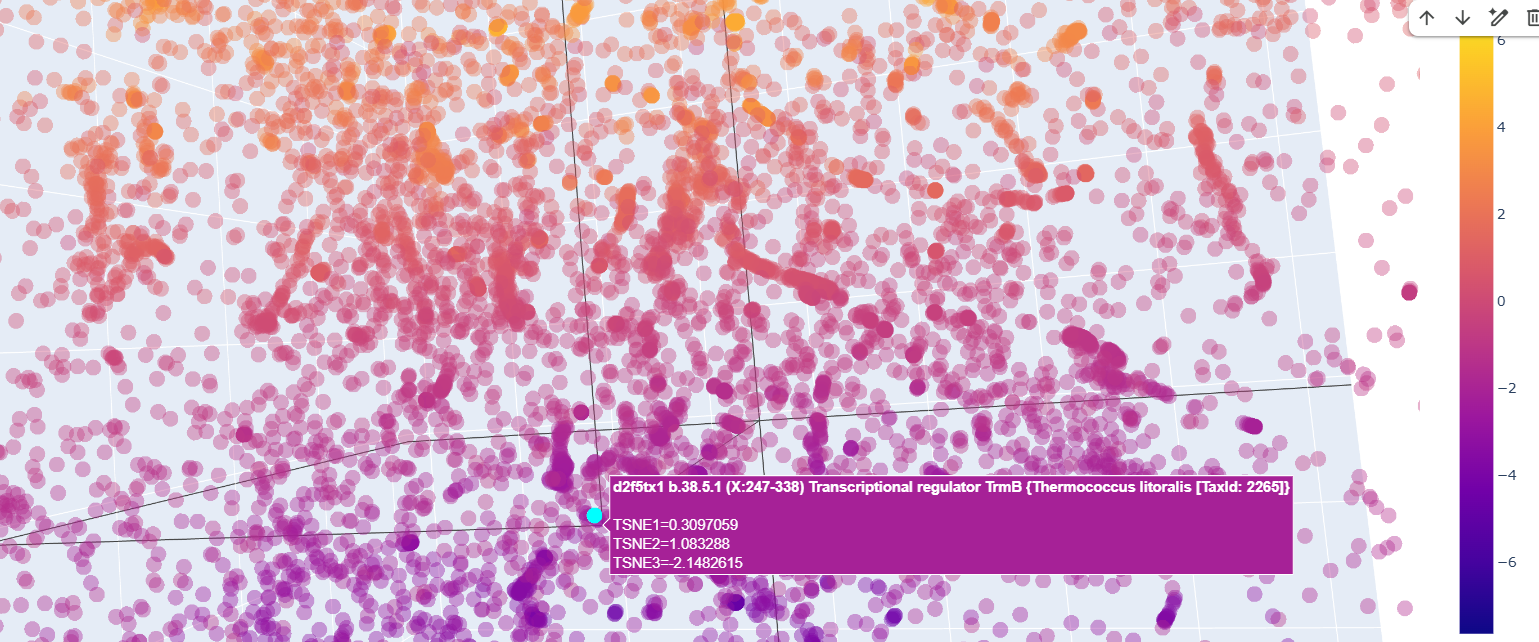
Also ther were transcriptional regulator factors of bacteria
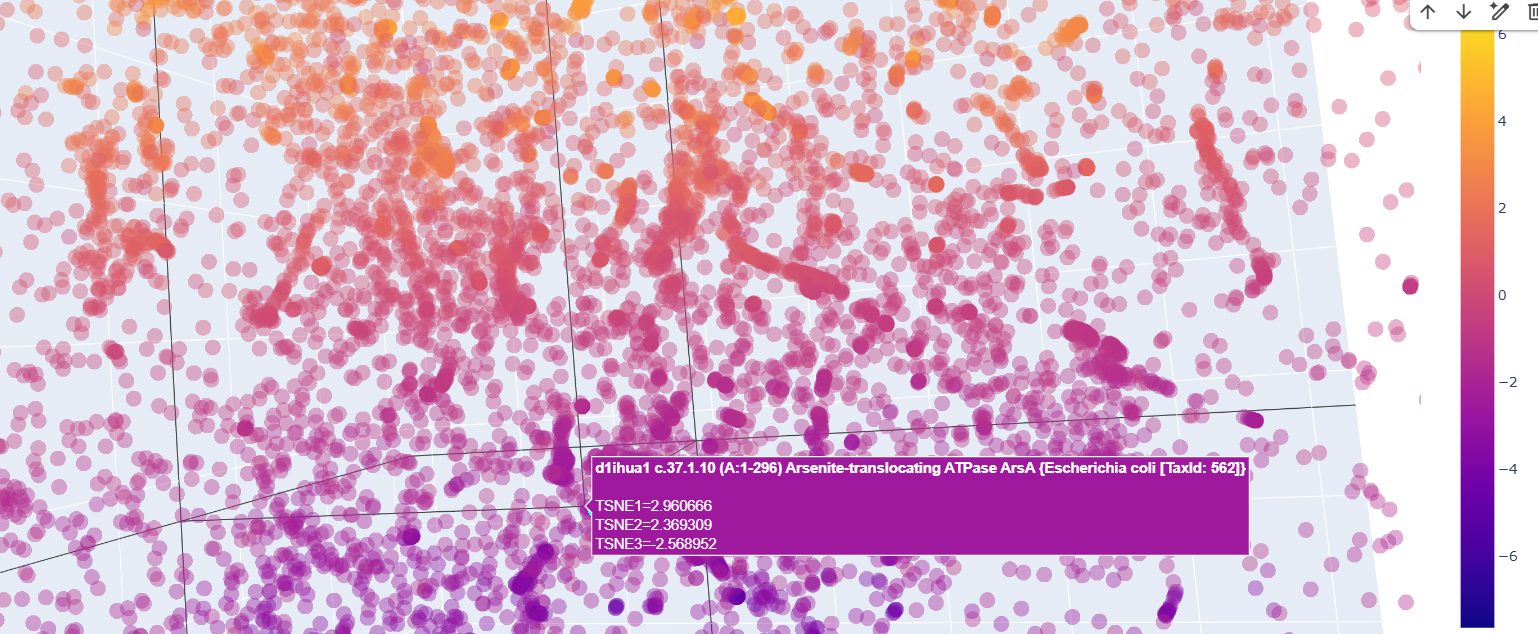
And quite surprisingly some functionally unrelated proteins like this arsenite translocating ATP-ase in E. colli
Curiously, proteins that at first glance were supposedly to be similr to Kfl4, were relativetely away from it.
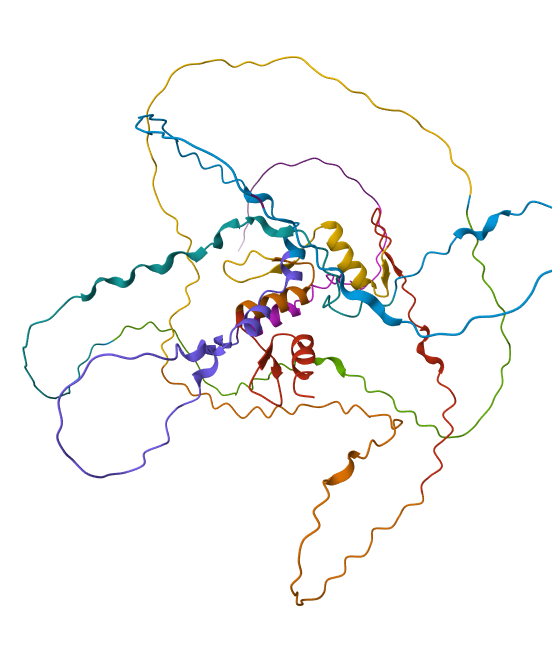
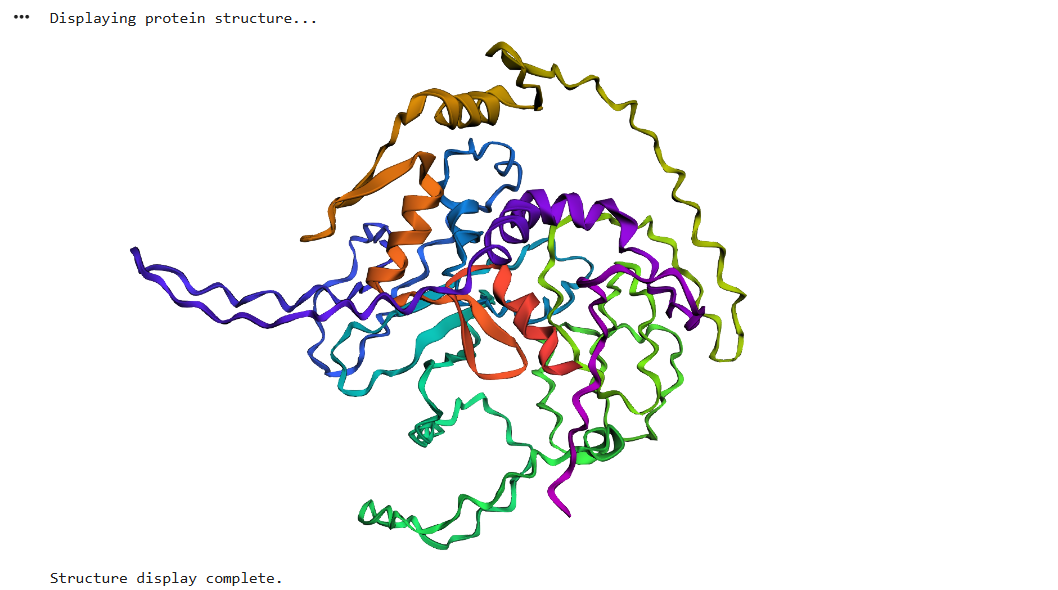
Protein Folding
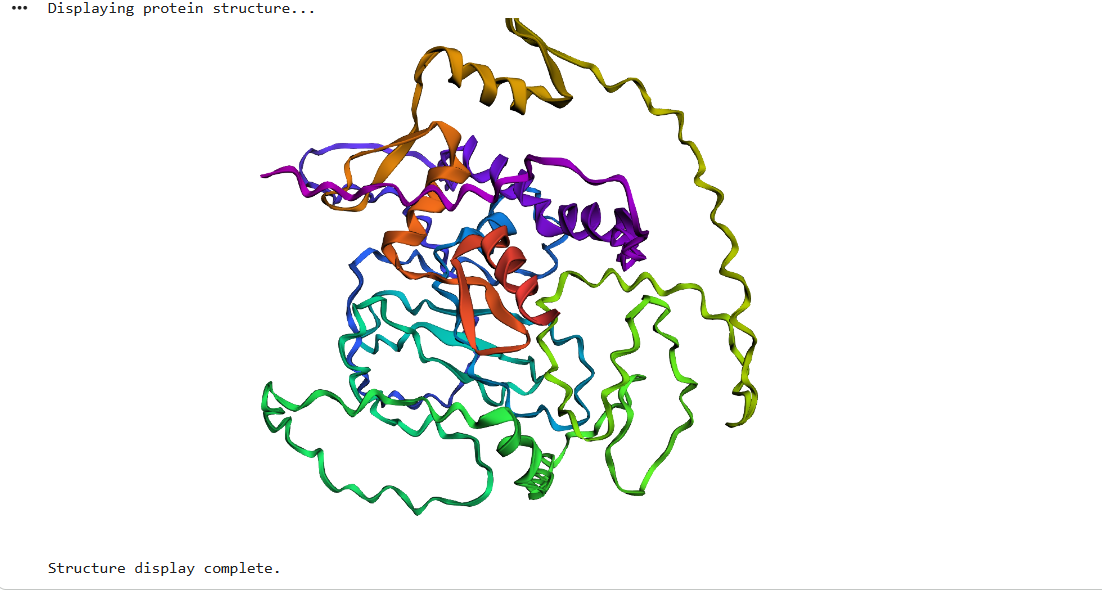
After running ESMFold with the Klf4
the result was quite similar to the RCSB 3D view of the same protein
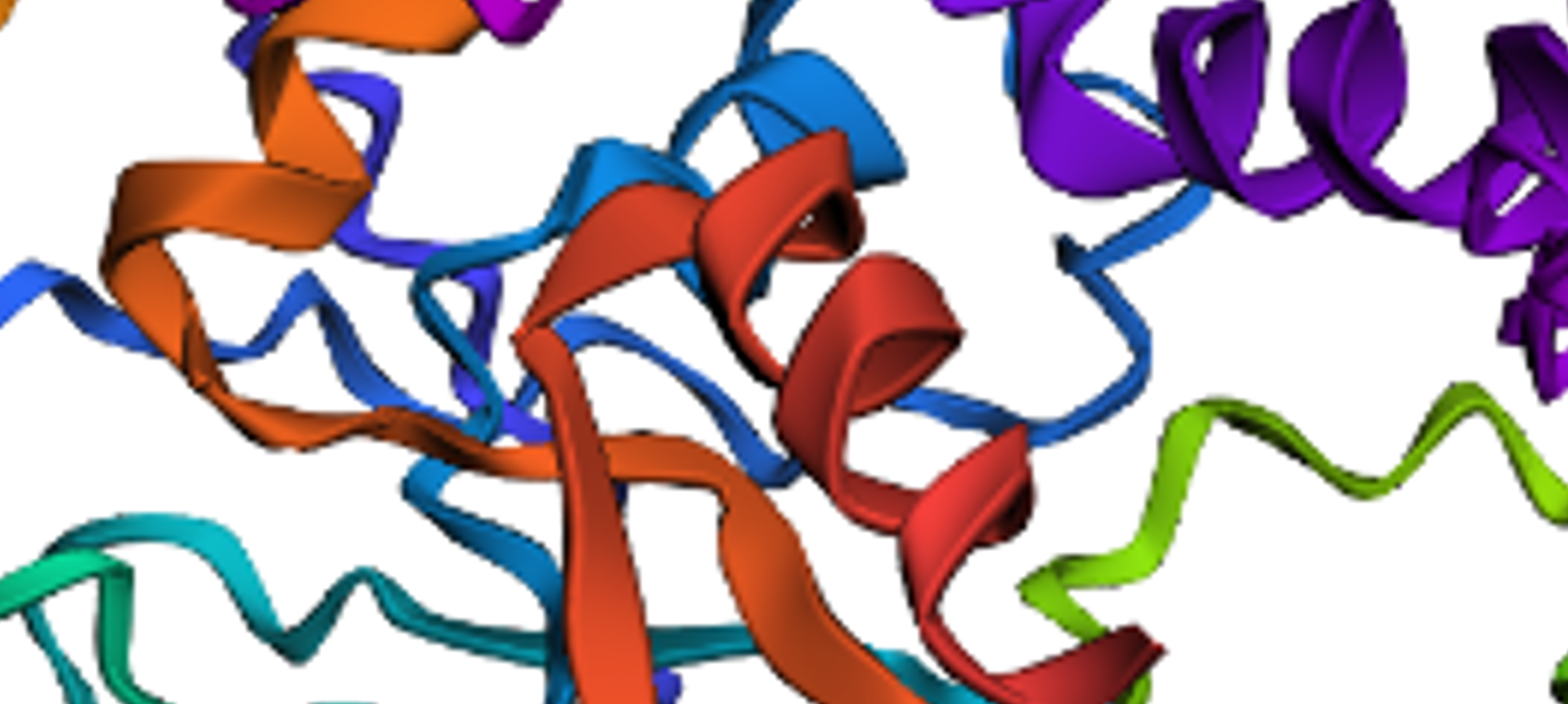
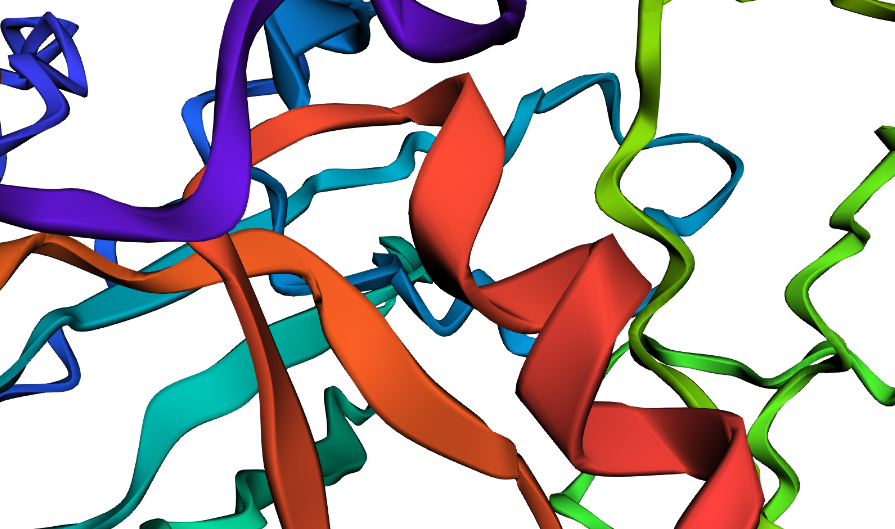
I did some puntual mutations at the end of the sequence (near the last alpha helix), replacing three amino acids with the last three prolines seen in the sequence below
The result at first glance appeared to be the same
But zooming in, you could se that the first turn of the last alpha helix was disrupted by the mutations
original
disrupted
Protein Generation (inverse-folding)
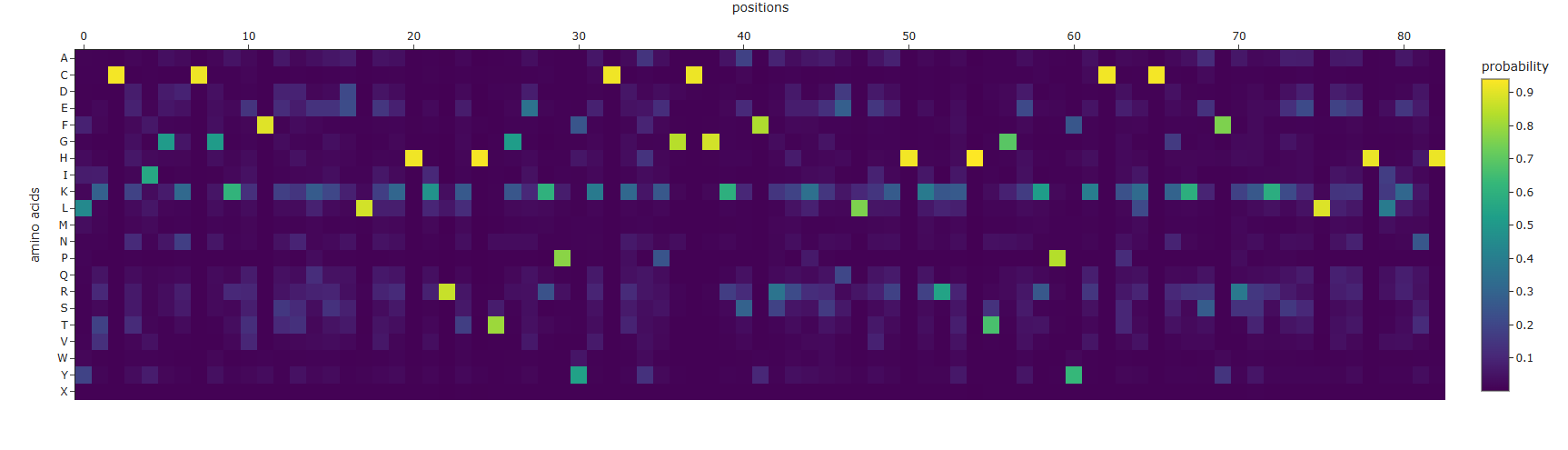
Using the code provided on this page, I inputted the PDB code for Klf4 (6VTX) to extract its sequence and generate a heatmap. I didn’t expect the resulting sequence to be this short compared to the original one
This heatmap is visually quite similar to the one we saw earlier. Indeed, while both the Deep Mutational Scan and this ProteinMPNN heatmap are probabilistic models that calculate conditional probabilities, their approaches are fundamentally different. The former uses a 1D array (as a masked language model) to predict an amino acid based on the sequence context of other amino acids. In contrast, ProteinMPNN uses a spatial graph to predict an amino acid based on the 3D structure of the protein, relying on the distances and angles between atoms.
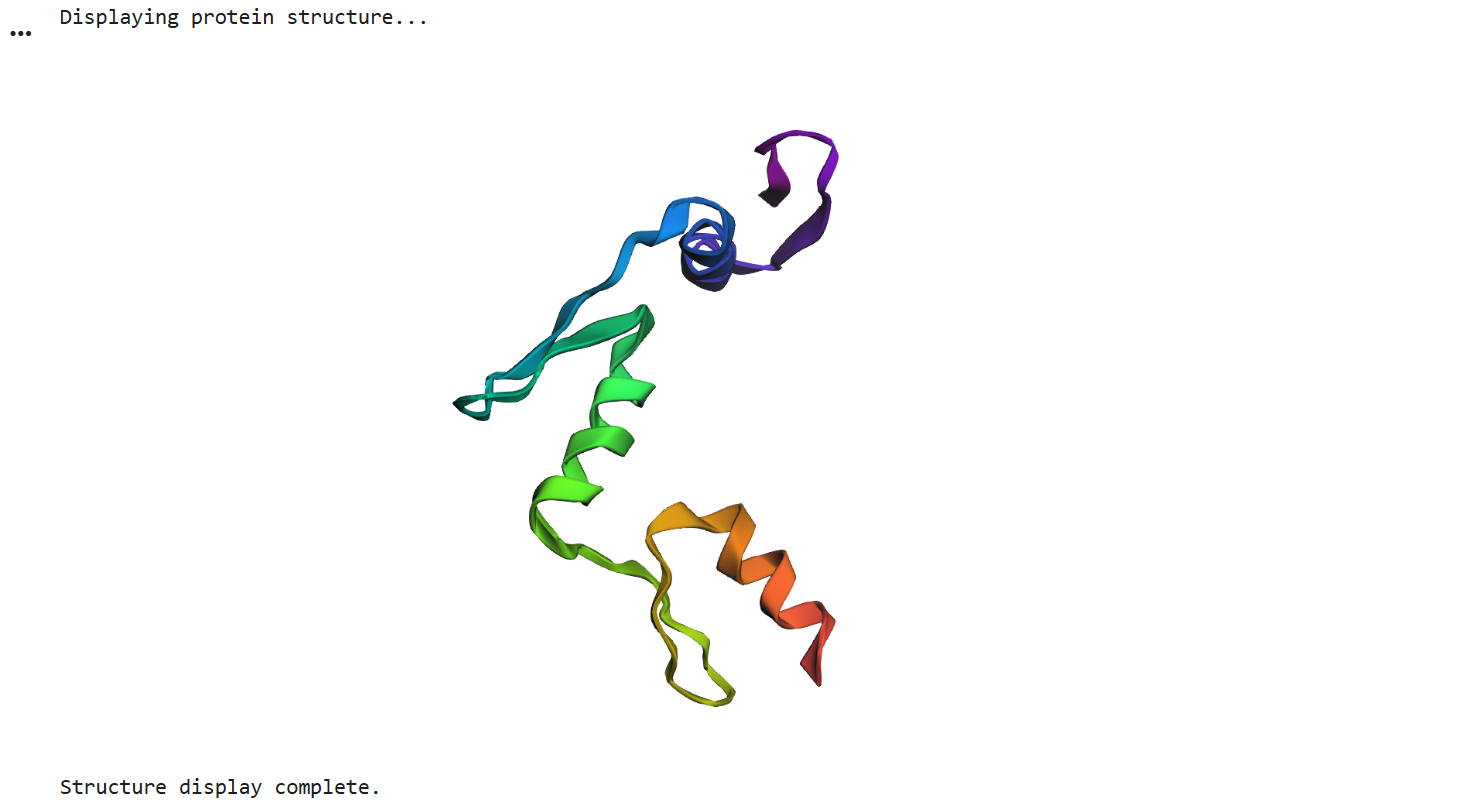
Finally, I ran ESMFold using the newly generated sequence and obtained this 3D structure. Although it’s visibly shorter than the original, the overall fold looks quite similar, and the alpha helices that predominantly define the structure have been successfully preserved