Using ML-Based Protein Design Tools
Protein Language Modeling
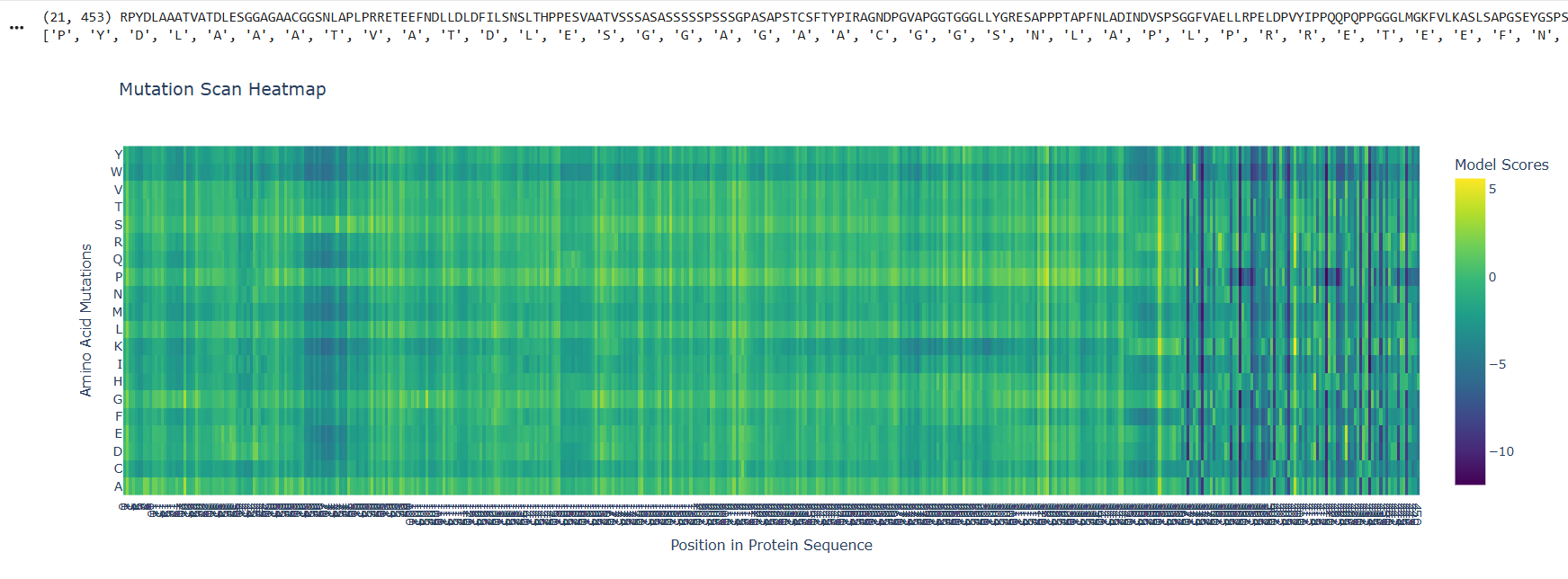
Deep Mutational Scans
At the beggining and the mid part of the protein there are visible and abundant green/yellow zones, indicating that the protein function its not likely to be compromised by a mutation on this parts, but there is also a lot of dark violet zones at the end of the sequences, indicating that the probability of a critical mutation increases in that zone
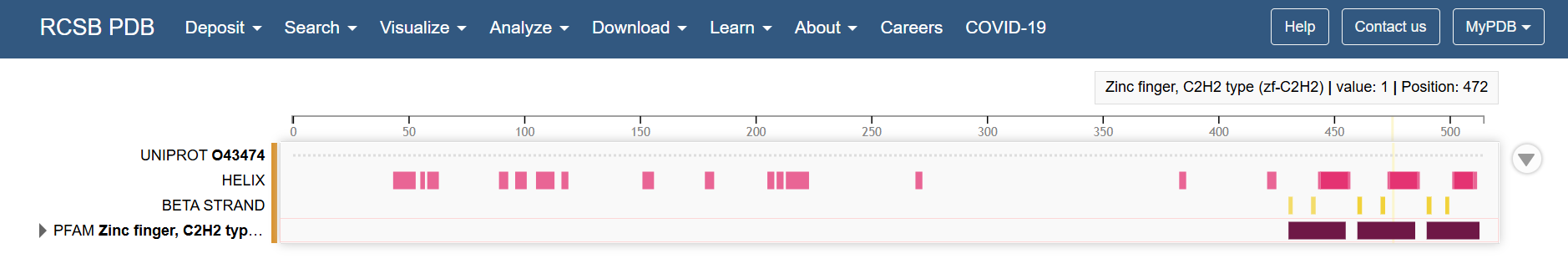
 Looking at the Klf4 in the RCSB PDB page, the structural features of the last part of the sequence corresponds to the zinc fingers
Looking at the Klf4 in the RCSB PDB page, the structural features of the last part of the sequence corresponds to the zinc fingers
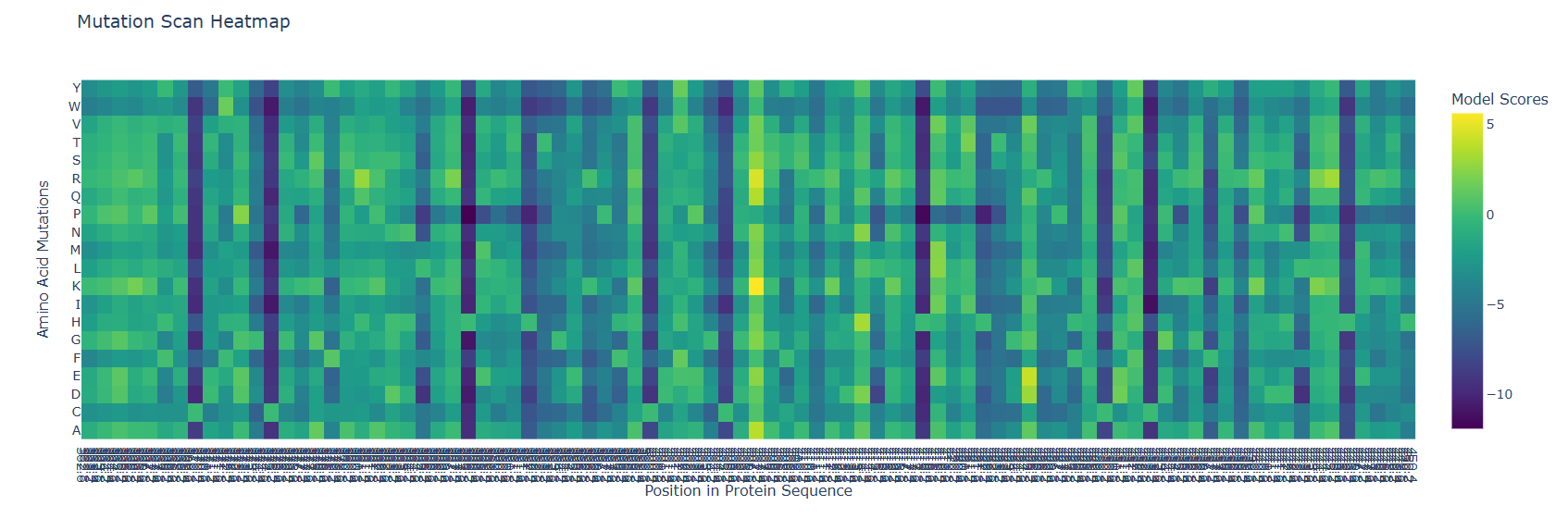
 So it makes sense that in a zoom in of the last part, the mutational scan looks like this
So it makes sense that in a zoom in of the last part, the mutational scan looks like this
Latent Space Analysis
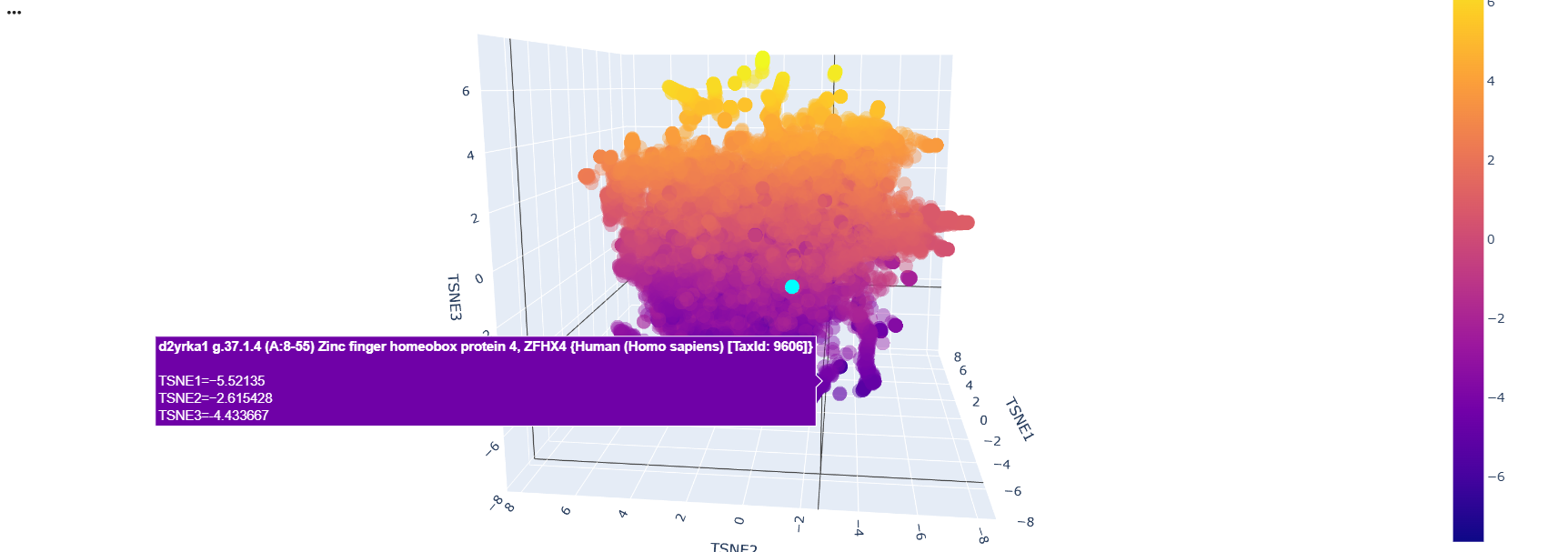
After running the code and analyzing the positions of tokens of sequences, I could find some clusters of similar proteins within the 3D depiction. just like these transcription factors whose position variability was almost constant on the TSNE2 axis
![]()
![]()
Then I added the Klf4 protein sequence to the “sequences” variable in the colab
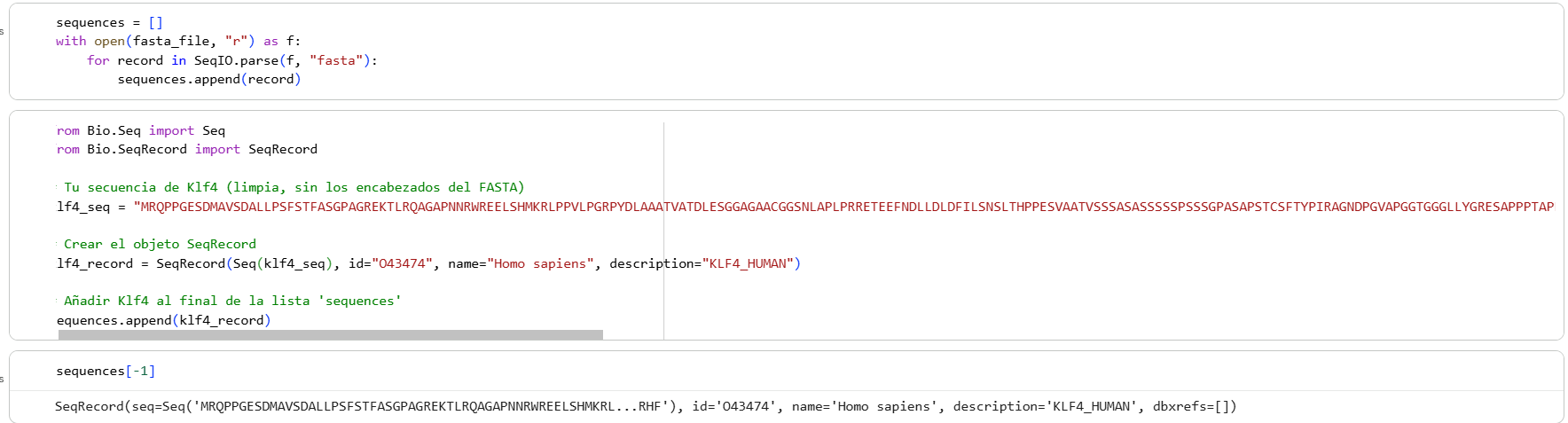
And modified some visualization components to highlight the Klf4
The neighbors of the protein were commongly also transcription factors.
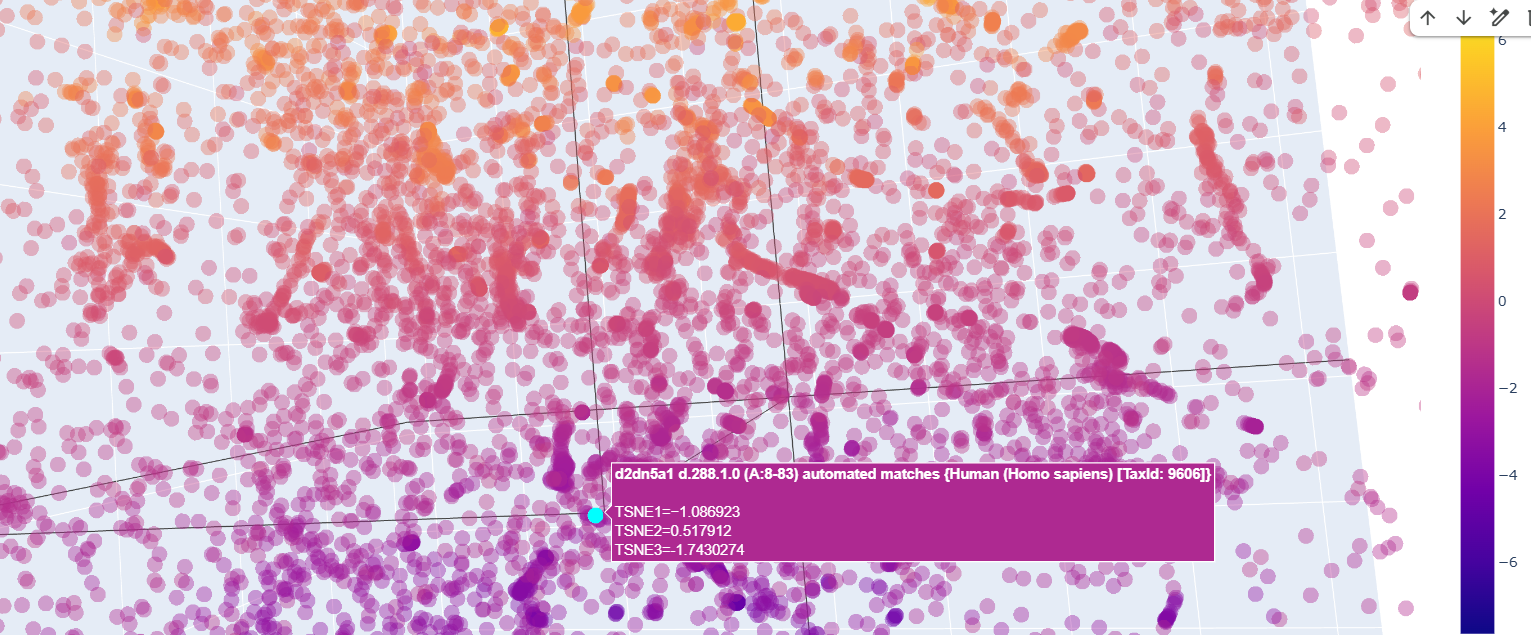
There was a lot of Homo sapiens general transcription factors, as the d2dn5a1 seen in the image below
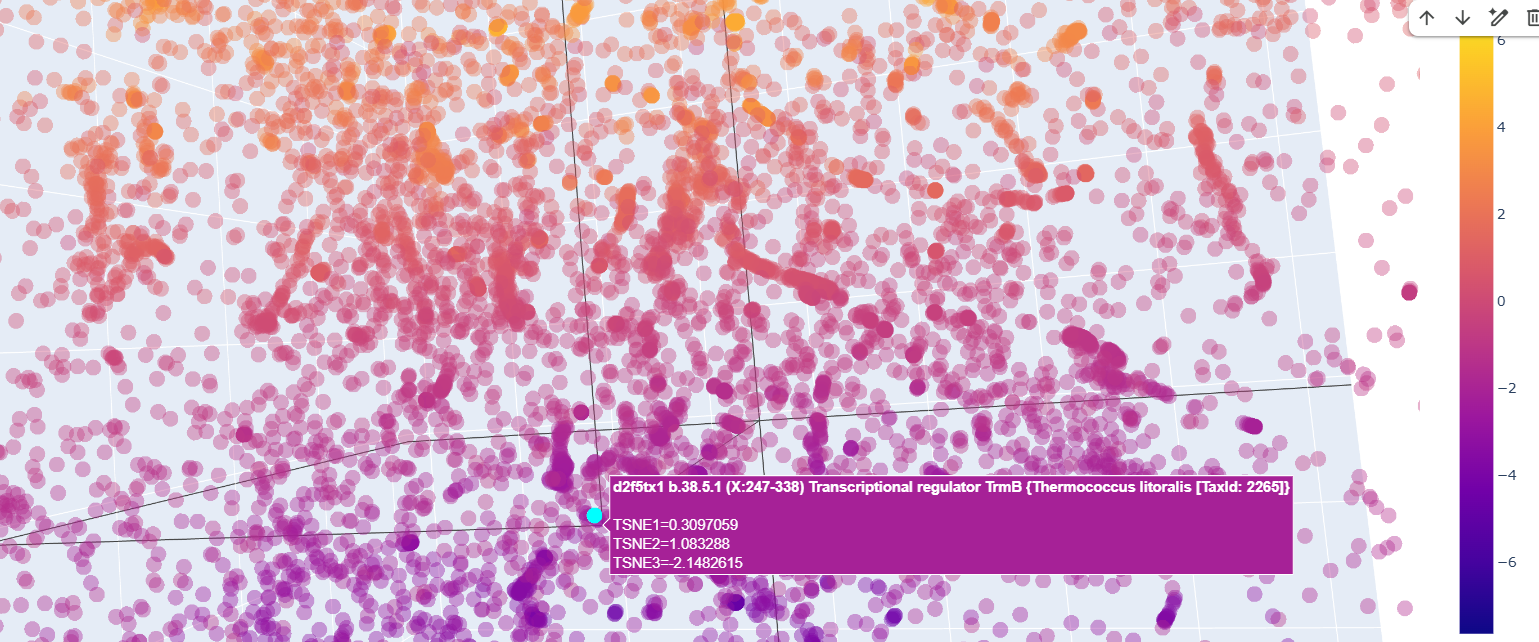
 Also ther were transcriptional regulator factors of bacteria
Also ther were transcriptional regulator factors of bacteria
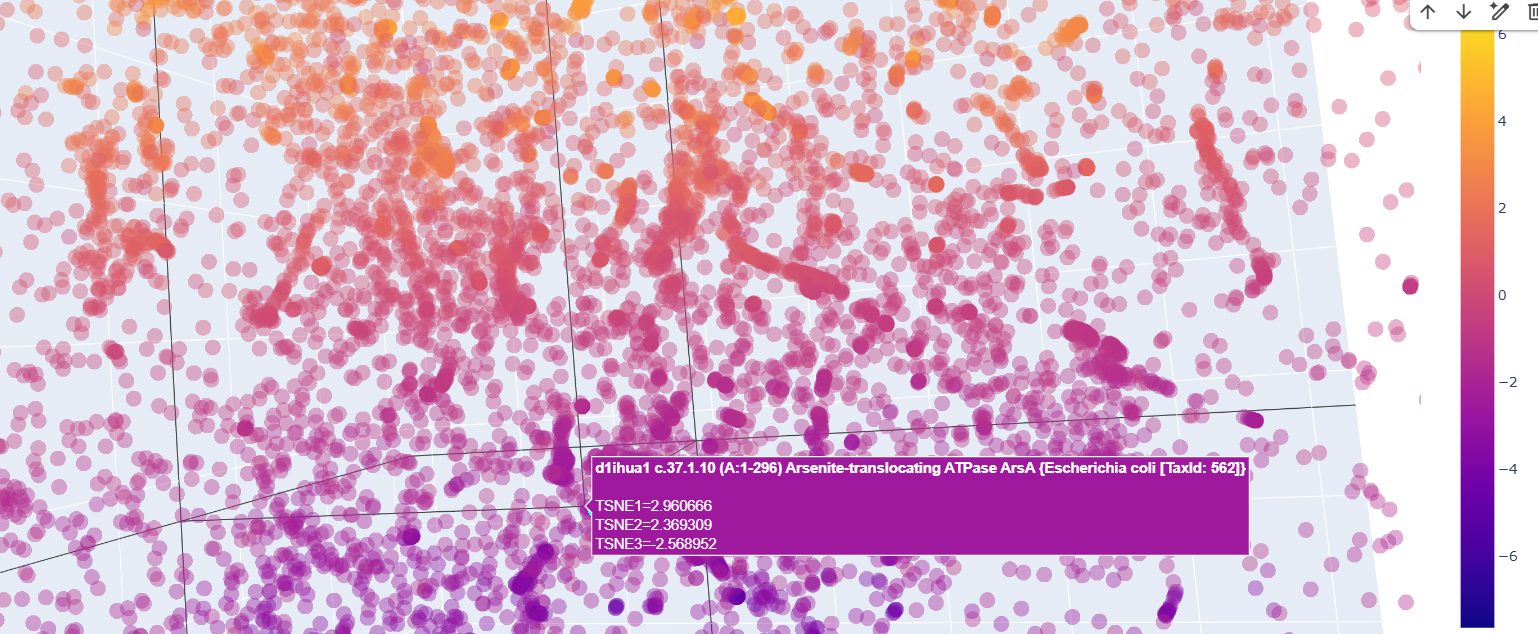
 And quite surprisingly some functionally unrelated proteins like this arsenite translocating ATP-ase in E. colli
And quite surprisingly some functionally unrelated proteins like this arsenite translocating ATP-ase in E. colli
Curiously, proteins that at first glance were supposedly to be similr to Kfl4, were relativetely away from it.
Protein Folding
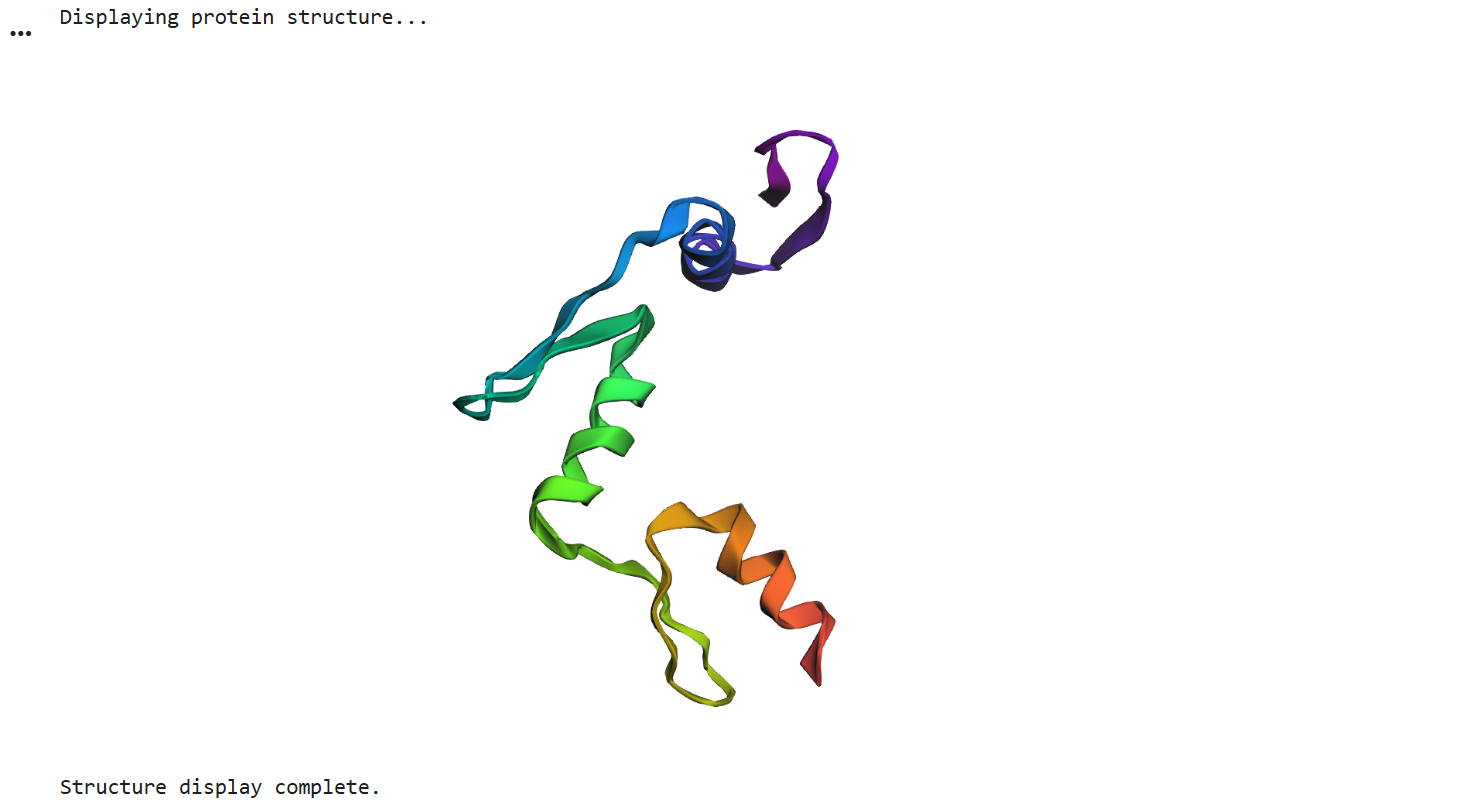
After running ESMFold with the Klf4
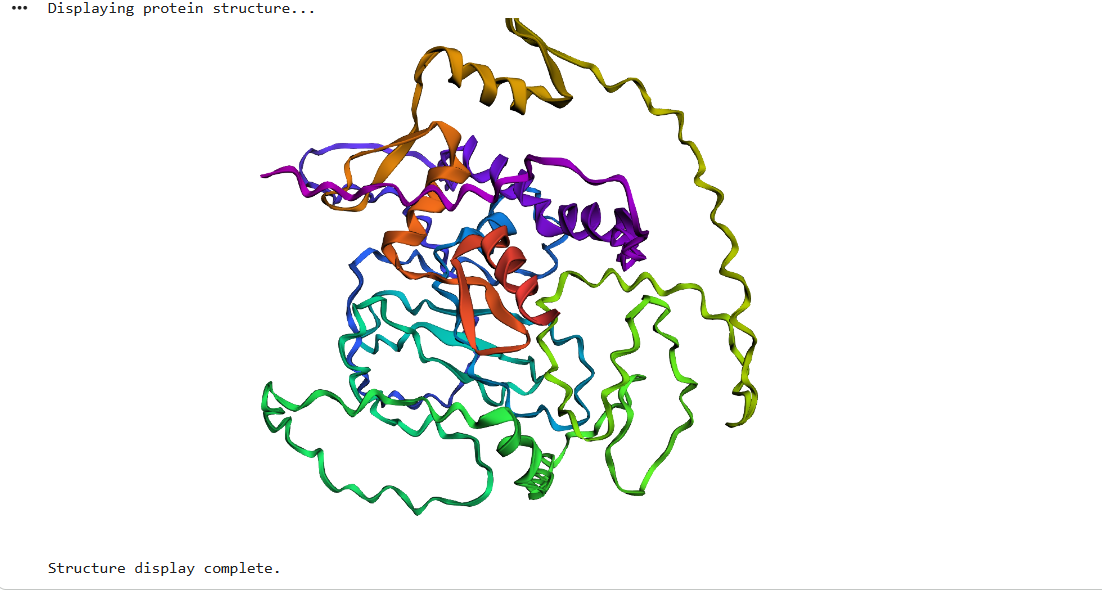 the result was quite similar to the RCSB 3D view of the same protein
the result was quite similar to the RCSB 3D view of the same protein
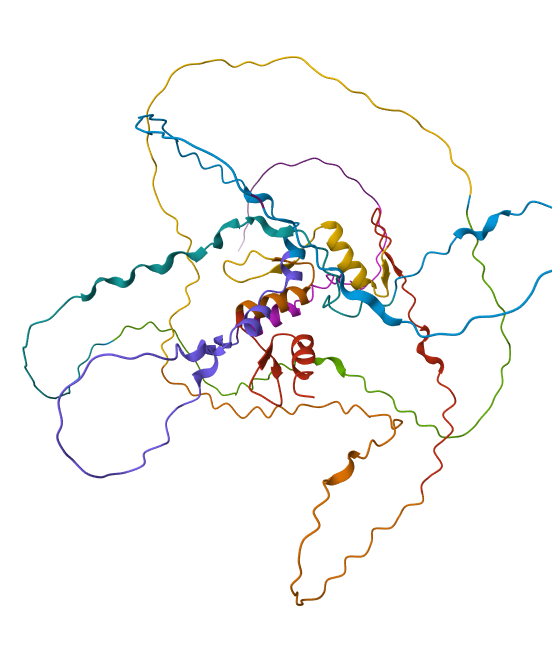
I did some puntual mutations at the end of the sequence (near the last alpha helix), replacing three amino acids with the last three prolines seen in the sequence below

The result at first glance appeared to be the same
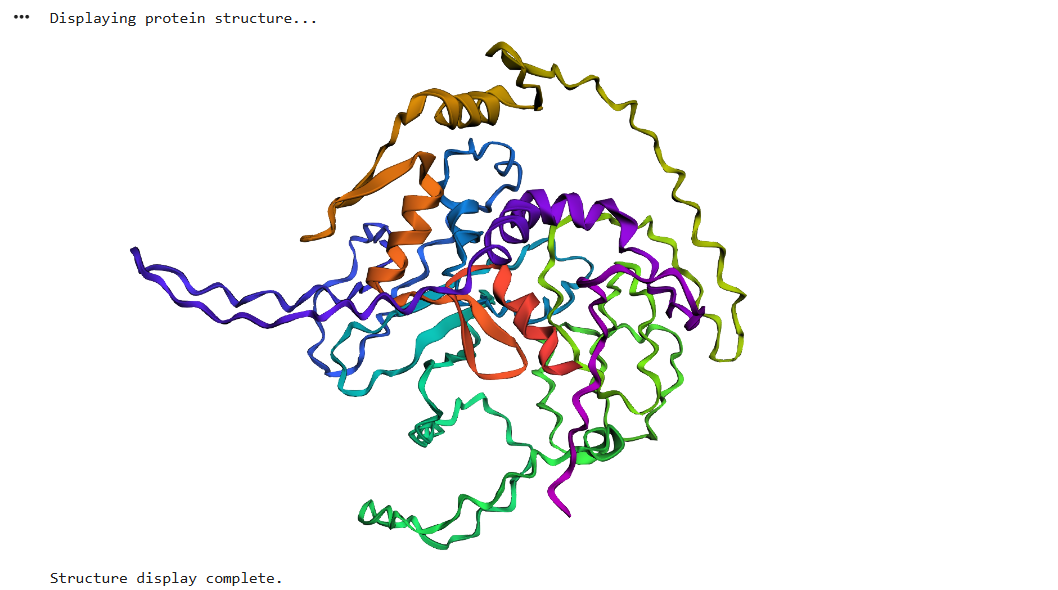
But zooming in, you could se that the first turn of the last alpha helix was disrupted by the mutations
original
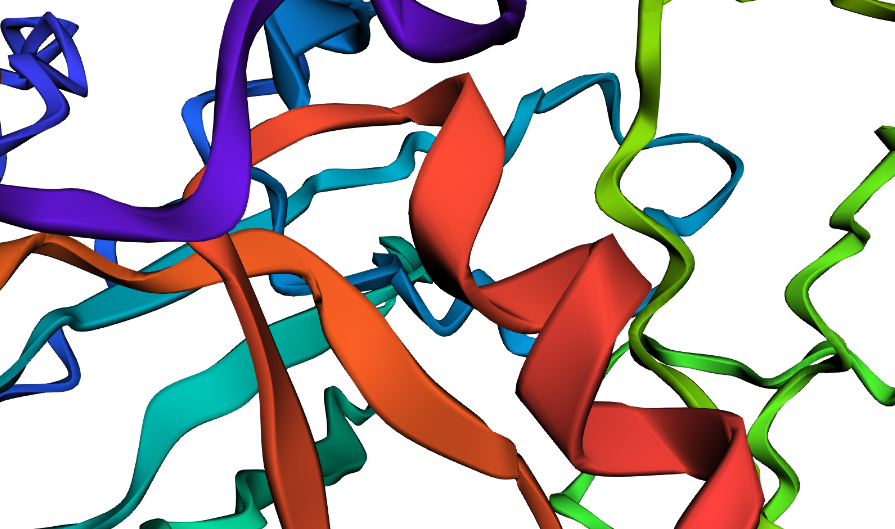
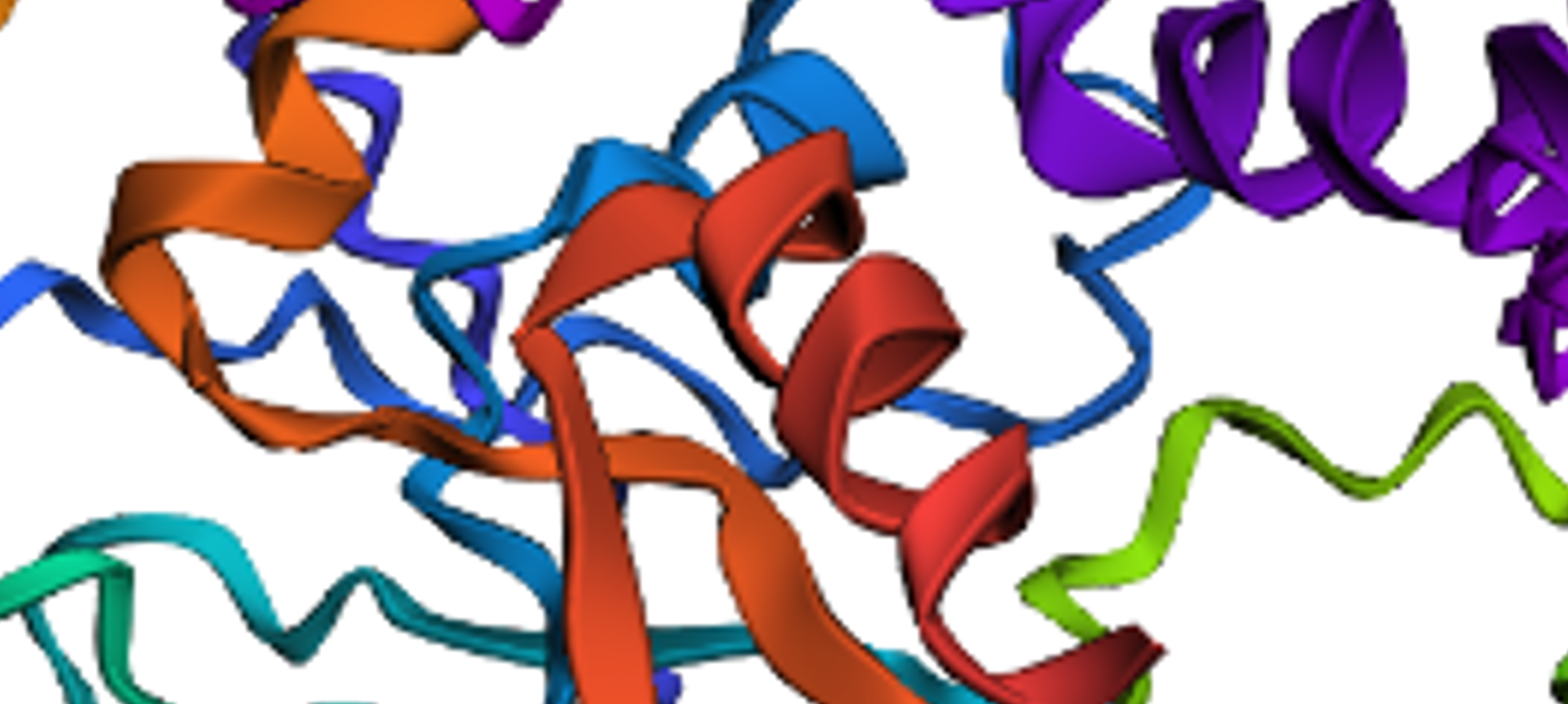 disrupted
disrupted
Protein Generation (inverse-folding)
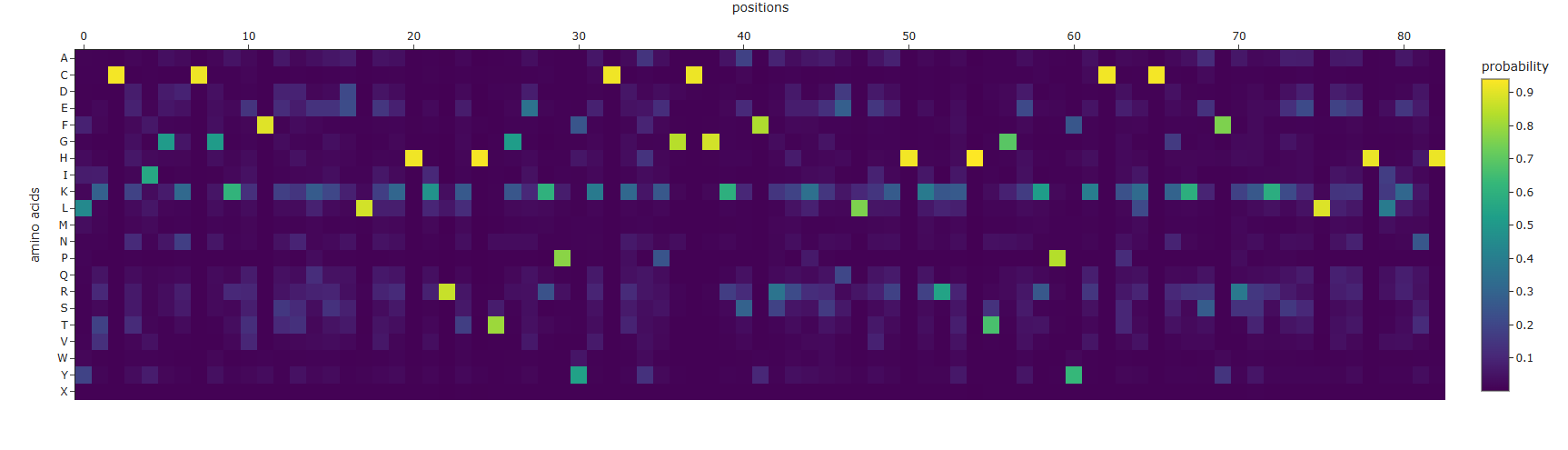
Using the code provided on this page, I inputted the PDB code for Klf4 (6VTX) to extract its sequence and generate a heatmap. I didn’t expect the resulting sequence to be this short compared to the original one

This heatmap is visually quite similar to the one we saw earlier. Indeed, while both the Deep Mutational Scan and this ProteinMPNN heatmap are probabilistic models that calculate conditional probabilities, their approaches are fundamentally different. The former uses a 1D array (as a masked language model) to predict an amino acid based on the sequence context of other amino acids. In contrast, ProteinMPNN uses a spatial graph to predict an amino acid based on the 3D structure of the protein, relying on the distances and angles between atoms.
Finally, I ran ESMFold using the newly generated sequence and obtained this 3D structure. Although it’s visibly shorter than the original, the overall fold looks quite similar, and the alpha helices that predominantly define the structure have been successfully preserved