Biotechnology graduate from the National University of Rosario (Argentina), with hands-on experience in molecular cloning, microbial systems, and experimental research.
I am particularly interested in genetic engineering, bioinformatics, and the application of AI-driven tools to improve experimental design.
Through HTGAA, I aim to strengthen my skills in AI-assisted experimental design and advanced biotechnological applications.
Tool Description Biosensor for the detection of stress or diseases in plants useing Escherichia coli chassis.
In this system, stress signals or specific markers associated with certain pathogens induce the production of a fluorescent protein like GFP. This biosensor could be used as a tool for the early detection of plant pathologies by exposing the bacteria to the plant extracts or exudates. T
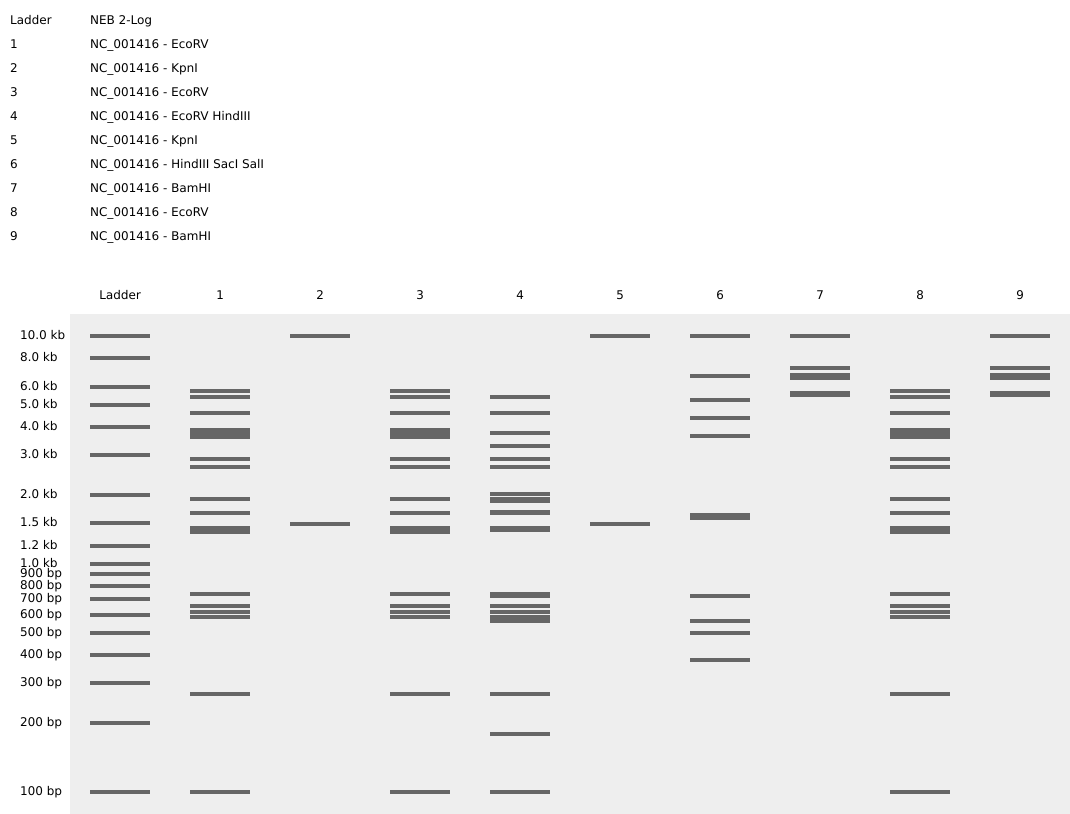
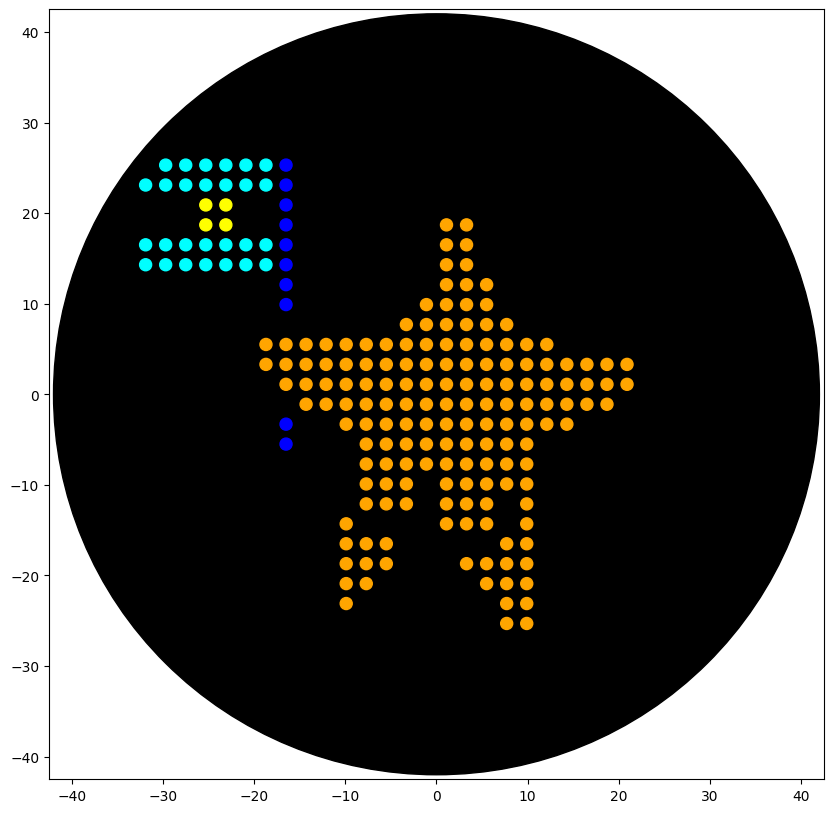
Part 1: Benchling & In-silico Gel Art I design two gels:
Gel A: ‘ART’
Gel B: ‘ATP’
Part 3: DNA Design Challenge 3.1. Choose a protein - TcHMGB For this homework, I chose the protein HMGB from Trypanosoma cruzi (TcHMGB), the parasite that causes Chagas disease. TcHMGB was characterized by the research group in which I carried out my undergraduate thesis. During this work, I constructed a DM28c cell line expressing TcHMGB fused to the biotin ligase TurboID, with the objective of performing a proximity labeling–based interactomics assay.
Python Script for Opentrons Artwork To create the Python file to run on an Opentrons liquid-handling robot, I used the scripts from the file downloaded from http://opentrons-art.rcdonovan.com/ as guide, to write the scripts unthe the ‘#YOUR CODE HERE’ line. I also edited the ‘well_color’ library to add some colors.
Google colab cell link: https://colab.research.google.com/drive/1Ke57DkO8O2jpCUDaYyK3krv4kUYFVgn8#scrollTo=pczDLwsq64mk&line=9&uniqifier=1
Simulation visualisation:
Post-Lab Questions Descrbe 3 proyects Proyect 1: Engineered Miniproteins to Disrupt and Prevent Bacterial Biofilm Formation THE PROBLEM Biofilms are structured microbial communities embedded in a self-produced extracellular polymeric substance (EPS) matrix composed of polysaccharides, proteins, extracellular DNA and lipids. They form on:
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) 2. Why do humans eat beef but do not become a cow, eat fish but do not become fish? 3. Why are there only 20 natural amino acids?
Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Subsections of Homework
Week 1 HW: Principles and Practices
Tool Description
Biosensor for the detection of stress or diseases in plants useing Escherichia coli chassis.
In this system, stress signals or specific markers associated with certain pathogens induce the production of a fluorescent protein like GFP. This biosensor could be used as a tool for the early detection of plant pathologies by exposing the bacteria to the plant extracts or exudates. T
he system could be build:
a. to respond to a group of markers related to plant infection or to a group of markers for a certain pathogen.
b. to detect if the plant is under abiotic stress or is infected, resulting in the production of different signals depending on the diaggnose.
Plants under abiotic stress and plants affected by pathogens often show similar external symptoms, but at a molecular level these two conditions can be differentiated. A biosensor capable of responding differentially to this conditions or only to one of these conditions could help distinguish between them, enabling more appropiate interventions
Governance Goals
Ambiental Security: we must avoid ambiental damage
Use a non pathogen strain as a chasis
the biosensor must be manipulated in a laboratory
propeer disposal of the material that has been in contact with the biosensor
Proper use of the tool
Inclusion
disminish costs
make the tool accesible for small productors
Responsible interpretation and use of results: missinterpretation of the output could lead to incorrect interventions
Ensure that the biosentor results are presented as screening tools
Provide clear guidance on the limitantions of the biosinsor´s accuracy
Governance Actions
Stablish requirements of use
Purpose:The biosensor is based on a genetically modified organism. This action proposes establishing its use exclusively in laboratories with the required biosafety level in order to avoid the accidental spread of the organism into the ecosystem.
Design: For this to work, a group of professionals must create a guide for use and establish requirements for purchasing the product. Users of the product must follow the provided guidelines and instructions.
Assumptions: It is assumed that trained personnel will follow the guide for use and will not misuse the product.
Risks of Failure & “Success”: Unresponsible personnel may misuse the biosensor, leading to the accidental release of the bacteria into the environment.
Propper disposal of the material
Purpose: Ensure that users have the appropriate means for the proper disposal of materials, and monitor correct disposal in order to prevent environmental contamination.
Design: Institutions and laboratories using the iosensor must provide approved disposal systems for biological waste, such as sterilization or inactivation procedures. Clear disposal protocols must be included in the user guide, and compliance should be overseen by institutional biosafety committees.
Assumptions: It is assumed that that users will follow established disposal protocols.
Risks of Failure & “Success”: disposal procedures may not be properly followed or enforced, leading to unintended release of genetically modified bacteria.
Result interpretation guidelines
Purpose:Prevent misinterpretation of biosensor outputs by ensuring that results are understood as indicative signals rather than definitive diagnoses.
Design:Develop standardized interpretation guidelines that accompany the biosensor, including clear explanations of what a positive or negative signal means and its limitations. These guidelines should be created by academic experts and included in the user manual.
Assumptions: It is assumed that users follow the interpretation guidelines when analyzing results.
Risks of Failure & “Success”: users may ignore or oversimplify the guidelines.
Does the option:
Requierments of use
Proper desposal
Result interpretation
Enhance Biosecurity
• By preventing incidents
1
2
n/a
• By helping respond
2
2
n/a
Foster Lab Safety
• By preventing incident
1
1
3
• By helping respond
2
2
3
Protect the environment
• By preventing incidents
1
1
n/a
• By helping respond
2
2
n/a
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
1
1
• Not impede research
2
2
1
• Promote constructive applications
2
2
1
Priorization of gocernance options
Based on the scoring, the governance options that should be prioritized are establishing requirements of use and result interpretation guidelines, while proper disposal of materials functions as a complementary measure.
Requirements of use are essential because they directly reduce risks related to biosecurity, laboratory safety, and environmental protection by limiting the biosensor to controlled laboratory settings. However, these requirements may increase costs and restrict access, particularly for smaller laboratories.
Result interpretation guidelines are a high-priority complementary action because they are easy to implement, low cost, and help prevent incorrect decisions based on biosensor outputs. Since the biosensor is intended as a screening tool, clear guidance is necessary to avoid inappropriate interventions.
Proper disposal of materials is also important but relies strongly on infrastructure and enforcement, which can vary between institutions.
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase.
What is the error rate of polymerase? 1/106
How does this compare to the length of the human genome? The human genome is approximately 3 × 10⁹ base pairs long. At this error rate, thousands of errors would be expected per replication cycle if no correction mechanisms were present
How does biology deal with that discrepancy?1. polymerases can detect and corect errors (proofreading activity); 2. Cells have repear sytems to correct DNA mutations (like MutS system)
How many different ways are there to code (DNA nucleotide code) for an average human protein? There are many ways to code for an average human protein due to the degeneracy of the genetic code: each aminocid is encoded by two or three codons.
In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? The use of codons differs between organisms; that is, some organisms commonly use codons that are rare in others. As a consequence, the translation efficiency of a human protein expressed in yeast or bacteria can be limited due to the low availability of the corresponding tRNAs. Another reason is the absence or differences in post-transcriptional and post-translational modifications between expression systems (such as bacteria or yeast) and human cells, which can affect protein folding, stability, and functionality.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently? Solid-phase chemical synthesis, specifically phosphoramidite chemistry, is the most widely used method for oligonucleotide synthesis.
Why is it difficult to make oligos longer than 200nt via direct synthesis? Because error in nucleotid addition accumulates with increasing length, leading to a sharp drop in yield and sequence fidelity for longer oligonucleotides.
Why can’t you make a 2000bp gene via direct oligo synthesis? Considerieng the avobe, direct syhthesis of a 2000bp gene would result in an unacceptably high error rate and extremely low yield.
Homework Question from George Church:
[Using Google & Prof. Church’s slide #4]
What are the 10 essential amino acids in all animals? **H, I, L K, M, F(Phe), T, W, V and R.
How does this affect your view of the “Lysine Contingency”? The “Lysine Contigency” could have made scence if the dinosaurs had have been genetically engineered to be able to produce all essential amino acids except lysine, so that they would survive only with lysine supplementation (and not the rest of the essential amino acids), and die when supplementation was withdrawn.
IA prompts:
Q: Which are the 10 essential aa in animals?
A:The 10 essential amino acids in animals are:
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Threonine
Tryptophan
Valine
Arginine
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
I design two gels:
Gel A: ‘ART’
Gel B: ‘ATP’
Part 3: DNA Design Challenge
3.1. Choose a protein - TcHMGB
For this homework, I chose the protein HMGB from Trypanosoma cruzi (TcHMGB), the parasite that causes Chagas disease. TcHMGB was characterized by the research group in which I carried out my undergraduate thesis. During this work, I constructed a DM28c cell line expressing TcHMGB fused to the biotin ligase TurboID, with the objective of performing a proximity labeling–based interactomics assay.
TcHMGB belongs to the HMG-B family, which plays a key role in chromatin organization and gene regulation in eukaryotic organisms. TcHMGB has been shown to modify chromatin structure, making it more accessible to the replication, transcription, repair, and recombination machinery. Additionally, TcHMGB is believed to be essential for T. cruzi survival and is therefore considered a potential therapeutic target.
Protein sequence
Found in UniProt
tr|Q4D714|Q4D714_TRYCC High mobility group protein, putative OS=Trypanosoma cruzi (strain CL Brener) OX=353153 GN=Tc00.1047053504431.64 PE=4 SV=1
Codon usage differs between organisms; that is, some organisms preferentially use codons that are rare in others. As a consequence, the translation efficiency of a T. cruzi gene expressed in yeast may be limited by the low availability of the corresponding tRNAs. Therefore, codon optimization is required to improve the efficiency of expression of the protein of interest.
tchmgb encodes a protein from Trypanosoma cruzi, a unicellular eukaryotic organism. Therefore, Saccharomyces cerevisiae was selected as the expression host because it provides an eukaryotic cellular context, including post-translational modification and protein-folding machinery, which is absent in prokaryotic systems such as Escherichia coli.
tcHMGB was codon-optimized for expression in Saccharomyces cerevisiae using the IDT Codon Optimization Tool (Integrated DNA Technologies; https://www.idtdna.com/CodonOpt)
ATG TCA ACT GAA CTG AAG AGT GGC CCA TTG CCT GCA GAC GTA GAA GAG GTC ATA GCT AAT ATT ATG AGG GAA GAA GGA GTG AAT TTT TTG ACT TCT AAG ATT CTT AGA CTT AGA CTA GAA GCA AGA TAC AGA ATG GAA TTT ACT TCA CAT AAA GCC GCT ATT GAA GGC ATC ATT ACA AAA TTA ATG CAG TTG CCT GAA TTT AAG AAG CAA TTA GAG AAC GCA GTA AAA GAA GAG AAG GCA GCG TCA TCT ATT GGA GGT AAA AAG AAA AAG AGA TCA GCT TCT GCG GCT GCA GAC GAA AGA AAT GCT AAG GTC AAC AAG AAG GAA AAA AAA CCT GAC GAT TAT CCA AAG GCA GCT TTG AGC CCA TAT ATT TTA TTC GGT AAT GAC CAT AGA GAT AAG GTG AAG GAA CAA AAT CCA GAA ATG AAA AAT ACT GAG ATT TTG CAA TCA TTG GGA AAA ATG TGG GCC GAG GCG TCT GAC GCA GTT AAA GAG AAA TAC AAG AAA TTA GCC GAA GAT GAT AAG AAA CGT TTC GAT AGA GAA TTA TCT GAA TAC AAA AAA AGT GGT GGT ACT GAA TAT AAA CGT GGA GGT GGC AAG GTT AAA GCT AAG GAT GAG AAC GCT CCT AAG CGT AGT ATG TCT GCA TAT TTT TTT TTC GTG AGT GAT TTC AGA AAA AAG CAT CCA GAC TTA AGT GTT ACA GAA ACC TCT AAA GCC GCC GGT GCT GCT TGG AAA GAA CTA TCC GAT GAG ATG AAA AAG CCA TAC GAA GCT ATG GCA CAG AAG GAT AAA GAA AGA TAT CAA AGA GAA ATG GCT GCT AGG GCG
3.4. You have a sequence! Now what?
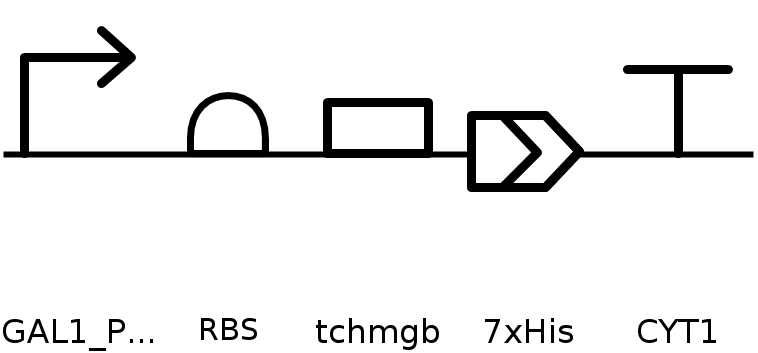
To produce this protein, it is first necessary to obtain a plasmid that enables expression of the protein of interest in the selected expression system, either cell-dependent or cell-free. For plasmid construction, there are two main options: (1) cloning the coding sequence into an existing expression vector, or (2) ordering a synthetic plasmid that includes regulatory elements selected according to the chosen expression system and experimental goals—such as a constitutive or inducible promoter, ribosome binding site, coding sequence, affinity tag, and transcription termination sequence—thereby improving expression efficiency and control.
Once the plasmid is obtained, it is introduced into the chosen expression system. In this case, Saccharomyces cerevisiae was selected as the host organism. The plasmid can be delivered into yeast cells by electroporation, followed by selection of successfully transformed cells. After selection, protein expression is induced under the appropriate conditions, allowing transcription of the gene and subsequent translation into the protein of interest.
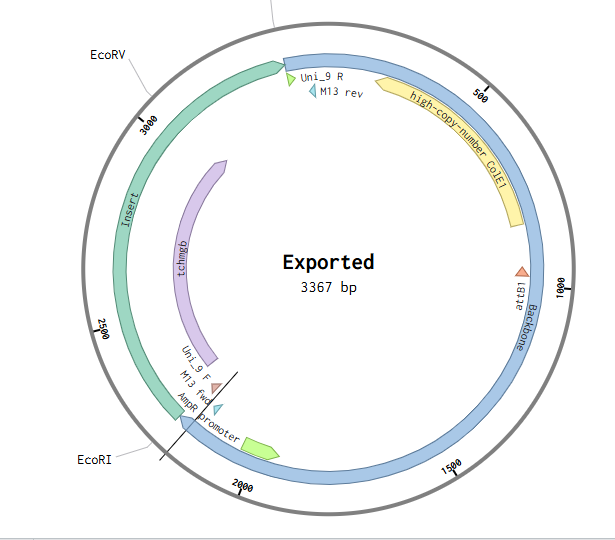
Since I could not use Twist directly for this exercise, I downloaded the pTwist Amp High Copy cloning vector map and imported it into Benchling. I then inserted the previously designed TcHMGB expression cassette into this backbone using Benchling’s sequence editing tools, allowing visualization and validation of the complete circular plasmid.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would choose to sequence DNA from hospital wastewater.Hospital effluents contain a high diversity of bacteria exposed to strong antibiotic selective pressure, making them important reservoirs of antimicrobial resistance genes. By sequencing this DNA, it is possible to identify and monitor resistance genes and their potential spread into the environment.
This information is highly relevant for public health, as it can contribute to early detection of emerging resistance patterns and support strategies to limit the dissemination of antibiotic resistance.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Oxford Nanopore to obtain long reads that provide structural and genetic context, and Illumina to obtain short, high-quality reads that allow more reliable genome assembly and error correction.
Is your method first-, second- or third-generation or other? How so?
Illumina is a second-generation sequencing technology, as it relies on sequencing-by-synthesis and generates large numbers of short, highly accurate reads in parallel. Oxford Nanopore is considered a third-generation sequencing technology because it sequences single DNA molecules directly and produces long reads without requiring amplificatio.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
My input would be total DNA extracted from hospital wastewater, which includes mixed microbial DNA from a metagenomic sample.
Essentially, Illumina requires DNA fragmentation and amplification, while Oxford Nanopore can sequence long, minimally processed DNA.
The essential steps are:
High-quality DNA extraction
Library preparation, including fragmentation (not necessary for Nanopore), amplification, and incorporation of adapters and indices
3.Sequencing
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Illumina decodes DNA by detecting fluorescent signals emitted when labeled nucleotides are incorporated during DNA synthesis. Each base is identified based on its specific fluorescence signal.
Oxford Nanopore decodes DNA by measuring changes in ionic current as a DNA strand passes through a nanopore. Different nucleotide sequences cause characteristic current disruptions, which are translated into base calls using computational algorithm.
What is the output of your chosen sequencing technology?
Illumina sequencing results in short, high-quality sequencing reads, while Oxford Nanopore produces long reads of variable length.
In both sequencing technologies, the output consists of FASTQ files containing all the reads, which can be processed using multiple platforms for downstream analysis
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs!
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
(ii) What technology or technologies would you use to perform these DNA edits and why?
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Week 3 HW: Lab automatation
Python Script for Opentrons Artwork
To create the Python file to run on an Opentrons liquid-handling robot, I used the scripts from the file downloaded from http://opentrons-art.rcdonovan.com/
as guide, to write the scripts unthe the ‘#YOUR CODE HERE’ line. I also edited the ‘well_color’ library to add some colors.
Proyect 1: Engineered Miniproteins to Disrupt and Prevent Bacterial Biofilm Formation
THE PROBLEM
Biofilms are structured microbial communities embedded in a self-produced extracellular polymeric substance (EPS) matrix composed of polysaccharides, proteins, extracellular DNA and lipids.
They form on:
Medical devices
Tissues
Industrial surfaces
Environmental niches.
Clinically, biofilms are highly problematic:
EPS matrix limits antibiotic penetration and there is a reduced metabolic activity of inner-layer cells leading to Antibiotic tolerance
Quorum sensing (QS) coordinates: cell-density (dependent gene regulation), virulence and matrix biosynthesis.
In pathogens such as Pseudomonas aeruginosa, QS regulators like LasR orchestrate biofilm maturation and virulence programs.
As a consequence, biofilm-associated infections are often chronic, recurrent, and difficult to eradicate.
THE VISION
Instead of bactericidal pressure (which accelerates resistance evolution), this project proposes a molecular destabilization strategy using de novo engineered miniproteins.
These programmable miniproteins will:
Bind and inhibit QS regulators
Target transcription factors such as LasR-type proteins
Block coordinated gene expression
Prevent structured biofilm formation
Interfere with EPS assembly
Bind structural matrix proteins
Disrupt polysaccharide–protein interactions
Destabilize mature biofilms
Be modular and retargetable
Scaffold-based design
Sequence redesign for new pathogens
Compatible with automated high-throughput screening
The goal is to convert biofilms from a protected, resistant state into a destabilized, antibiotic-sensitive state.
AUTOMATIZATION
Automation will rely on an Opentrons OT-2 liquid-handling robot to execute the design–build–test cycle. Miniprotein variants will be assembled through automated cloning workflows using liquid-handling robotics, the the constructed plasmids will be transformed and cultured in multiwell plate formats and finally biofilm formation and inhibition assays will be performed using automated pipetting and plate-based quantification.
%%{init: {'themeVariables': { 'fontSize': '15px' }}}%%
flowchart LR
A["<b>1. Computational Design.</b><br/>Use de novo protein design algorithms to design small, stable binders targeting QS regulators or matrix components"]
B["<b>2. Gene Synthesis and Expression</b><br/>DNA synthesis and cloning<br/>Expresion in bacteria or-cellfree systems for screening"]
C["<b>3. Functional Validation"]
D["<b>4. Delivery Strategy"</b><br/>A. Engineered probiotic chassis for local secretion.
B. Virus-like particles for targeted protein delivery.]
A --> B --> C --> D
Proyect 2: Biomarkers for Endometriosis Detection
(still developing this idea)
THE PROBLEM
Endometriosis is a chronic, estrogen-dependent inflammatory disease affecting ~10% of reproductive-age women and causing chronic pelvic pain and infertility.
Despite its prevalence, diagnosis is delayed by 7–10 years due to social, clinical, and technical factors, including normalization of severe menstrual pain and the lack of validated non-invasive screening biomarkers.
This delay contributes to disease progression, chronic pain sensitization, reduced fertility, increased surgical burden, and significant psychological distress — highlighting the need for early, accessible detection tools.
THE VISION
Develop a standardized, automated, non-invasive screening tool — such as a blood-based microRNA panel and/or a combined inflammatory profile — to enable earlier risk identification of endometriosis through circulating molecular biomarkers.
The goal is not to provide a definitive diagnosis, but to develop a tool capable of indicating the probability of endometriosis. This would help identify women at higher risk, support earlier referral to specialists, reduce diagnostic delay, and ultimately improve long-term clinical outcomes.
FIRST APROACH
Circulating microRNAs (miRNAs) are among the most promising non-invasive biomarker candidates for endometriosis. Additionally, altered inflammatory markers such as IL-6, IL-8, and TNF-α further support the presence of a detectable circulating molecular signature.
However, no single biomarker has demonstrated sufficient sensitivity and specificity for clinical implementation due to inter-study variability and lack of methodological standardization.
Therefore, the initial aim is to standardize and automate a reproducible biomarker panel, primarily based on previously reported circulating miRNAs and potentially complemented by selected inflammatory markers. By integrating these candidates into a controlled, automated laboratory workflow, the project seeks to reduce technical variability, improve reproducibility, and enable scalable early risk stratification for endometriosis.
Proyect 3: Paper-Based Biosensor for Early Detection of Plant Water Stress
THE PROBLEM
Water stress is one of the main causes of crop yield loss worldwide. Current monitoring strategies rely primarily on soil moisture sensors, satellite imaging, or visible plant symptoms.
However:
Soil moisture does not necessarily reflect plant physiological status.
Visual symptoms appear after stress is already advanced.
Molecular stress markers require laboratory-based analysis.
There is currently no simple, field-deployable tool to detect early physiological stress directly from plant tissue.
THE VISION
Develop a portable, paper-based biosensor capable of detecting early molecular markers of water stress from leaf extracts.
Instead of measuring environmental parameters, this system will assess the plant’s internal physiological response.
The device will:
Detect stress-associated biomarkers (e.g., ROS-related signals or osmotic stress markers).
First sensor constructs will be designed and prototyped using automated cloning workflows on an Opentrons liquid-handling platform. Then variants will be assembled in parallel and expressed in bacterial or cell-free systems. Screening of biosensor performance will be performed in 96-well plate formats prior to integration into the paper-based device. Finally the experimental data will guide iterative redesign of sensing circuits.
Opentrons-Based Pipeline
Automated Golden Gate / Gibson assembly
Parallel promoter variant library construction
Transformation or TXTL assembly
96-well high-throughput screening
Quantitative readout (plate reader)
WORK FLOW
%%{init: {'themeVariables': { 'fontSize': '15px' }}}%%
flowchart LR
A["<b>1. Biomarker Selection</b><br/>Identify and select early molecular indicators of plant water stress."]
B["<b>2. Biosensor Design</b>
a. Genetic circuit design
b. Automated assembly and screening
c. Sensitivity and specificity validation<br/>"]
C["<b>3. Prototype Development</b>
a. Freeze-drying optimization
b. Paper device fabrication
c. Device integration"]
D["<b>4. Field Validation</b><br/>Correlate sensor output with physiological plant stress measurements"]
A --> B --> C --> D
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
3. Why are there only 20 natural amino acids?
There are only 20 natural (canonical) amino acids that can be synthesized by the biological machinery of cells and incorporated into tRNAs to make proteins. The ribosome, tRNAs, and aminoacyl-tRNA synthetases recognize and use these 20 building blocks.
Evolution led to this number because, although it may seem low, it is sufficient to generate enormous structural and functional diversity. Even short sequences composed of 20 different amino acids can produce an astronomical number of possible combinations. Moreover, these amino acids have diverse physicochemical properties — hydrophobic, hydrophilic, charged, aromatic, etc. — allowing proteins to fold into complex tertiary and quaternary structures and perform highly specific functions.
Therefore, 20 amino acids provide enough chemical and structural diversity to build the wide range of proteins required for life.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, non-natural amino acids can be made. In fact, many have already been synthesized and incorporated into proteins using engineered tRNA–aminoacyl-tRNA synthetase systems or expanded genetic codes.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life began, amino acids likely formed through abiotic chemical processes. At that time, Earth’s conditions were extreme (high temperature, volcanic activity, lightning, and a reducing atmosphere), which allowed reactions such as Miller–Urey–type experiments to occur, producing organic molecules including amino acids. In addition, mineral-catalyzed synthesis at hydrothermal vents may have contributed to their formation.
There is also the possibility that these essential molecules had an extraterrestrial origin. In recent decades, meteorites have been found to contain amino acids, suggesting that some of the building blocks of life may have been delivered to early Earth from space.
6.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? (and 8. Why are most molecular helices right-handed?)
Since natural proteins contain right-handed α-helices, I would expect a left-handed helix if it were made with D-amino acids. This can be explained as follows:
Except for glycine, most amino acids are chiral: they exist as two non-superimposable mirror-image forms (enantiomers):
L-amino acids, with the amino group on the left in the Fischer projection.
D-amino acids, with the amino group on the right.
The translational machinery of life is stereospecific for L-amino acids. Hence, L-amino acids are the standard in the genetic code and are normally used to build proteins in living organisms. D-amino acids are rare forms, usually not recognized by the ribosomal machinery, but they can be found in some organisms such as bacteria (for example, in bacterial cell walls and certain antibiotics).
L and D enantiomers have opposite stereochemistry (and opposite optical rotation), resulting in different backbone geometry. Therefore, an α-helix made up of L-amino acids will have the opposite handedness of one made up of D-amino acids. Since natural proteins contain right-handed α-helices, a helix composed of D-amino acids would be expected to be left-handed.
7.Can you discover additional helices in proteins?
According to what was discussed in the lecture, the structural space of proteins is not yet fully explored, and advances in computational protein design suggest that new structural motifs could be discovered or designed. With modern computational design tools and advances in AI and structure prediction methods, it is possible to identify sequences that could stabilize new structural motifs and generate novel conformations. Therefore, alternative backbone geometries and new helical motifs could potentially be discovered or designed if they are energetically stable.
9.Why do β-sheets tend to aggregate?
- What is the driving force for β-sheet aggregation?
10. Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials?
11.Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
I selected the human High Mobility Group Box 1 (HMGB1) protein because if find it realy interesting, it is a multifunctional chromatin-associated protein with both nuclear and extracellular roles. In the nucleus, HMGB1 acts as a DNA-binding architectural protein involved in transcriptional regulation, DNA replication, recombination, and repair. However, under conditions of cellular stress or damage, HMGB1 can be released into the extracellular space, where it functions as a pro-inflammatory signaling molecule (alarmin), contributing to immune activation and chronic inflammation.
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Does your protein belong to any protein family?
Homo Sapiens HMGB1 sequence (UNIPROT ID: P09429): MGKGDPKKPRGKMSSYAFFVQTCREEHKKKHPDASVNFSEFSKKCSERWKTMSAKEKGKFEDMAKADKARYEREMKTYIPPKGETKKKFKDPNAPKRPPSAFFLFCSEYRPKIKGEHPGLSIGDVAKKLGEMWNNTAADDKQPYEKKAAKLKEKYEKDIAAYRAKGKPDAAKKGVVKAEKSKKKKEEEEDEEDEEDEEEEEDEEDEDEEEDDDDE
length
Most frequent AA
Homologs
Protein Family
Domains
Reference
250 AA
K (20.0%)
250 hits*
High mobylity Group B family
HMG Box A and B
*BLAST search on UniProt (default parameters) returned 250 hits (the maximum), all with identity > 80%, e-value < 0.0001, and score > 1000. These parameters indicate that the proteins found are homologous to human HMGB1 and correspond to HMGB proteins in other organisms, wich is consistent with the fact that the this family is highly conserved among eukaryotes.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
I searched the UniProt ID of the protein in the RCSB PDB and found 13 structures corresponding to Homo sapiens. There is no structure of the full-length protein available; the most complete structure lacks the last ~45 amino acids corresponding to the acidic C-terminal tail.
2YRQ sequence:adittional AA | lacking AAGSSGSSGMGKGDPKKPRGKMSSYAFFVQTCREEHKKKHPDASVNFSEFSKKCSERWKTMSAKEKGKFEDMAKADKARYEREMKTYIPPKGETKKKFKDPNAPKRPPSAFFLFCSEYRPKIKGEHPGLSIGDVAKKLGEMWNNTAADDKQPYEKKAAKLKEKYEKDIAAYRAKGKPDAAKKGVVKAEKSKKKKEEEEDEEDEEDEEEEEDEEDEDEEEDDDDE
The selected structure was deposited in April 2007 and released in February 2008. It was solved by solution NMR, therefore a crystallographic resolution in Ångström is not reported. Instead, the quality of the structure is evaluated using NMR-specific validation parameters:
Conformers Calculated: 100
Conformers Submitted: 20
Clashscore: 18
Ramachandran outliers: 1.8%
Side-chains outliers: 4.6%
- Are there any other molecules in the solved structure apart from protein?
No, the structure contains only the protein.
- Does your protein belong to any structure classification family?
Yes. HMGB1 belongs to the High Mobility Group Box (HMG-box) fold, a well-characterized DNA-binding structural family composed mainly of α-helices. The structure contains HMG Box A and HMG Box B domains, which are conserved among eukaryotes.
Open the structure of your protein in any 3D molecule visualization software:
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon visualization. HMG box A in violet, HMG box B in redRibbon visualization. HMG box A in violet, HMG box B in redBall and stick visualization. HMG box A in violet, HMG box B in red
Color the protein by secondary structure. Does it have more helices or sheets?
The human HMGB1 structure contains α-helices and disordered regions, but no β-sheets. The helices form the structured HMG-box domains that mediate DNA binding, while the disordered regions provide the flexibility required for its function.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The amino acid composition shows a relatively balanced proportion of positively charged, negatively charged, polar, and hydrophobic residues. These residues are also distributed relatively uniformly along the protein sequence, rather than being strongly clustered in specific regions.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Protein Surface. HMG box A in violet, HMG box B in redCartoon visualization HMG box A in violet, HMG box B in red.Protein surface colored by residue type.
Between the HMG boxes, there is a “pocket” where the protein interacts with DNA. The blue residues correspond to positively charged amino acids, which are important for binding to the negatively charged DNA backbone.
Part C. Using ML-Based Protein Design Tools
For this part of the homework, I chose the HMGB protein of T. cruzi (TcHMGB)
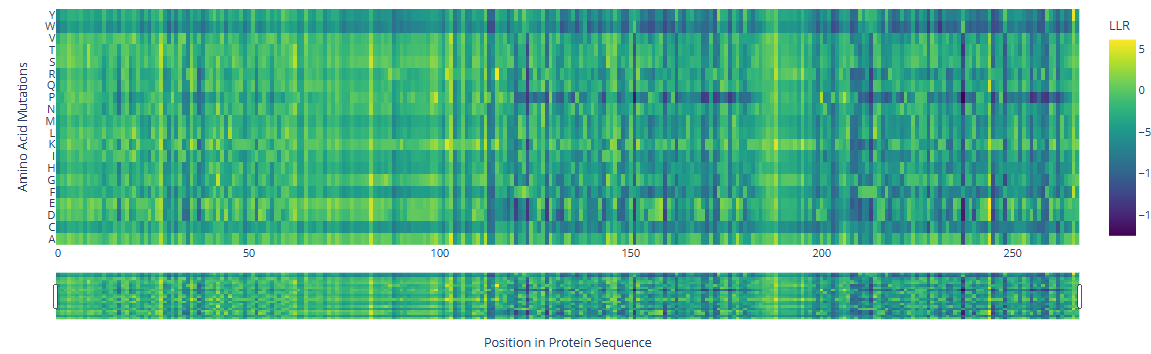
Deep Mutational Scansa. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out.)
There are three specific regions where the heatmap shows predominantly blue colors, indicating negative LLR values and therefore unfavorable mutations. These regions correspond to the two HMG box domains, which are highly conserved among eukaryotes and are critical for DNA binding, and the Deck-C domain, which is conserved among trypanosomatids.
In contrast, the regions in between the domain display less dark-blue colors and more greenish-yellowish colors, corresponding to positive LLR values. This suggests that mutations in this region are better tolerated, consistent with a lower functional or structural constraint.
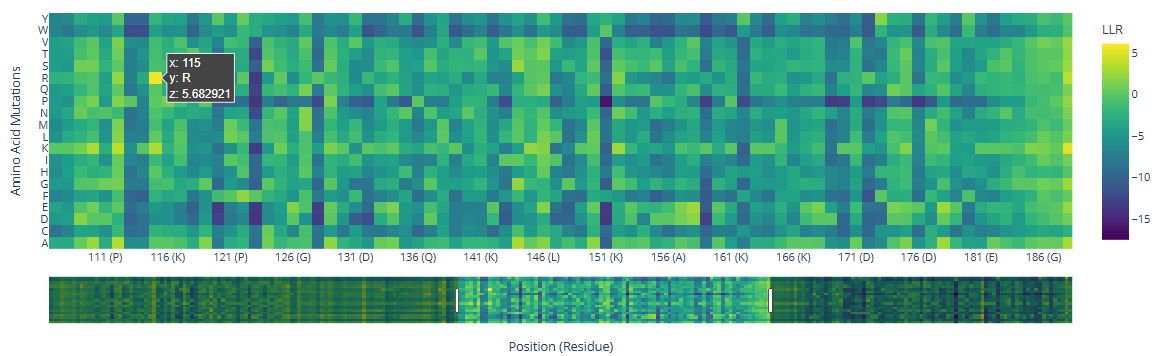
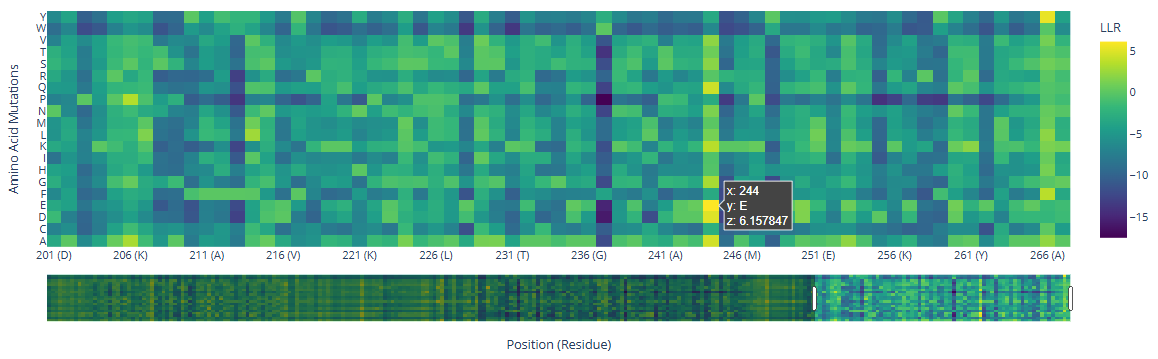
There are a few mutations that stand out in the HMG Box domains:
Position
wt
mutated
LLR value
115
R ()
5.68
244
()
E ()
6.16
c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to the experiment.
Latent Space Analysisa. Use the provided sequence dataset to embed proteins in reduced dimensionality.
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
a. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
b. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
c. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
To calculate the reference binder pseudo-perplexity, I used the compute_pseudo_perplexity function defined in the Load Model cell. For this purpose, I created an additional cell in the Colab notebook and computed the pseudo-perplexity of the known SOD1-binding peptide FLYRWLPSRRGG under the same conditions used for the generated binders.
The resulting pseudo-perplexity values are shown in the table below. Lower pseudo-perplexity values indicate higher model confidence in the corresponding binder sequences. Notably, several PepMLM-generated binders exhibited lower pseudo-perplexity than the reference peptide, suggesting that the model assigns higher confidence to these sequences when conditioned on the mutant SOD1 context.
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
ipTM value
Binder structure
Bindeing site
SOD1-Reference
0.33
Disordered
The C-terminus is located between two small α-helices (aa 51–67 and 134–136), while the N-terminus lies near the bottom of the β-barrel, opposite to the V4 residue.
SOD1-Binder0
0.48
Disordered
Binding in SOD1 surface near a β-sheet region of the SOD1 β-barrel (aa 90–102).
SOD1-Binder1
0.2
Small α-helice in the middle
Burried between two small α-helices (aa51-67 and 134-136) next-to the barrel. Not close to V4
SOD1-Binder2
0.34
Disordered
Same binding that Binder 1
SOD1-Binder3
0.42
Disordered
Binding in the barre surface, far from amino acid V4 of SOD1
The predicted protein–peptide complexes exhibit relatively low ipTM values, ranging from 0.20 to 0.48, indicating limited confidence in the modeled interfaces. Among the PepMLM-generated peptides, Binder0 displays the highest ipTM score (0.48), exceeding that of the known SOD1-binding reference peptide (0.33). Most peptides bind to surface-exposed regions of the SOD1 β-barrel or near small α-helical elements rather than localizing close to the N-terminal region harboring the A4V mutation. Additionally, several peptides remain largely disordered upon binding, suggesting flexible and potentially weak interactions. Overall, one generated binder shows improved predicted interface confidence relative to the reference, although the low ipTm value indicates a limited confidence in the modeled interfaces.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes:
a. Predicted binding affinity
b. Solubility
c. Hemolysis probability
d. Net charge (pH 7)
e. Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.