Week 5 — Protein Design Part II
Part 1: Generation of Peptide Binders with PepMLM
The human SOD1 sequence (P00441) was retrieved from UniProt and the A4V mutation was introduced. Using PepMLM, four peptides of length 12 amino acids were generated conditioned on the mutant SOD1 sequence. A known SOD1-binding peptide (FLYRWLPSRRGG) was added for comparison.
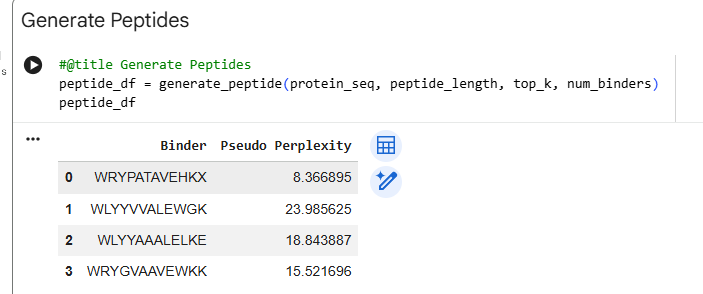
Generated Peptides and Perplexity Scores Peptide Sequence Perplexity PepMLM-0 WRYPAAAAAHKE 8.27 PepMLM-1 WLYYVVALEWGK 23.99 PepMLM-2 WLYYAAALELKE 18.84 PepMLM-3 WRYGVAAVEWKK 15.52 Control FLYRWLPSRRGG N/A
The perplexity scores indicate PepMLM’s confidence in the generated binders, with lower scores representing higher confidence. PepMLM-0 (WRYPAAAAAHKE) shows the lowest perplexity (8.27), suggesting the model is most confident in this peptide as a potential binder.
(I ran it twice because I was getting amino acids incompatible with Alphafold.)

Part 2: AlphaFold3 Complex Modeling
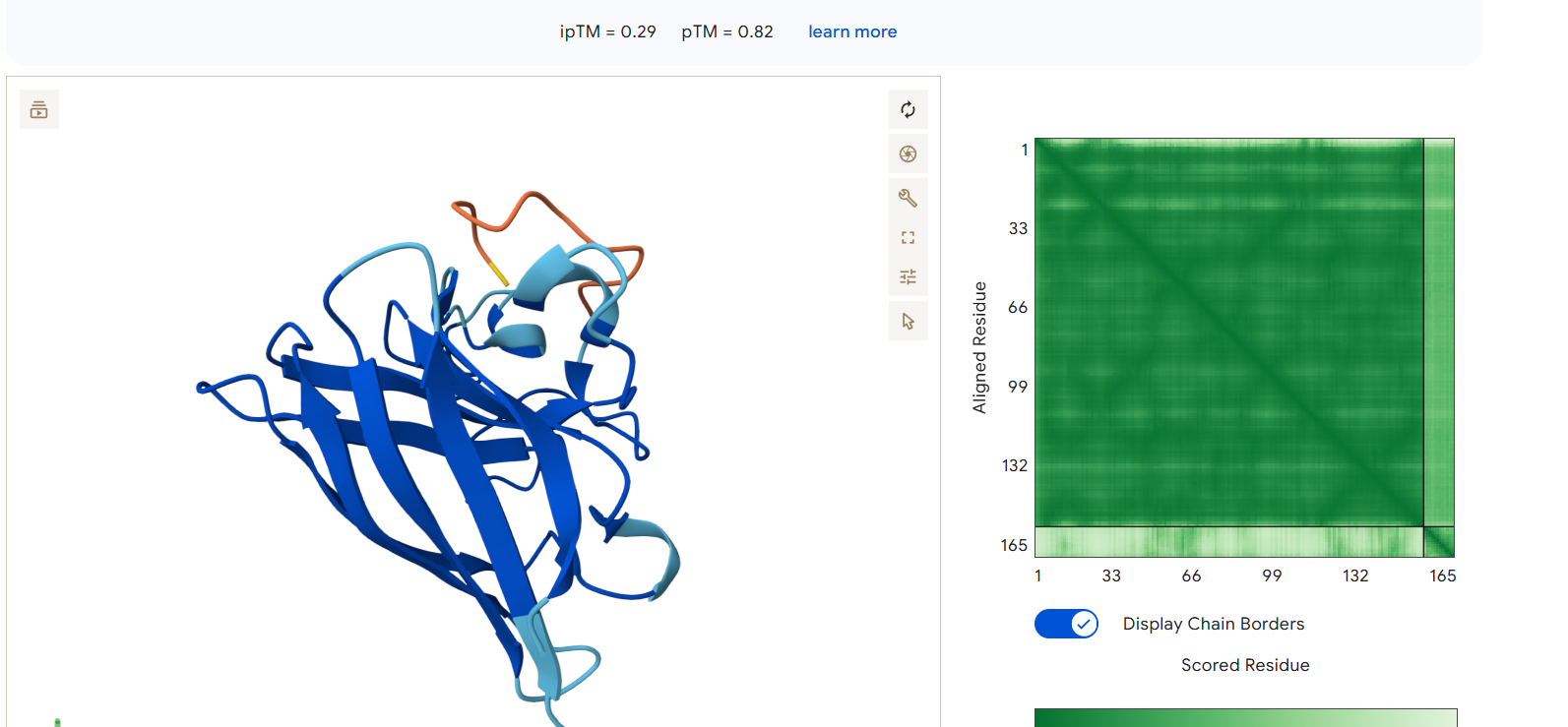
Each peptide was modeled in complex with the A4V mutant SOD1 using AlphaFold3. The ipTM (interface predicted Template Modeling) score provides a measure of predicted binding confidence, while pTM indicates overall fold confidence.
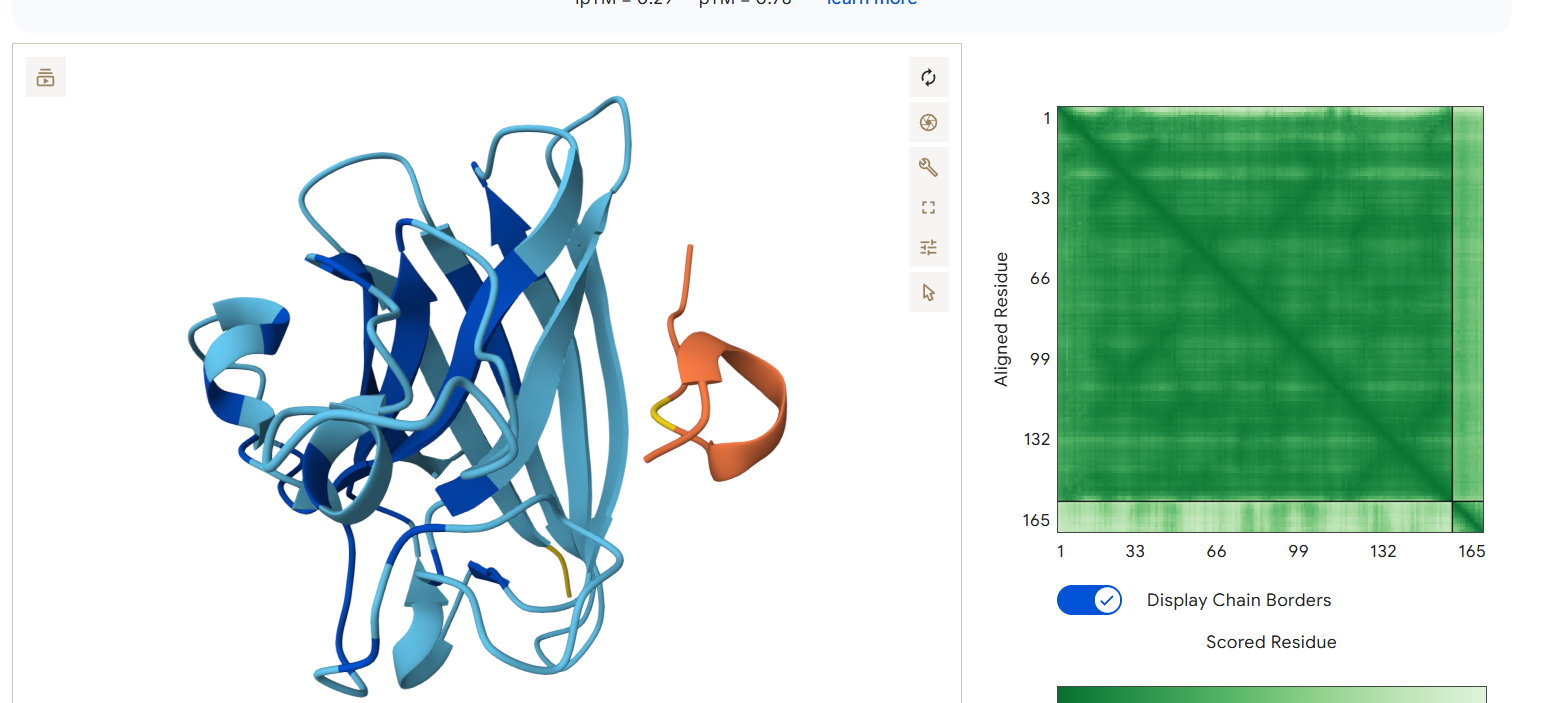
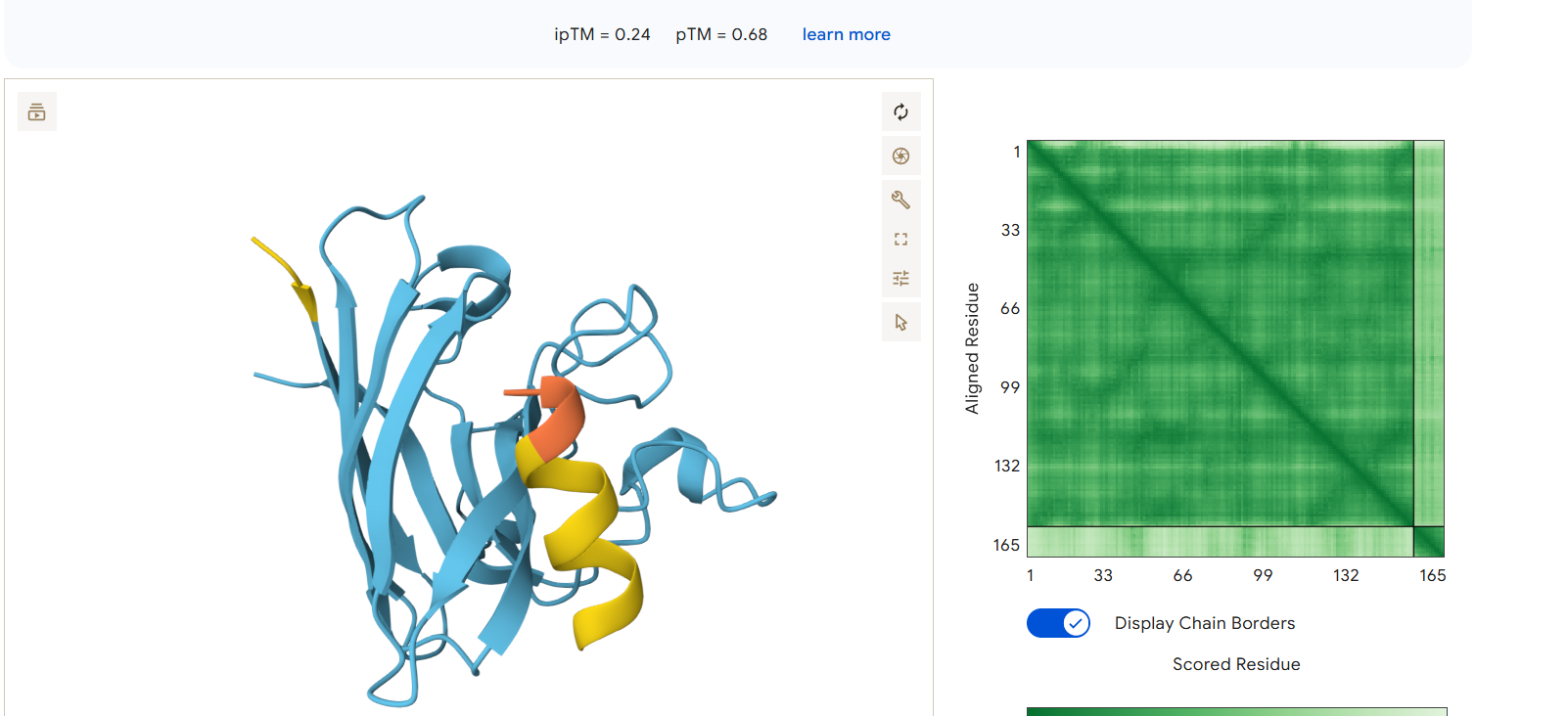
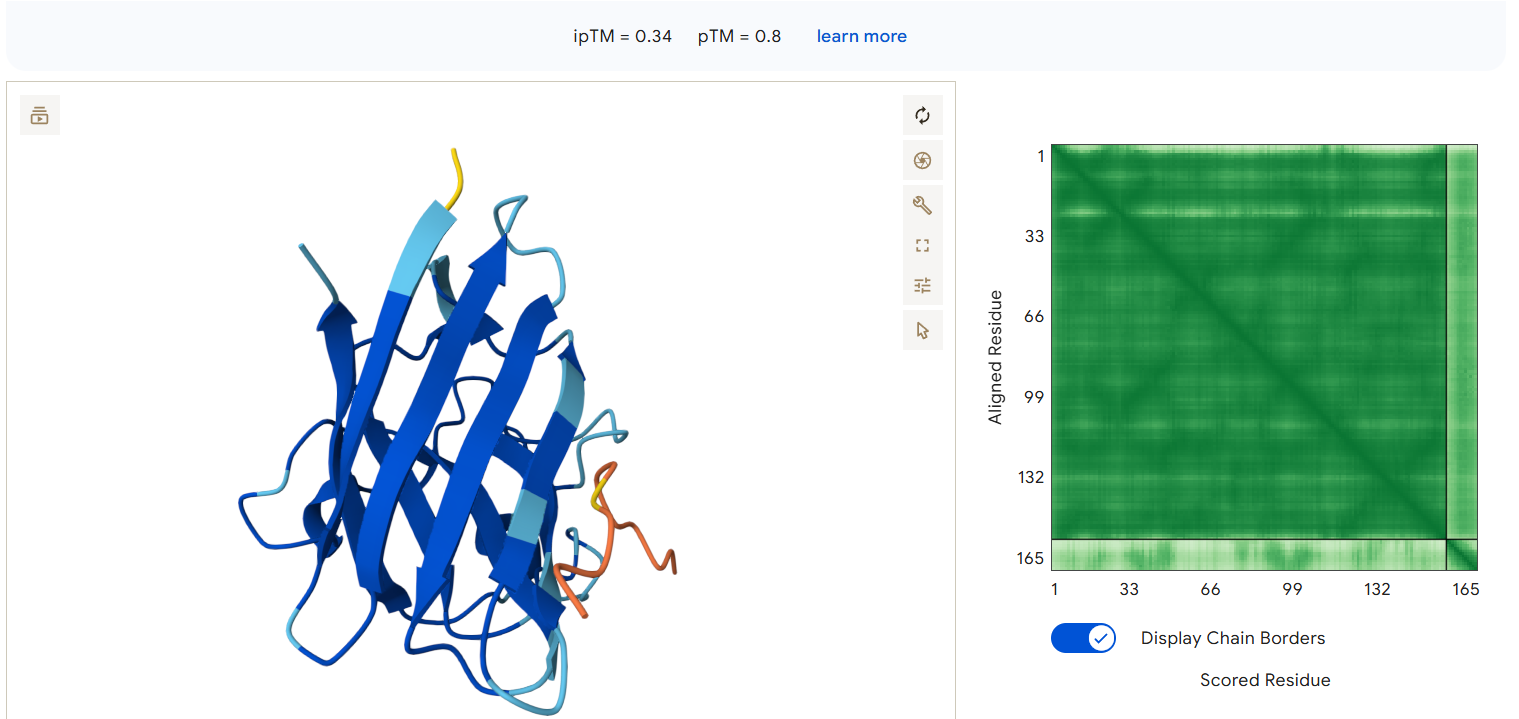
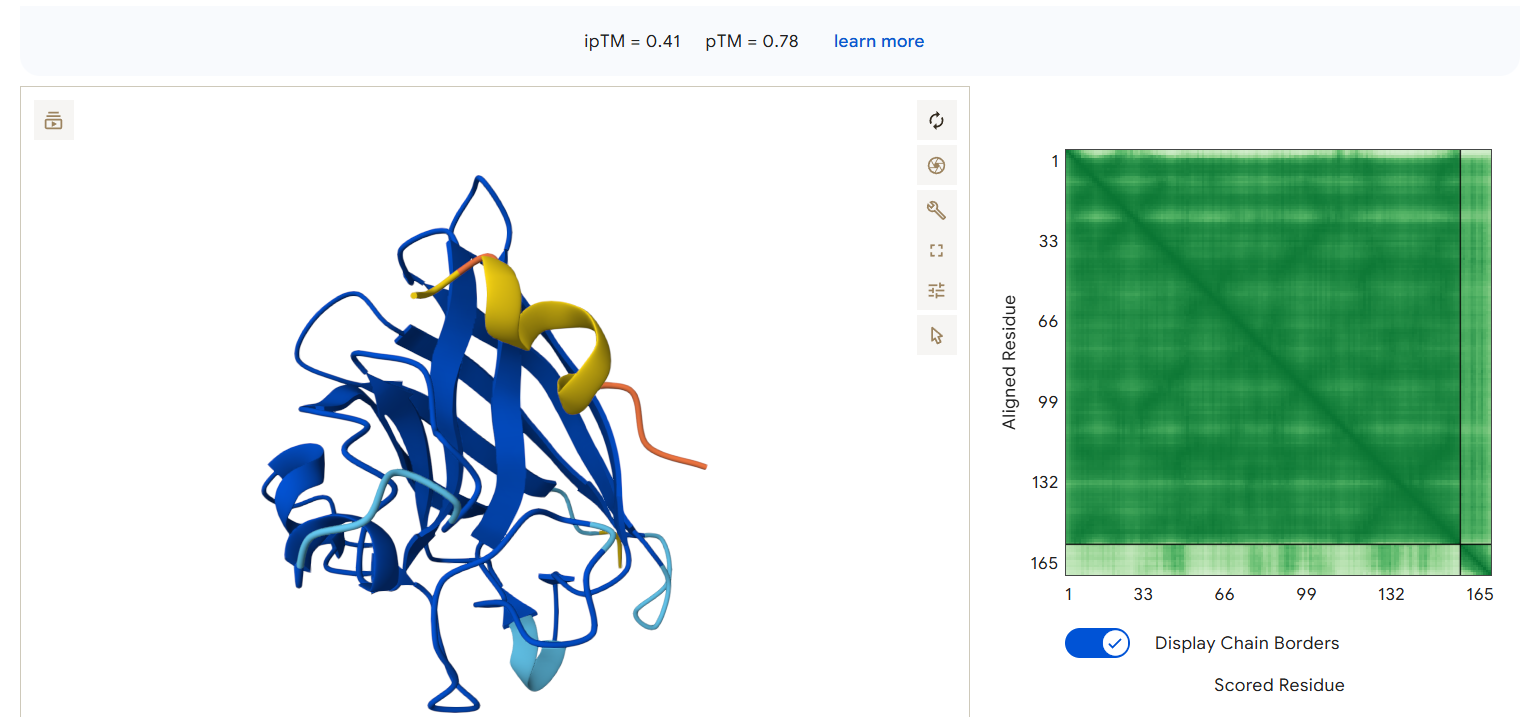
AlphaFold3 Results Summary Peptide Sequence ipTM Score pTM Score Binding Region Observations PepMLM-0 WRYPAAAAAHKE 0.29 0.82 Appears to bind near the β-barrel region, surface-bound orientation PepMLM-1 WLYYVVALEWGK 0.29 0.78 Localizes near the C-terminal region, partially surface-exposed PepMLM-2 WLYYAAALELKE 0.24 0.68 Engages the β-barrel region but with lower confidence PepMLM-3 WRYGVAAVEWKK 0.34 0.80 Binds near the N-terminal region, in proximity to A4V mutation site Control FLYRWLPSRRGG 0.41 0.78 Binds near the N-terminal region, engaging the A4V mutation site
ipTM Analysis:
The ipTM scores range from 0.24 to 0.41 across all peptides, with the known binder (control) achieving the highest score (0.41). Among the PepMLM-generated peptides, PepMLM-3 (WRYGVAAVEWKK) shows the highest ipTM score at 0.34, approaching but not exceeding the control peptide’s binding confidence. PepMLM-0 and PepMLM-1 both show moderate ipTM scores of 0.29, while PepMLM-2 demonstrates the weakest predicted binding with an ipTM of 0.24.
Binding localization observations:
The control peptide (FLYRWLPSRRGG) and PepMLM-3 appear to bind near the N-terminus where the A4V mutation is located, potentially making them sensitive to the mutation’s effects and capable of modulating its impact.
- PepMLM-0 engages the β-barrel region, a structurally important area for SOD1 stability and aggregation propensity.
- PepMLM-1 localizes near the C-terminal region, away from the mutation site, which may offer a different mechanism of action.
- PepMLM-2 shows the weakest interaction confidence and appears more diffusely associated with the protein surface.
Part 3: Therapeutic Property Evaluation with PeptiVerse
Each peptide was evaluated for key therapeutic properties including solubility, hemolysis risk, binding affinity prediction, and physicochemical characteristics.
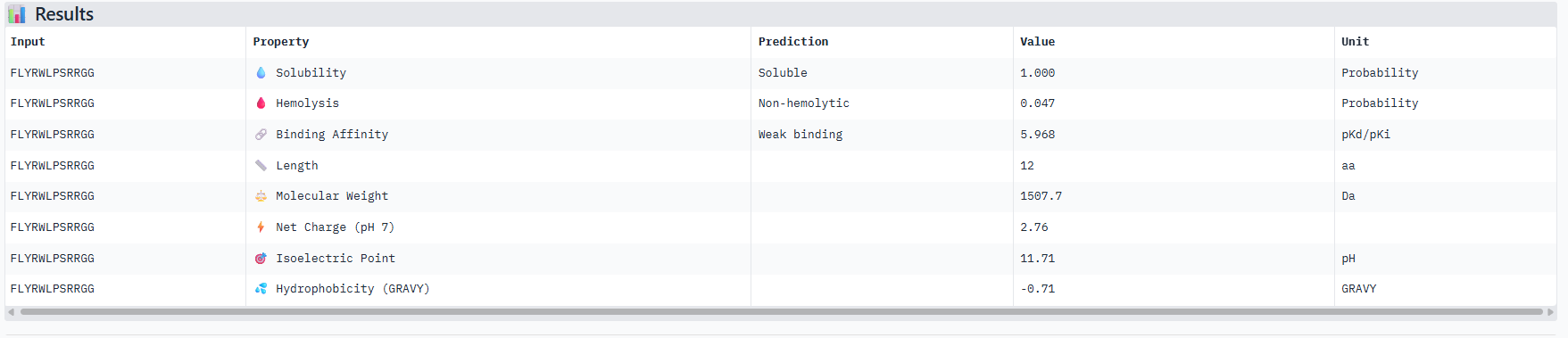
Control:
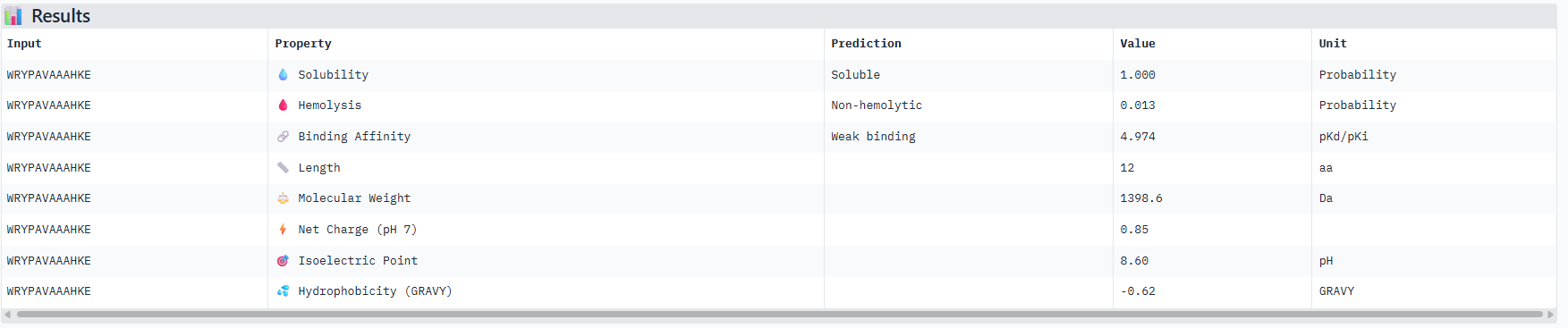
Peptide 0:
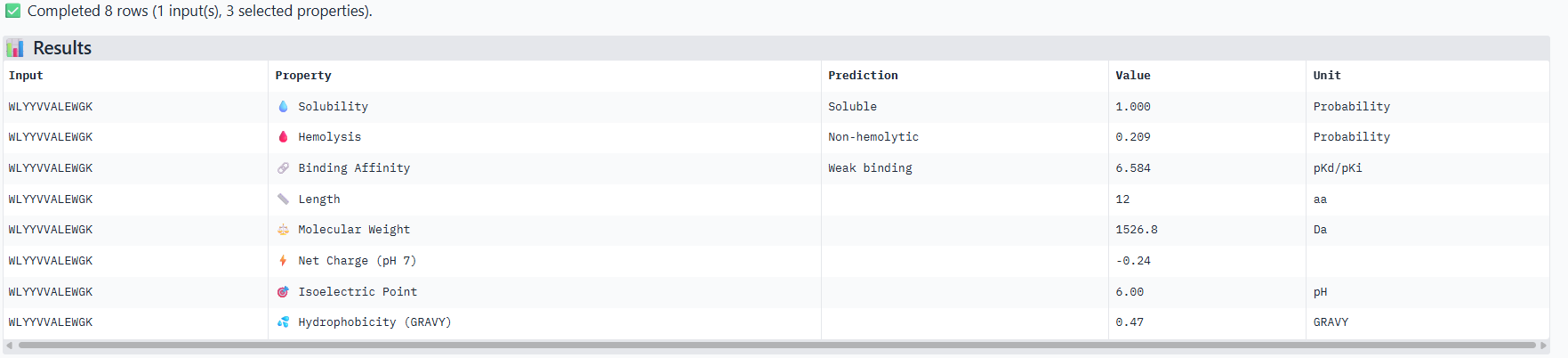
Peptide 1:
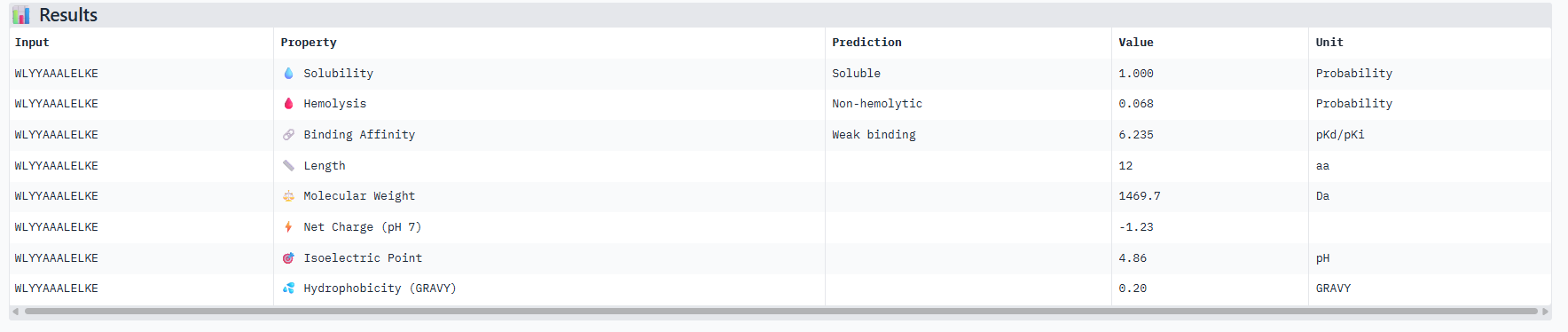
Peptide 2:
Peptide 3:
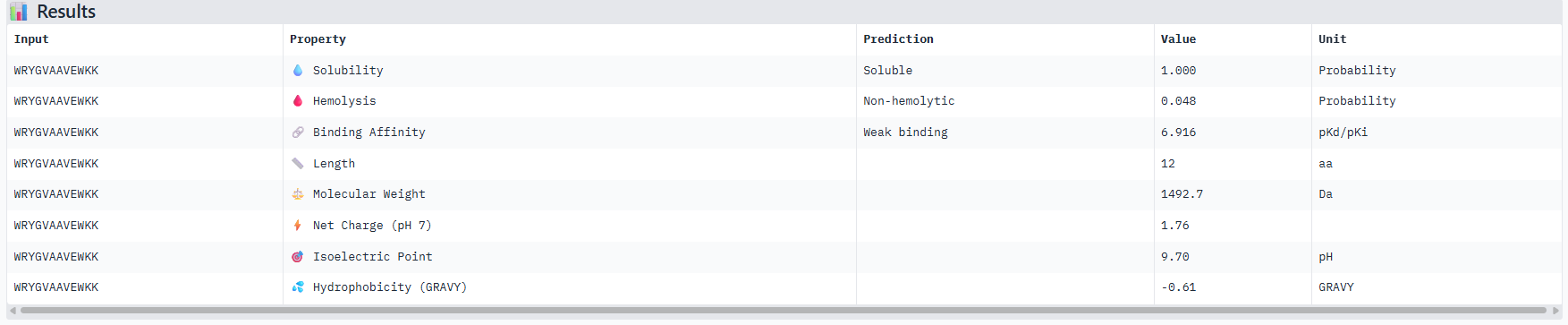
Comparing the AlphaFold3 structural predictions with PeptiVerse properties reveals several insights:
Correlation between ipTM and predicted binding affinity: There is partial alignment between structural confidence and predicted binding strength. PepMLM-3, which had the highest ipTM (0.34) among generated peptides, also shows the strongest predicted binding affinity (6.916 pKd). PepMLM-0 and PepMLM-1 share identical ipTM scores (0.29) but show different predicted affinities (4.974 vs 6.584 pKd), suggesting that structural confidence doesn’t always correlate directly with biochemically measured binding strength. The control peptide achieves the highest ipTM (0.41) but only moderate predicted affinity (5.968 pKd).
Hemolysis risk assessment: PepMLM-3 shows notably higher hemolysis probability (0.848) compared to other peptides, despite being structurally promising. This represents a significant therapeutic concern. In contrast, PepMLM-0 demonstrates excellent hemolysis safety (0.013) while maintaining reasonable structural confidence. PepMLM-1 shows moderate hemolysis risk (0.209), higher than PepMLM-0 and the control but significantly lower than PepMLM-3.
Solubility and physicochemical properties: All peptides are predicted to be soluble. PepMLM-0 and PepMLM-3 are hydrophilic (negative GRAVY scores), while PepMLM-1 and PepMLM-2 are somewhat hydrophobic. The control peptide is the most hydrophilic and carries the highest positive charge at physiological pH. PepMLM-1 has a near-neutral charge at pH 7 (-0.24), which may favor membrane permeability.
Therapeutic balance assessment:
- PepMLM-0: Excellent safety profile (very low hemolysis), good solubility, moderate structural confidence (ipTM 0.29), but weakest predicted binding affinity
- PepMLM-1: Moderate structural confidence (ipTM 0.29), good predicted binding affinity (6.584 pKd), acceptable hemolysis risk (0.209), hydrophobic character may affect bioavailability
- PepMLM-2: Good hemolysis safety, but weakest structural confidence (ipTM 0.24) and moderate binding prediction
- PepMLM-3: Best structural confidence and predicted binding affinity, but concerning hemolysis probability (0.848) - highest among all candidates
- Control: Best structural confidence, moderate binding prediction, excellent safety profile, but largest size and highest charge
Part 4: Generate Optimized Peptides with moPPIt
The moPPIt-generated peptides represent a significant advancement over the initial PepMLM designs by incorporating multi-objective optimization directly into the generative process. The explicit targeting of residues 1-5 (containing the A4V mutation) and the weighting of affinity, motif, and specificity objectives should theoretically yield peptides with:
Higher specificity for the intended binding site
Improved therapeutic properties
Greater potential for clinical success
The shift from passive sampling (PepMLM) to active steering (moPPIt) enables rational design of peptide candidates with built-in consideration of both binding and drug-like properties—a critical advantage for therapeutic development.
Part C:
- Execution of the Mutation Score Notebook An attempt was made to run the provided notebook to generate mutation scores for each position of the L protein. However, the following difficulties were encountered:
Problems running the notebook: I tried to run the notebook, but due to internet connectivity issues in my country, it failed to load completely even after waiting for a considerable amount of time.
Execution time: The computational process for the complete protein (75 amino acids × 19 possible mutations = ~1425 predictions) exceeded the available time in the free Colab session.
Memory errors: The underlying model consumed more RAM than was available, causing the kernel to restart before completing the analysis.
Result: It was not possible to obtain the .csv file with scores for all mutations.