Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
3. Why are there only 20 natural amino acids?
There are only 20 natural (canonical) amino acids that can be synthesized by the biological machinery of cells and incorporated into tRNAs to make proteins. The ribosome, tRNAs, and aminoacyl-tRNA synthetases recognize and use these 20 building blocks.
Evolution led to this number because, although it may seem low, it is sufficient to generate enormous structural and functional diversity. Even short sequences composed of 20 different amino acids can produce an astronomical number of possible combinations. Moreover, these amino acids have diverse physicochemical properties — hydrophobic, hydrophilic, charged, aromatic, etc. — allowing proteins to fold into complex tertiary and quaternary structures and perform highly specific functions.
Therefore, 20 amino acids provide enough chemical and structural diversity to build the wide range of proteins required for life.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, non-natural amino acids can be made. In fact, many have already been synthesized and incorporated into proteins using engineered tRNA–aminoacyl-tRNA synthetase systems or expanded genetic codes.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life began, amino acids likely formed through abiotic chemical processes. At that time, Earth’s conditions were extreme (high temperature, volcanic activity, lightning, and a reducing atmosphere), which allowed reactions such as Miller–Urey–type experiments to occur, producing organic molecules including amino acids. In addition, mineral-catalyzed synthesis at hydrothermal vents may have contributed to their formation.
There is also the possibility that these essential molecules had an extraterrestrial origin. In recent decades, meteorites have been found to contain amino acids, suggesting that some of the building blocks of life may have been delivered to early Earth from space.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? (and 8. Why are most molecular helices right-handed?)
Since natural proteins contain right-handed α-helices, I would expect a left-handed helix if it were made with D-amino acids. This can be explained as follows:
Except for glycine, most amino acids are chiral: they exist as two non-superimposable mirror-image forms (enantiomers):
L-amino acids, with the amino group on the left in the Fischer projection.
D-amino acids, with the amino group on the right.
The translational machinery of life is stereospecific for L-amino acids. Hence, L-amino acids are the standard in the genetic code and are normally used to build proteins in living organisms. D-amino acids are rare forms, usually not recognized by the ribosomal machinery, but they can be found in some organisms such as bacteria (for example, in bacterial cell walls and certain antibiotics).
L and D enantiomers have opposite stereochemistry (and opposite optical rotation), resulting in different backbone geometry. Therefore, an α-helix made up of L-amino acids will have the opposite handedness of one made up of D-amino acids. Since natural proteins contain right-handed α-helices, a helix composed of D-amino acids would be expected to be left-handed.
7. Can you discover additional helices in proteins?
According to what was discussed in the lecture, the structural space of proteins is not yet fully explored, and advances in computational protein design suggest that new structural motifs could be discovered or designed. With modern computational design tools and advances in AI and structure prediction methods, it is possible to identify sequences that could stabilize new structural motifs and generate novel conformations. Therefore, alternative backbone geometries and new helical motifs could potentially be discovered or designed if they are energetically stable.
9. Why do β-sheets tend to aggregate? - What is the driving force for β-sheet aggregation?
10. Why do many amyloid diseases form β-sheets? - Can you use amyloid β-sheets as materials?
11. Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
- Briefly describe the protein you selected and why you selected it.
I selected the human High Mobility Group Box 1 (HMGB1) protein because if find it realy interesting, it is a multifunctional chromatin-associated protein with both nuclear and extracellular roles. In the nucleus, HMGB1 acts as a DNA-binding architectural protein involved in transcriptional regulation, DNA replication, recombination, and repair. However, under conditions of cellular stress or damage, HMGB1 can be released into the extracellular space, where it functions as a pro-inflammatory signaling molecule (alarmin), contributing to immune activation and chronic inflammation.
- Identify the amino acid sequence of your protein.
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
- How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
- Does your protein belong to any protein family?
Homo Sapiens HMGB1 sequence (UNIPROT ID: P09429): MGKGDPKKPRGKMSSYAFFVQTCREEHKKKHPDASVNFSEFSKKCSERWKTMSAKEKGKFEDMAKADKARYEREMKTYIPPKGETKKKFKDPNAPKRPPSAFFLFCSEYRPKIKGEHPGLSIGDVAKKLGEMWNNTAADDKQPYEKKAAKLKEKYEKDIAAYRAKGKPDAAKKGVVKAEKSKKKKEEEEDEEDEEDEEEEEDEEDEDEEEDDDDE
| length | Most frequent AA | Homologs | Protein Family | Domains | |
|---|---|---|---|---|---|
| Reference | 250 AA | K (20.0%) | 250 hits* | High mobylity Group B family | HMG Box A and B |
*BLAST search on UniProt (default parameters) returned 250 hits (the maximum), all with identity > 80%, e-value < 0.0001, and score > 1000. These parameters indicate that the proteins found are homologous to human HMGB1 and correspond to HMGB proteins in other organisms, wich is consistent with the fact that the this family is highly conserved among eukaryotes.
- Identify the structure page of your protein in RCSB
Identyfied sturcture: 2YRQ | pdb_00002yrq
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
I searched the UniProt ID of the protein in the RCSB PDB and found 13 structures corresponding to Homo sapiens. There is no structure of the full-length protein available; the most complete structure lacks the last ~45 amino acids corresponding to the acidic C-terminal tail.
2YRQ sequence:
adittional AA | lacking AA
GSSGSSGMGKGDPKKPRGKMSSYAFFVQTCREEHKKKHPDASVNFSEFSKKCSERWKTMSAKEKGKFEDMAKADKARYEREMKTYIPPKGETKKKFKDPNAPKRPPSAFFLFCSEYRPKIKGEHPGLSIGDVAKKLGEMWNNTAADDKQPYEKKAAKLKEKYEKDIAAYRAKGKPDAAKKGVVKAEKSKKKKEEEEDEEDEEDEEEEEDEEDEDEEEDDDDE
The selected structure was deposited in April 2007 and released in February 2008. It was solved by solution NMR, therefore a crystallographic resolution in Ångström is not reported. Instead, the quality of the structure is evaluated using NMR-specific validation parameters:
| Conformers Calculated: 100 | Conformers Submitted: 20 | Clashscore: 18 | Ramachandran outliers: 1.8% | Side-chains outliers: 4.6% |
- Open the structure of your protein in any 3D molecule visualization software:
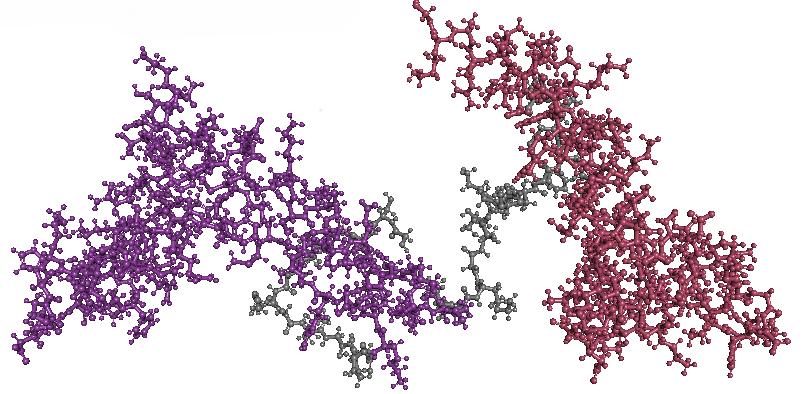
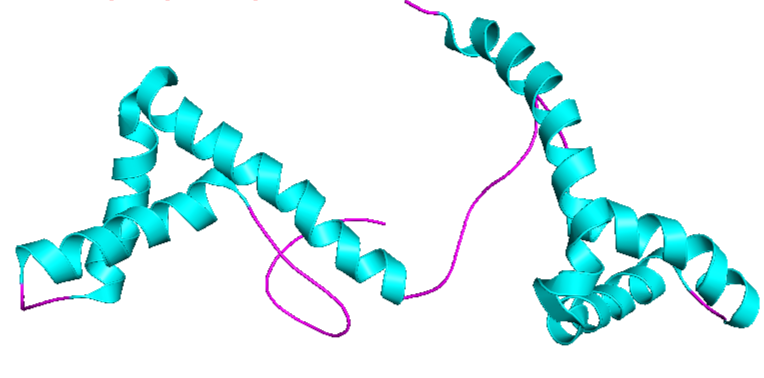
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
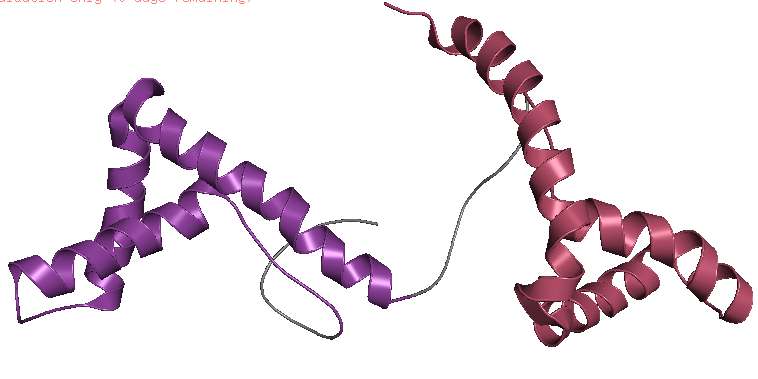
- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
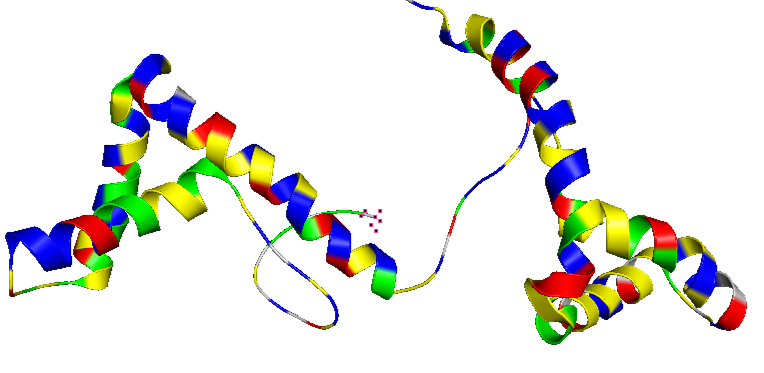
The amino acid composition shows a relatively balanced proportion of positively charged, negatively charged, polar, and hydrophobic residues. These residues are also distributed relatively uniformly along the protein sequence, rather than being strongly clustered in specific regions.
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
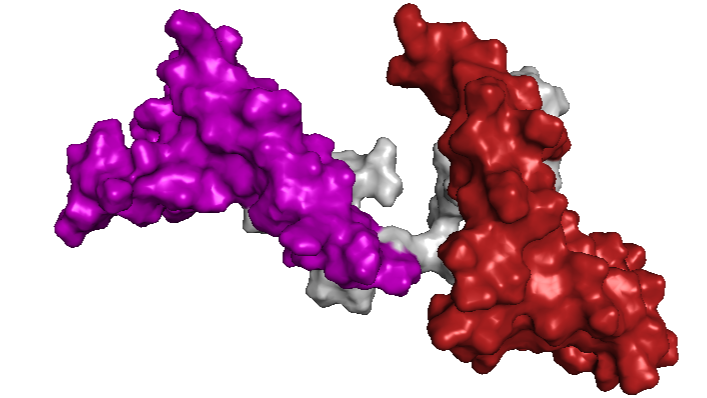
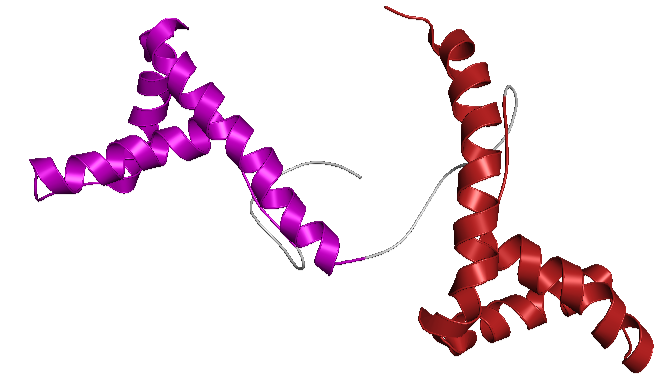
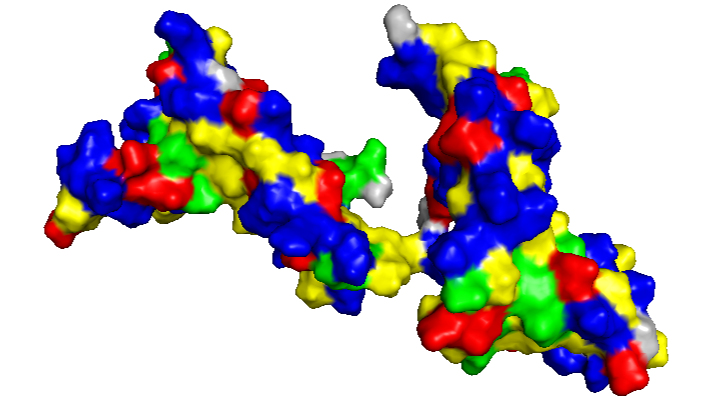
Between the HMG boxes, there is a “pocket” where the protein interacts with DNA. The blue residues correspond to positively charged amino acids, which are important for binding to the negatively charged DNA backbone.
Part C. Using ML-Based Protein Design Tools
For this part of the homework, I chose the HMGB protein of T. cruzi (TcHMGB)
Sequence: (TriTripDB ID:TcCLB.507951.1141) MSTELKSGPLPADVEEVIANIMREEGVSFLTSKILRLRLEARYRMEFTSHKAAIEGIITKLMQLPEFKKQLENAVKEEKAASSIGGKKKKRSASAAADERNAKVNKKEKKPDDYPKAALSPYILFGNDHRDKVKEQNPGMKNTEILQSLGKMWAEASDAVKEKYKKLAEDDKKRFDRELSEYKKSGGTEYKRGGGKVKAKDENAPKRSMSAYFFFVSDFRKKHPDLSVTETSKAAGAAWKALSDDMKKPYEAMAQKDKERYQREMAARAS
Domais:
- Deck-C terminal: 10-61
- HMG Box A: 110-179
- HMG Box B: 214-266
C1. Protein Language Modeling
- Deep Mutational Scans a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
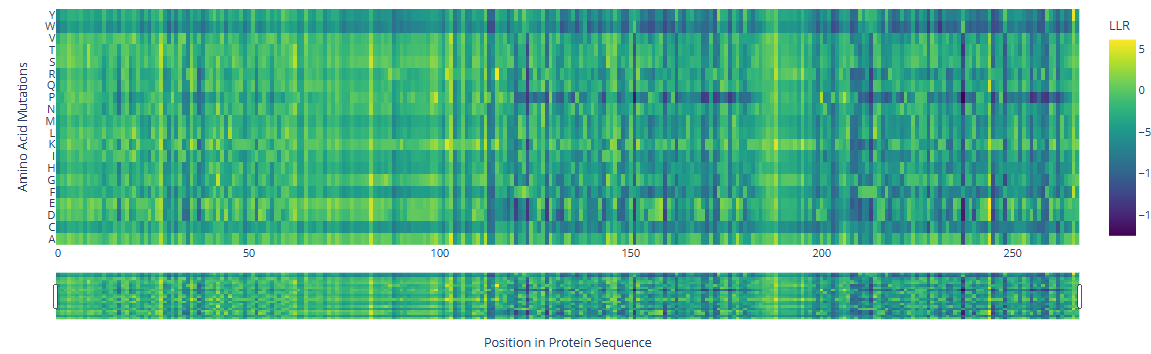
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out.)
There are three specific regions where the heatmap shows predominantly blue colors, indicating negative LLR values and therefore unfavorable mutations. These regions correspond to the two HMG box domains, which are highly conserved among eukaryotes and are critical for DNA binding, and the Deck-C domain, which is conserved among trypanosomatids.
In contrast, the regions in between the domain display less dark-blue colors and more greenish-yellowish colors, corresponding to positive LLR values. This suggests that mutations in this region are better tolerated, consistent with a lower functional or structural constraint.
There are a few mutations that stand out in the HMG Box domains:
| Position | wt | mutated | LLR value |
|---|---|---|---|
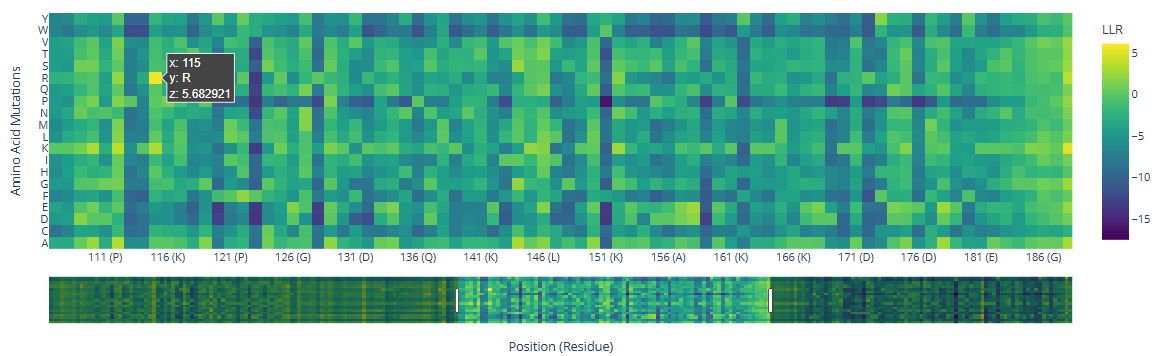
| 115 | R () | 5.68 | |
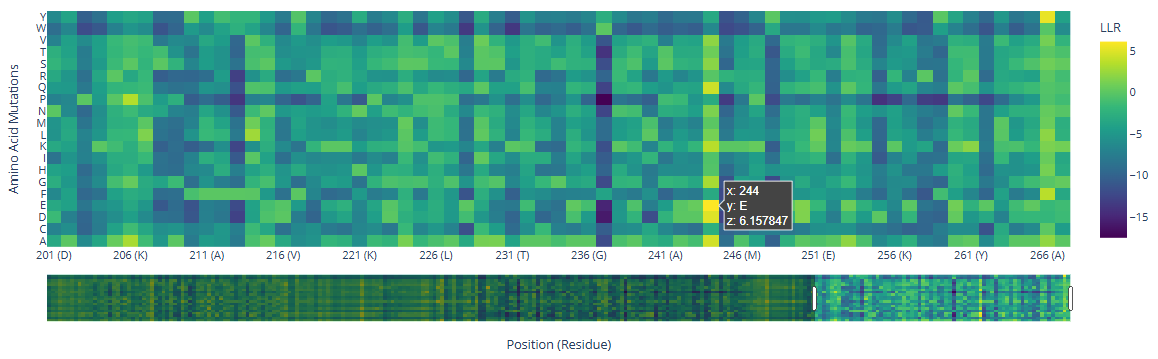
| 244 | () | E () | 6.16 |
c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to the experiment.
- Latent Space Analysis a. Use the provided sequence dataset to embed proteins in reduced dimensionality. b. Analyze the different formed neighborhoods: do they approximate similar proteins? c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
- Input this sequence into ESMFold and compare the predicted structure to your original.