Week 2 HW: dna read write and edit
WEEK 2 Assignment
AI Assistance Disclosure
For this week’s assignments, I continued using ChatGPT as a learning support tool to help clarify key concepts that were not yet fully formed in my understanding. I used it primarily to better comprehend the questions and to guide my reasoning process while developing my responses.
I uploaded and referenced the lecture slides materials directly within ChatGPT so that they could serve as the primary foundation for the explanations and guidance provided. ChatGPT was used to support conceptual clarification, provide structural guidance, and help organize the final responses in a clear and coherent manner.
Part 1: Benchling & In-silico Gel Art
Part 1 - answer (click to expand)
First, I obtained the genome sequence from the
NCBI website:
Escherichia phage Lambda, complete genome
GenBank accession: J02459.1
https://www.ncbi.nlm.nih.gov/nuccore/J02459.1?report=genbank&log$=seqview
I imported the Lambda phage genome into Benchling and performed in-silico restriction digests using only the enzymes listed in the assignment. The final enzymes of choice used were:
- EcoRV
- SacI
- SalI
For gel visualization, I selected the Life 1 kb Plus ladder in Benchling as the molecular weight reference.
I was not able to determine which ladder was used in the online gel-art simulation tool (https://rcdonovan.com/gel-art). Therefore, I used Donovan’s website primarily as a visual reference to explore possible banding patterns and compositions. When comparing the simulated gel in Benchling using the Life 1 kb Plus ladder, the patterns appeared slightly different from those shown in the Donovan tool. However, it was still possible to approximate the general distribution, number of fragments, and relative band intensity.
Concept and Strategy
My conceptual goal was to create a face-like figure using the gel band patterns.
My strategy was:
- In the outer lanes, I used enzymes that produced a larger number of fragments distributed across the gel. These denser patterns formed a visual “frame,” resembling hair around the face.
- For the central features (eyes and mouth), I selected enzymes that produced fewer fragments and simpler band patterns. These cleaner lanes helped define the facial features with only lines in different positions
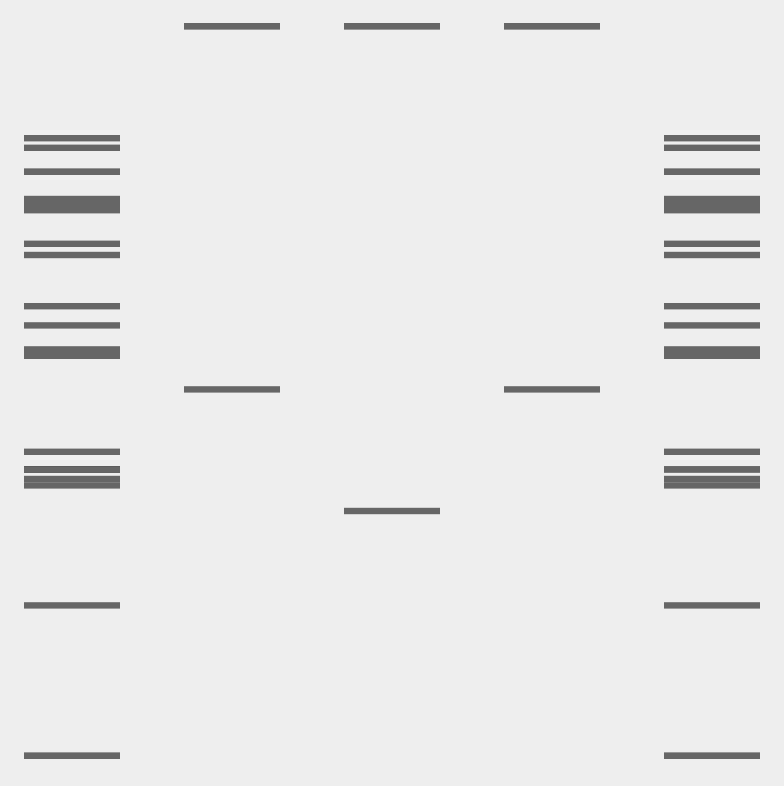
Here is the print of the Benchling:

Below is the image of the gel pattern I intended to generated.
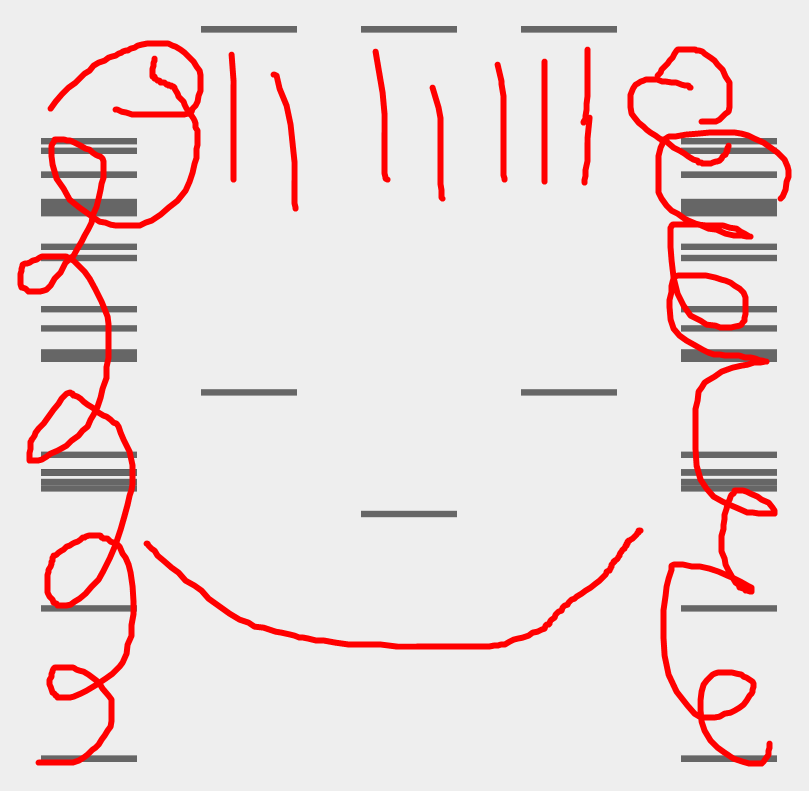
Additionally, I created an overlay sketch to better illustrate and clarify the face-like figure that I intended to form.
Part 3: DNA Design Challenge
3.1. Choose your protein.
3.1. - answer (click to expand)
Protein Chosen: AlgG – Mannuronan C5-epimerase
Alginate C-5 epimerase AlgG
Organism: Azotobacter vinelandii
UniProtKB: Reviewed (Swiss-Prot)
Accession: P70805
Length: 525 amino acids
I chose the AlgG (mannuronan C-5-epimerase) protein from Azotobacter vinelandii because it plays a key role in alginate biosynthesis. AlgG catalyzes the epimerization of β-D-mannuronic acid (M) residues into α-L-guluronic acid (G) residues within the alginate polymer. The M/G ratio directly influences the mechanical properties and calcium crosslinking behavior of alginate-based biomaterials. Since my research interests focus on alginate structural materials and biofabrication, this enzyme is conceptually aligned with my project goals.
AlgG protein sequence - FASTA
sp|P70805|ALGG_AZOVI Mannuronan C5-epimerase AlgG OS=Azotobacter vinelandii OX=354 GN=algG PE=1 SV=1
MNVQRKLASTQLKPVLLGVLLATSAWSQAAPPEQARQSAPPTLSSKQYSVTSASIEALKL DPPKLPDLSGYTHAAVEAKIRRKPGGRIAAAMLQQTALKDFTGGSGRLREWIVRQGGMPH AIFIEGGYVELGQLARQLPANQFAETTPGVYVARVPIVVAPGATLHIGKNVKELRLSEER GAFLVNDGKLFITDTKLVGWSEKNNAPSAYRGPESFWAFLVSWGGTETYISRRPVASLGY NTSKAYGVSITQYTPEMHKRLKRPRPTGWLIDSVFEDIYYGFYCYEADDVVLKGNTYRDN IIYGIDPHDRSERLVIAENHVYGTKKKHGIIVSREVNNSWIINNRTHDNKLSGIVLDRNS EHNLVAYNEVYQNHSDGITLYESSNNLIWGNRLINNARHGIRMRNSVNIRIYENLSVVNQ LTGIYGHIKDLSSTDRDFKLDPFDTKVSMIVVGGQLTGNGSSPISVDSPLSLELYRVEML APTKSSGLTFTGILEDKQEEILDLLVRRQKAVLIDPVVDLAQAEL
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
3.2. - answer (click to expand)
I did not use any reverse translation tool, because I retrieved the native coding sequence (CDS) of the algG gene directly from the NCBI GenBank database (Accession: X87973).
Because the AlgG protein sequence is already experimentally characterized and linked to a published genomic record, obtaining the authentic CDS from GenBank provides the biologically accurate nucleotide sequence rather than a hypothetical reverse-translated version. Reverse translation can generate multiple possible DNA sequences due to codon degeneracy, whereas the GenBank entry corresponds to the naturally occurring gene sequence from Azotobacter vinelandii.
The CDS spans nucleotides 68–1645 of the deposited genomic record, corresponding to a 1578 bp coding region, including the stop codon. This sequence encodes the 525–amino-acid AlgG protein and matches the reviewed UniProt entry (P70805).
FASTA CDS: 68 to 1645
lcl|X87973.1_cds_CAA61231.1_2 [gene=algG][db_xref=GOA:P70805,InterPro:IPR006626,InterPro:IPR006633,InterPro:IPR011050,InterPro:IPR012334,InterPro:IPR022441,UniProtKB/Swiss-Prot:P70805][protein=epimerase] [protein_id=CAA61231.1] [location=68..1645] [gbkey=CDS]ATGAACGTGCAAAGAAAACTTGCATCCACCCAGCTGAAACCCGTGTTGCTCGGCGTGCTGCTGGCCACCA GCGCCTGGAGCCAGGCCGCCCCGCCGGAGCAGGCGAGGCAGTCCGCGCCCCCCACCCTGAGTTCGAAGCA GTACAGCGTCACCAGCGCCTCGATCGAAGCCTTGAAGCTGGACCCGCCCAAACTGCCGGATCTCTCCGGC TACACTCACGCGGCGGTGGAGGCCAAGATCCGGCGCAAGCCCGGTGGACGCATCGCTGCGGCCATGCTGC AGCAGACCGCCCTGAAGGACTTCACCGGTGGCAGCGGACGTCTGCGCGAGTGGATCGTCCGCCAGGGCGG TATGCCTCACGCGATCTTCATAGAAGGCGGCTATGTCGAGCTGGGTCAGTTGGCCAGGCAGTTGCCGGCC AATCAGTTCGCCGAGACCACGCCGGGCGTCTACGTGGCGCGGGTGCCGATCGTCGTCGCCCCCGGCGCGA CCCTGCACATCGGCAAGAACGTCAAGGAGCTGCGCCTCTCCGAGGAGCGCGGCGCCTTCCTGGTCAACGA TGGCAAGCTGTTCATCACCGACACCAAGCTGGTCGGCTGGAGCGAGAAGAACAACGCTCCGTCCGCCTAC CGCGGCCCGGAAAGCTTCTGGGCCTTCCTGGTGTCCTGGGGCGGCACCGAGACCTACATCTCGCGCAGAC CCGTCGCCAGCCTGGGCTACAACACCAGTAAGGCCTACGGCGTGAGCATCACCCAGTACACCCCGGAAAT GCACAAGCGCCTCAAGCGCCCGCGCCCGACCGGCTGGCTGATCGACTCGGTATTCGAGGACATCTACTAC GGCTTCTACTGCTACGAAGCCGACGACGTGGTGCTCAAGGGCAATACCTACCGCGACAACATCATCTACG GCATCGACCCCCACGACCGCTCGGAACGCCTGGTCATCGCCGAGAACCACGTCTACGGGACGAAGAAGAA GCACGGCATCATCGTCTCGCGGGAGGTCAACAACAGTTGGATCATCAACAACCGCACCCACGACAACAAG CTGTCGGGCATCGTTCTCGACCGTAACAGCGAACACAACCTGGTCGCCTACAACGAGGTGTACCAGAACC ACTCCGACGGCATCACCCTCTACGAGAGTTCGAACAACCTGATCTGGGGCAACCGGCTCATCAACAACGC GCGCCACGGCATCCGCATGCGCAACAGCGTGAACATCCGGATCTACGAGAACCTGTCCGTCGTCAACCAG TTGACCGGTATCTACGGTCACATCAAGGACCTCAGCAGCACCGACCGTGACTTCAAGCTCGACCCCTTCG ACACCAAGGTGTCGATGATCGTGGTCGGTGGCCAACTGACCGGCAACGGTTCGTCGCCGATCTCCGTGGA CTCGCCGCTGAGCCTCGAACTCTACCGCGTGGAGATGCTCGCCCCGACCAAGAGTTCCGGCCTCACCTTC ACCGGCATCCTCGAGGACAAACAAGAAGAGATCCTCGATCTGCTGGTGCGCCGCCAGAAGGCCGTGCTGA TCGACCCCGTCGTCGATCTCGCCCAGGCCGAGCTGTAG
3.3. Codon optimization
3.3. - answer (click to expand)
I selected Escherichia coli as the host organism for codon optimization because it is a widely used bacterial system for recombinant protein production.
Codon optimization is necessary because different organisms exhibit codon bias, meaning they preferentially use certain codons to encode specific amino acids. Although multiple codons can encode the same amino acid, translation efficiency depends on the availability and abundance of corresponding tRNAs within the host organism.
Since the native algG gene originates from Azotobacter vinelandii, its codon usage may not be optimal for efficient expression in E. coli. Differences in codon preference could result in reduced translation efficiency or lower protein yield. Therefore, the nucleotide sequence was optimized for E. coli to enhance translation efficiency, improve mRNA stability, and increase recombinant protein production while preserving the original 525-amino-acid sequence of AlgG.
I used the GenSmart Codon Optimization Tool:
Here is the GenSmart Optimization PDF results:
and the algG for Escherichia coli Optimized sequence:
ATGAATGTACAAAGGAAACTAGCTTCAACACAACTGAAGCCGGTATTGCTGGGCGTTTTACTGGCCACCTCGGCGTGGTCACAAGCGGCTCCGCCAGAGCAGGCGCGTCAAAGCGCGCCTCCGACCCTGTCTAGCAAGCAGTATAGCGTTACCTCTGCGAGCATTGAAGCGCTGAAGCTCGACCCGCCCAAACTGCCAGATCTGTCCGGTTACACCCACGCCGCGGTGGAAGCTAAAATCCGCCGTAAACCGGGTGGGCGCATCGCGGCCGCTATGCTGCAGCAGACCGCACTGAAGGACTTCACCGGTGGCAGCGGCCGTCTGCGTGAATGGATTGTTCGTCAAGGTGGCATGCCGCACGCAATTTTTATCGAAGGTGGTTATGTTGAACTTGGCCAGTTAGCGAGACAACTGCCGGCAAATCAGTTTGCAGAAACCACGCCGGGCGTTTACGTTGCCCGCGTGCCGATAGTGGTGGCGCCGGGTGCTACCCTTCACATTGGTAAGAACGTGAAAGAACTGCGTTTGTCTGAGGAACGTGGTGCGTTCTTGGTCAACGATGGCAAGCTGTTCATCACCGATACCAAGCTGGTTGGTTGGTCGGAGAAAAACAATGCCCCGAGCGCGTATCGTGGTCCGGAGAGCTTTTGGGCATTCCTGGTATCCTGGGGTGGTACGGAAACGTACATTTCTCGTCGTCCGGTTGCGAGCCTGGGTTATAACACCAGCAAAGCGTATGGCGTGAGCATCACGCAGTACACCCCGGAGATGCATAAACGTCTGAAGCGCCCGCGTCCGACAGGCTGGCTGATCGACAGCGTTTTTGAAGATATTTACTATGGTTTTTACTGCTATGAAGCTGATGATGTTGTTTTGAAGGGCAACACCTACCGCGACAATATTATCTACGGCATCGACCCGCATGATCGTAGCGAGCGCTTGGTCATCGCCGAGAACCACGTTTACGGTACGAAGAAGAAACATGGTATCATCGTGAGCCGTGAGGTTAACAACAGCTGGATTATCAACAACCGCACTCATGATAACAAACTCTCTGGCATCGTGCTGGACCGTAATTCCGAGCACAACCTGGTGGCCTACAATGAAGTGTATCAGAATCACTCCGATGGTATCACCCTGTACGAGAGCAGTAATAACTTGATCTGGGGCAACCGTCTGATTAATAACGCGCGGCACGGCATCCGCATGCGTAATTCCGTGAACATACGGATTTACGAGAACCTTAGCGTGGTTAACCAGTTGACCGGCATCTACGGCCATATTAAAGACCTGTCTTCGACCGATAGAGATTTCAAATTAGACCCATTCGATACTAAGGTGAGCATGATTGTGGTCGGTGGTCAATTGACTGGCAATGGTAGCTCTCCGATTAGCGTTGACAGTCCGCTGTCCCTGGAGCTGTATCGTGTTGAGATGCTGGCTCCGACCAAGTCCTCTGGTCTGACGTTCACCGGCATCCTGGAGGACAAGCAAGAGGAAATCTTGGACTTGCTGGTACGCCGTCAAAAAGCGGTTCTGATTGACCCGGTGGTGGACCTTGCGCAGGCGGAACTGTAA
3.4. You have a sequence! Now what?
3.4. - answer (click to expand)
Once the codon-optimized algG DNA sequence is obtained, the protein can be produced using either cell-dependent or cell-free expression systems.
Cell-Dependent Expression
In a cell-dependent system, the optimized algG gene would be cloned into an expression plasmid and introduced into Escherichia coli.
The essential steps include:
- Cloning the gene into a plasmid under a suitable promoter (e.g., T7).
- Transforming the plasmid into E. coli.
- Growing the cells and inducing gene expression.
- Transcription of DNA into mRNA.
- Translation of mRNA into the AlgG protein.
- Protein purification for further use.
In this system, the bacterial cell performs transcription and translation using its native molecular machinery.
Cell-Free Expression
Alternatively, the DNA could be added to a cell-free transcription–translation system, where purified ribosomes and enzymes synthesize the protein in vitro. This method is faster but more expensive and less scalable.
Conclusion
For AlgG production, cell-dependent expression in E. coli seems to be the most appropriate method. AlgG is a bacterial enzyme and does not require complex post-translational modifications, making E. coli a cost-effective and scalable host for recombinant protein production.
Part 4: Prepare a Twist DNA Synthesis Order (UNABLE TO ACESS TWIST SITE AND TOOLS)
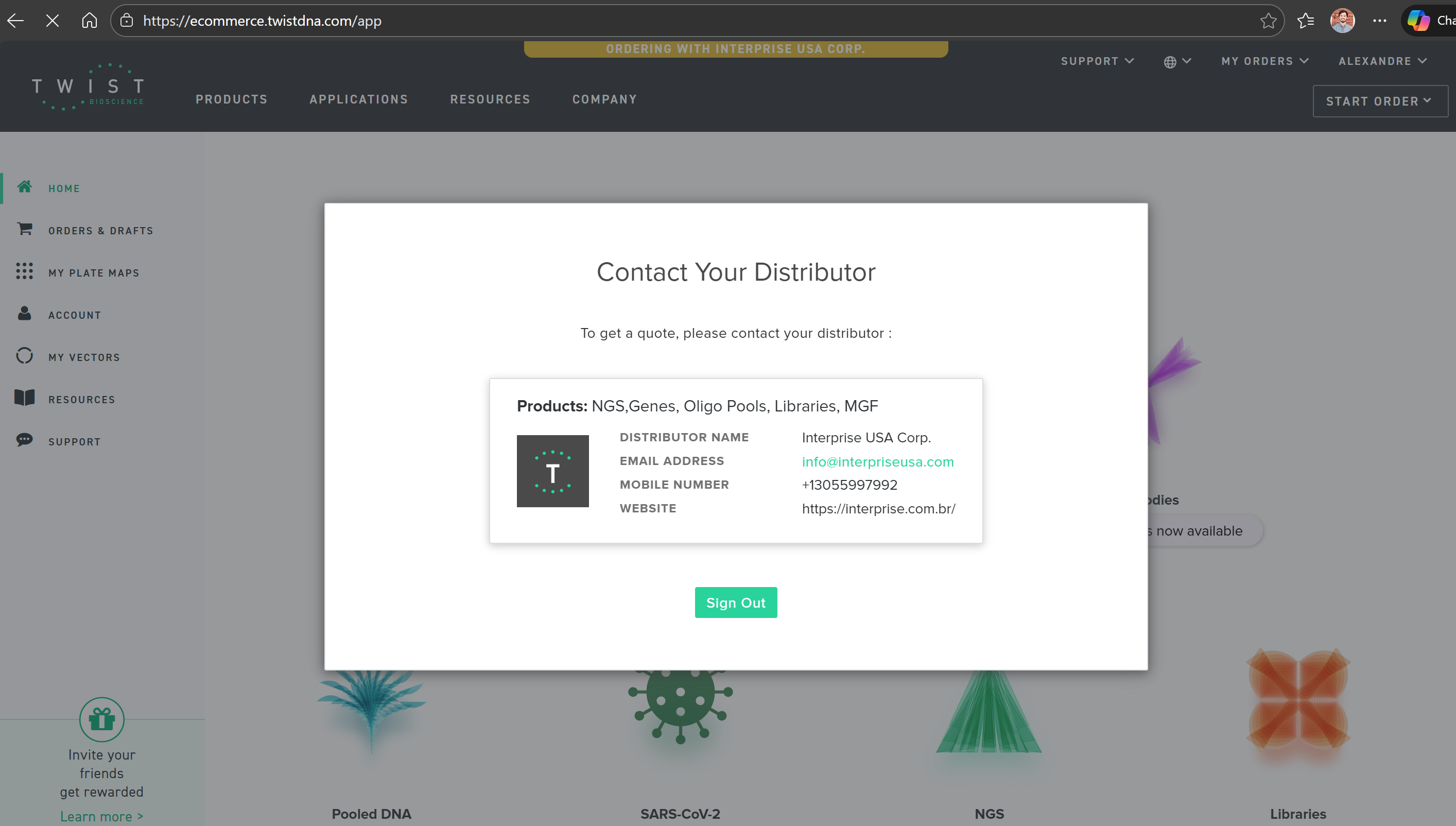
I created my account on Twist; however, I am not able to access the website or any of its tools because the site requests that I contact the distributor in my country. Please see the attached screenshot.
{kind=link}
Part 5: DNA Read/Write/Edit
5.1 DNA Read
5.1.1 What DNA would you want to sequence and why?
5.1.1 - answer (click to expand)
Although the algG gene from Azotobacter vinelandii has already been characterized, I would sequence environmental isolates of Azotobacter vinelandii and related alginate-producing bacteria to investigate naturally occurring variants of the alginate biosynthesis gene cluster.
The alginate biosynthesis pathway is encoded by a biosynthetic gene cluster, which includes multiple adjacent genes responsible for polymer synthesis, modification, and export. Variations within this cluster—particularly in genes such as algG (mannuronan C-5-epimerase)—may influence the M/G ratio, block distribution, polymer length, and degree of modification of alginate.
Since the mechanical properties of calcium-crosslinked alginate materials depend strongly on G-block continuity and molecular weight, identifying naturally evolved variants of the biosynthetic cluster could inform the engineering of alginate-based structural biomaterials. By sequencing environmental strains, it may be possible to discover variants that produce alginates with enhanced calcium-binding capacity and improved mechanical strength, which is directly relevant to my broader research goal of developing calcium-precipitated alginate-based construction materials.
5.1.2 In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
5.1.2 - answer (click to expand)
To analyze the full alginate biosynthesis gene cluster and its structural organization, I would use third-generation long-read sequencing technology, possibly a platform from Oxford Nanopore Technologies.
Because the alginate biosynthesis cluster spans multiple adjacent genes and may contain repetitive or regulatory regions, long-read sequencing is particularly suitable for capturing entire gene clusters in single continuous reads. This reduces assembly ambiguity and allows detection of structural variations, insertions, or rearrangements that could influence alginate production.
- Oxford Nanopore sequencing is a third-generation sequencing technology. It sequences single DNA molecules in real time without requiring extensive amplification and produces long reads that can span tens of kilobases.
- Input: High-molecular-weight genomic DNA extracted from environmental isolates of Azotobacter vinelandii.
Essential preparation steps:
- Genomic DNA extraction from bacterial cultures.
- DNA purification to remove proteins and contaminants.
- Quality control (concentration and fragment length assessment).
- End repair and preparation of DNA ends.
- Adapter ligation to attach nanopore-compatible sequencing adapters.
- Loading the prepared DNA library onto a nanopore flow cell.
PCR amplification is typically minimized to preserve long fragment length.
- In nanopore sequencing:
- A single DNA molecule passes through a protein nanopore embedded in a membrane.
- An electrical current flows through the pore.
- Each nucleotide (A, T, G, C) causes a characteristic change in electrical current.
- The device records these signal disruptions.
- A computational algorithm performs base calling by translating electrical signal patterns into nucleotide sequences.
Unlike fluorescence-based sequencing, nanopore technology decodes DNA by measuring changes in electrical current in real time.
- The output includes:
- Raw electrical signal files (FAST5 format).
- Processed sequence files (FASTQ), containing:
- DNA sequence
- Quality scores for each base.
- Assembled genome sequences (FASTA format) after bioinformatic processing.
- Identification and structural analysis of the alginate biosynthesis gene cluster.
Long-read output enables reconstruction of full biosynthetic clusters and comparison between environmental variants.
5.2 DNA Write
5.2.1 What DNA would you want to synthesize (e.g., write) and why?
5.2.1 - answer (click to expand)
As an initial step, I would synthesize a codon-optimized version of the algG gene (1578 bp), designed for expression in Escherichia coli.
Rather than engineering the entire alginate biosynthesis cluster at once, focusing on algG provides a targeted and controlled strategy to modulate the M/G ratio of alginate. Since AlgG (mannuronan C-5 epimerase) directly converts mannuronic acid (M) residues into guluronic acid (G) residues, altering its activity can influence G-block continuity, which is a key determinant of calcium-mediated crosslinking strength.
Because the mechanical properties of alginate-based materials strongly depend on G-block length and distribution, engineering algG represents a rational first step toward designing alginate biomaterials with enhanced structural performance. Once the effect of epimerase modulation is characterized, additional regulatory elements or biosynthetic genes could be engineered in subsequent stages.
5.2.2 What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
- What are the essential steps of your chosen sequencing methods?
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.2.2 - answer (click to expand)
To synthesize the codon-optimized algG gene (1578 bp), I would use commercial gene synthesis technology from Twist Bioscience.
Twist uses high-throughput silicon-based DNA synthesis platforms that chemically synthesize short oligonucleotides and assemble them into full-length gene constructs. This method allows precise, sequence-verified DNA synthesis with scalable production capacity.
Because the algG sequence is within a typical gene-length range (~1.6 kb), it is well suited for commercial gene synthesis and can be delivered cloned into a plasmid vector for immediate expression in Escherichia coli.
The essential steps of synthetic gene production include:
- In silico DNA design and codon optimization.
- Chemical synthesis of short DNA oligonucleotides (typically 60–200 bp).
- Assembly of overlapping oligos into larger fragments.
- Enzymatic assembly into the full-length gene (e.g., Gibson Assembly or similar methods).
- Cloning into a plasmid vector.
- Sequence verification (usually by next-generation sequencing).
- Delivery of the verified construct.
Limitations of this method:
- Speed: Gene synthesis typically requires several days to weeks depending on sequence complexity and production queue.
- Accuracy: Although modern synthesis platforms are highly accurate, errors can occur during oligonucleotide synthesis or assembly. Therefore, sequence verification is required.
- Scalability: Single-gene synthesis is routine and scalable. However, synthesizing very large constructs (e.g., entire operons or genomes) increases cost and technical complexity.
- Sequence Constraints: Extreme GC content, repetitive elements, or regions prone to forming strong secondary structures may reduce synthesis efficiency. However, the GC content of the codon-optimized algG gene (~53%) falls within an optimal range for DNA synthesis and does not present significant technical constraints.
5.3 DNA Edit
5.3.1 What DNA would you want to edit and why? What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
5.3.1 - answer (click to expand)
Initially I would focus on editing the alginate biosynthesis pathway in Azotobacter vinelandii.
Specifically, I would edit the algG gene to:
- Increase epimerase activity
- Modify substrate specificity
- Enhance G-block continuity
Since AlgG controls the conversion of mannuronic acid (M) to guluronic acid (G), modifying its catalytic efficiency could directly influence the mechanical properties of calcium-crosslinked alginate.
Because calcium crosslinking strength depends strongly on guluronic acid block length, precise genome editing of algG could enable the production of alginate with enhanced structural performance for biomaterial applications.
This represents a controlled microbial engineering approach rather than editing higher organisms.
5.3.2 What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
5.3.2 - answer (click to expand)
I would use the CRISPR-Cas9 genome editing system. CRISPR-Cas9 enables precise, targeted modifications in bacterial genomes and is widely used for microbial metabolic engineering.
CRISPR-Cas9 works through:
- A guide RNA (gRNA) designed to match a specific DNA sequence.
- The Cas9 nuclease binds to the guide RNA.
- The complex scans the genome.
- When the guide RNA matches the target sequence, Cas9 creates a double-strand break.
- The cell repairs the break via:
- Non-homologous end joining (NHEJ) → small insertions/deletions
- Homology-directed repair (HDR) → precise edits using a repair template
For precise modification of algG, a donor DNA template would be provided to introduce specific mutations via HDR.
Preparation steps include:
- Designing a guide RNA targeting a specific region of algG
- Designing a donor DNA template containing the desired mutation
- Cloning CRISPR components into a plasmid system
Inputs required:
- Cas9 enzyme (expressed from plasmid)
- Guide RNA sequence
- Donor repair template (if precise edit is desired)
- Competent bacterial cells
- Selection markers for screening edited colonies
Limitations:
- Efficiency: HDR efficiency in bacteria can be variable.
- Off-target effects: Cas9 may cut unintended genomic regions if guide RNA specificity is imperfect.
- Cellular stress: Double-strand breaks can reduce cell viability.
- Regulatory concerns: Genome-edited organisms may face biosafety and regulatory restrictions.