Week 5 HW: Protein design part 2
Part 1: Generate Binders with PepMLM
For this exercise, I used the human SOD1 target protein and introduced the A4V mutation. I then used PepMLM to generate four candidate 12-amino-acid peptide binders against the mutant target sequence. As requested in the assignment, I also included the known binder peptide FLYRWLPSRRGG for comparison.
What is a A4V mutation:
- A = alanine
- 4 = position 4
- V = valine
- So it means the alanine at that position is replaced by valine. . In SOD1, A4V is a famous mutation. It is often described as one of the more aggressive SOD1-linked variants.
*What is SOD1:
- Stands for superoxide dismutase 1. It is the gene/protein for an enzyme that helps protect cells from oxidative damage by breaking down superoxide radicals, which are harmful oxygen byproducts of normal metabolism. Human SOD1 is the well-known copper/zinc superoxide dismutase found in the cytoplasm
Target sequence used
A4V mutant SOD1 sequence:
Interpretation
- PepMLM produced four short peptide candidates for the mutant SOD1 target. Based on the perplexity values, PepMLM-2 (WDWDSAAAAAAK) is the most promising candidate, because it has the lowest perplexity, which indicates the highest model confidence among the generated sequences. PepMLM-3 ranked second, while PepMLM-1 and PepMLM-4 had higher perplexity and are therefore less favored by the model.
- It is also interesting that the generated peptides are quite different in composition from the known binder FLYRWLPSRRGG. The PepMLM outputs are enriched in small, polar, and acidic residues such as A, G, D, H, and S, while the known binder contains more hydrophobic and basic residues such as F, L, W, R, and Y. This suggests that the model explored a different part of sequence space while still proposing candidate binders for the same target.
- Overall, the strongest candidate from this step is PepMLM-2, which I would prioritize for the next stage of structural evaluation.
Part 2: Evaluate Binders with AlphaFold3
I evaluated each peptide by submitting the A4V mutant SOD1 sequence together with each peptide as separate chains in AlphaFold Server. For each prediction, I recorded the ipTM score and visually inspected where the peptide appeared to bind on SOD1. The goal was to see whether the peptide localized near the N-terminus/A4V region, the β-barrel surface, or the dimer interface. AlphaFold Server reports ipTM as a confidence measure for predicted interfaces in complexes, so higher values suggest a more confident protein–peptide interaction.
What is??
ipTM stands for interface predicted TM-score. It is a confidence score for the relative positioning of the chains basically, how believable the predicted interaction interface is between the protein and the peptide. Higher is better. A commonly used rough interpretation is: above 0.8 = strong confidence, below 0.6 = likely weak or failed prediction, and 0.6–0.8 = gray zone where the pose may or may not be right.
N-terminus / A4V region is the beginning of the protein chain. In SOD1, the A4V mutation is right near that beginning region: alanine is replaced by valine close to the N-terminal end. In the A4V mutant, the overall SOD1 structure is mostly preserved, studies report increased disorder around the N-terminus and a shift in how the two SOD1 subunits sit together. Reff
β-barrel is a protein fold made from multiple β-strands that wrap around into a barrel-like shape. SOD1’s monomer is built around an eight-stranded antiparallel β-barrel, and SOD1 is a dimer of two such β-barrels. The β-barrel surface means the outside exposed face of that folded barrel.
Dimer interfaceSOD1 normally functions as a homodimer, meaning two identical SOD1 subunits bind together. The dimer interface is the set of surfaces and contacts where those two subunits touch each other Reff
AlphaFold results
| Peptide ID | Sequence | Top ipTM | Interpretation of binding pose |
|---|---|---|---|
| PepMLM-1 | WSDDAVVDAVHA | 0.52 | Weak-to-moderate interface. The peptide sits near the protein surface, but the pose is not tightly packed and looks only loosely associated. |
| PepMLM-2 | WDWDSAAAAAAK | 0.49 | Weak interface. The peptide appears offset from the SOD1 surface and does not form a convincing bound complex. |
| PepMLM-3 | WHSGPGAAAAAK | 0.64 | Strongest of the five tested peptides. The peptide lies across the surface of SOD1 in a more continuous contact pose than the others. |
| PepMLM-4 | HHSGSGGAAGKH | 0.39 | Weak interface. The peptide touches one side of the protein but remains extended and low-confidence. |
| Known binder | FLYRWLPSRRGG | 0.33 | Weakest result in this AlphaFold screen. The peptide remains mostly detached and does not form a convincing bound pose in the top-ranked model. |
Structural observations
PepMLM-1
The top-ranked model for PepMLM-1 gave an ipTM of 0.52, which was moderate but not especially convincing. In the chain-colored view, the peptide is close to SOD1 but still looks somewhat detached rather than tightly docked. I interpreted this as a weak or ambiguous interaction, not a strongly defined binding mode.
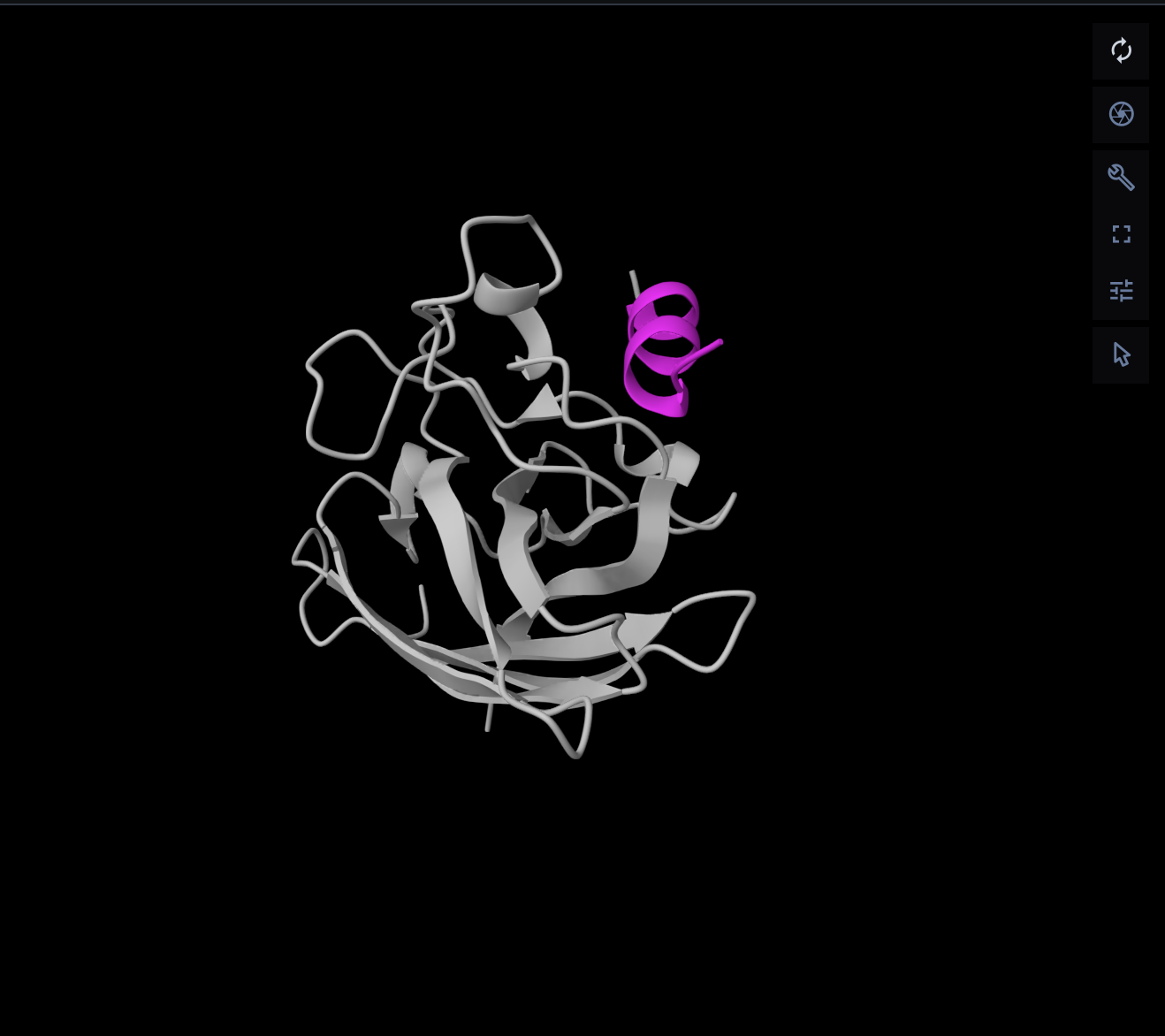
PepMLM-2
Although PepMLM-2 had the best PepMLM perplexity score in Part 1, the AlphaFold result was less convincing. Its top-ranked model had an ipTM of 0.49, and the peptide appears offset from the protein surface rather than packed into a clear binding site. This suggests that sequence plausibility from PepMLM did not translate into the strongest structural interface.
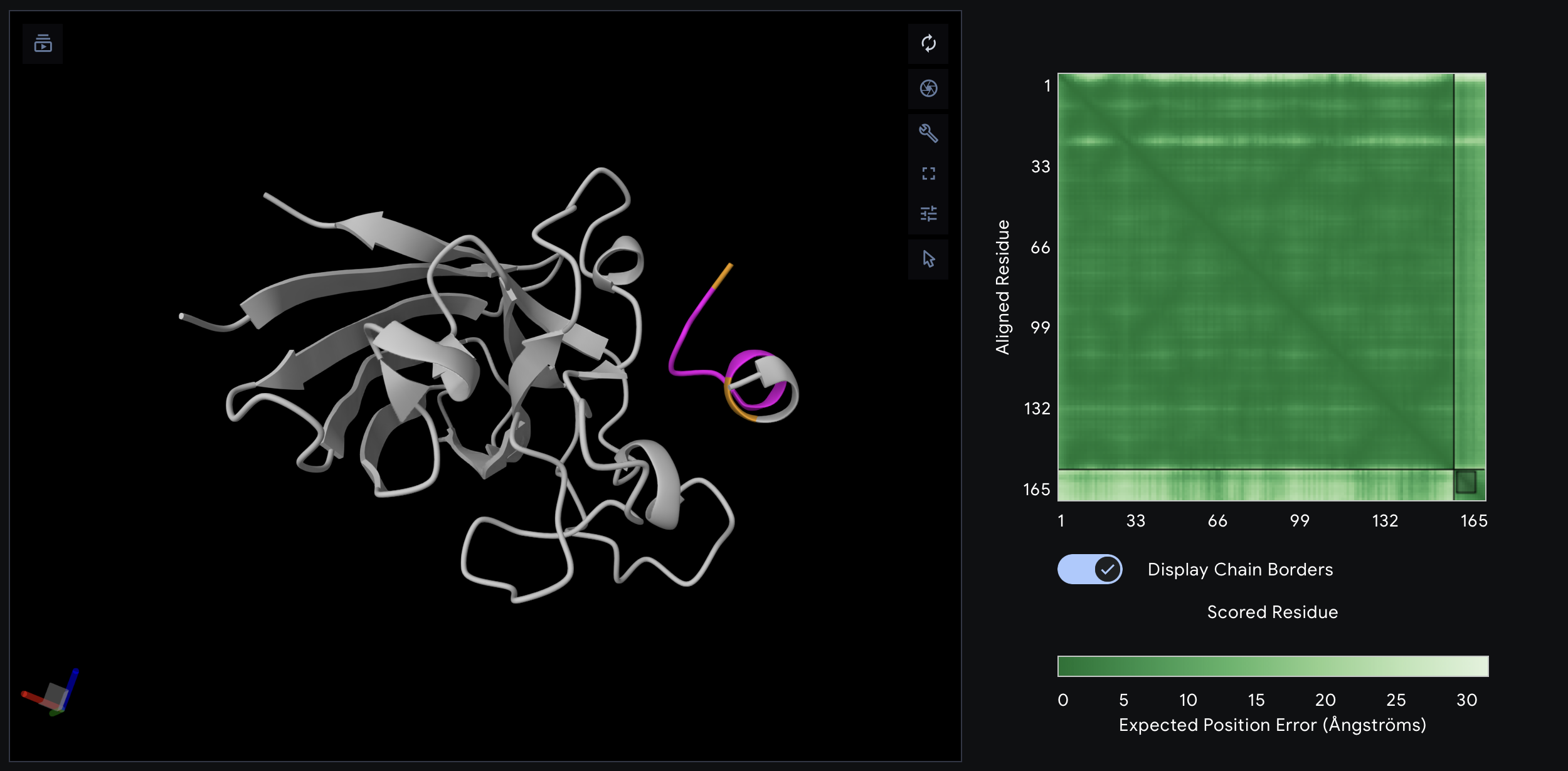
PepMLM-3
PepMLM-3 performed best in the AlphaFold comparison, with a top-ranked ipTM of 0.64. Visually, this peptide follows the SOD1 surface much more closely than the others and appears to form a broader, more continuous contact region. Even though this is still not an extremely high-confidence interface, it is the most convincing binding pose among the five peptides tested.
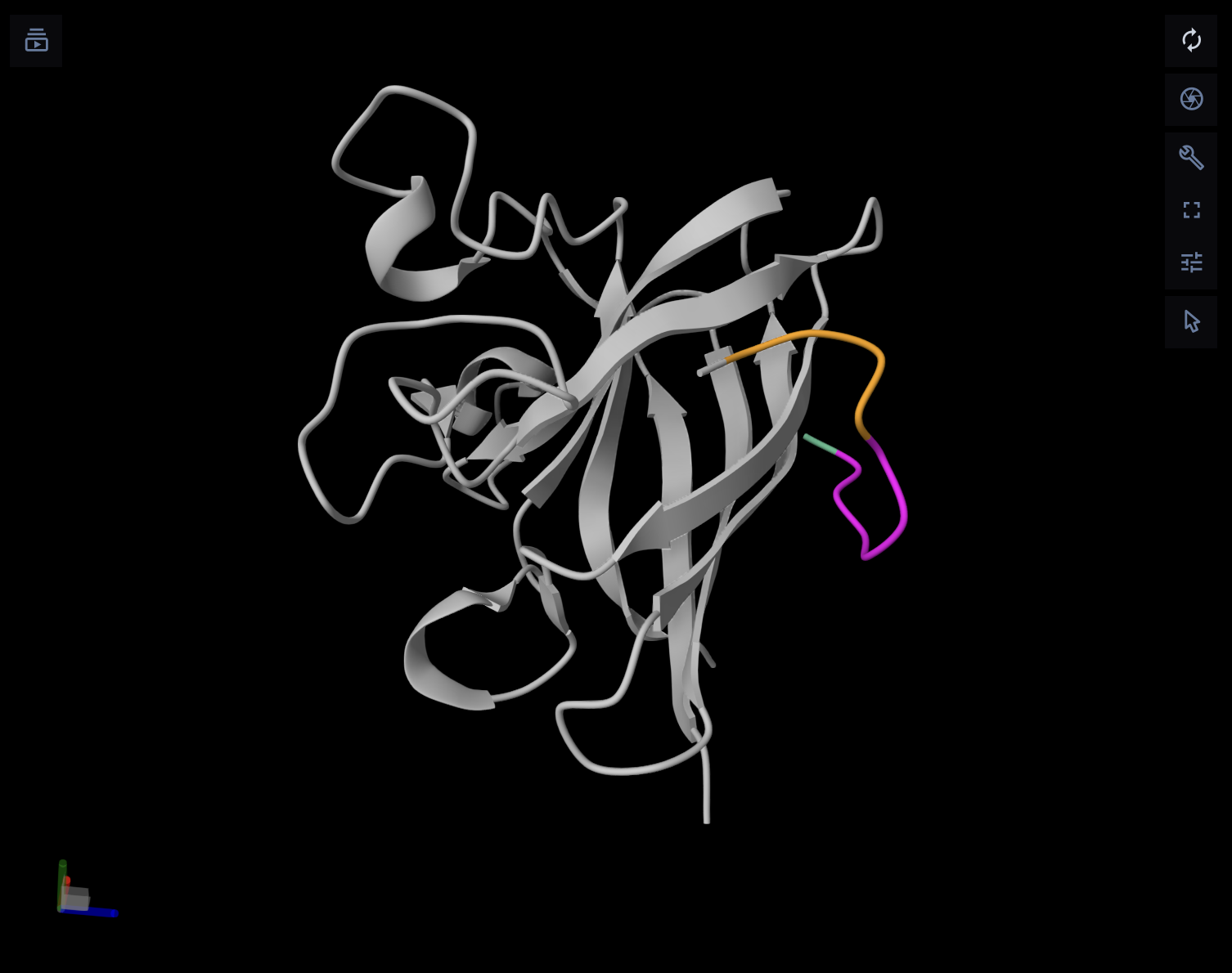
PepMLM-4
For PepMLM-4, the top-ranked model had an ipTM of 0.39. The peptide touches the protein surface, but the interaction looks elongated and weak, without a compact docking geometry. I therefore considered this a poor candidate relative to PepMLM-1 and especially PepMLM-3.
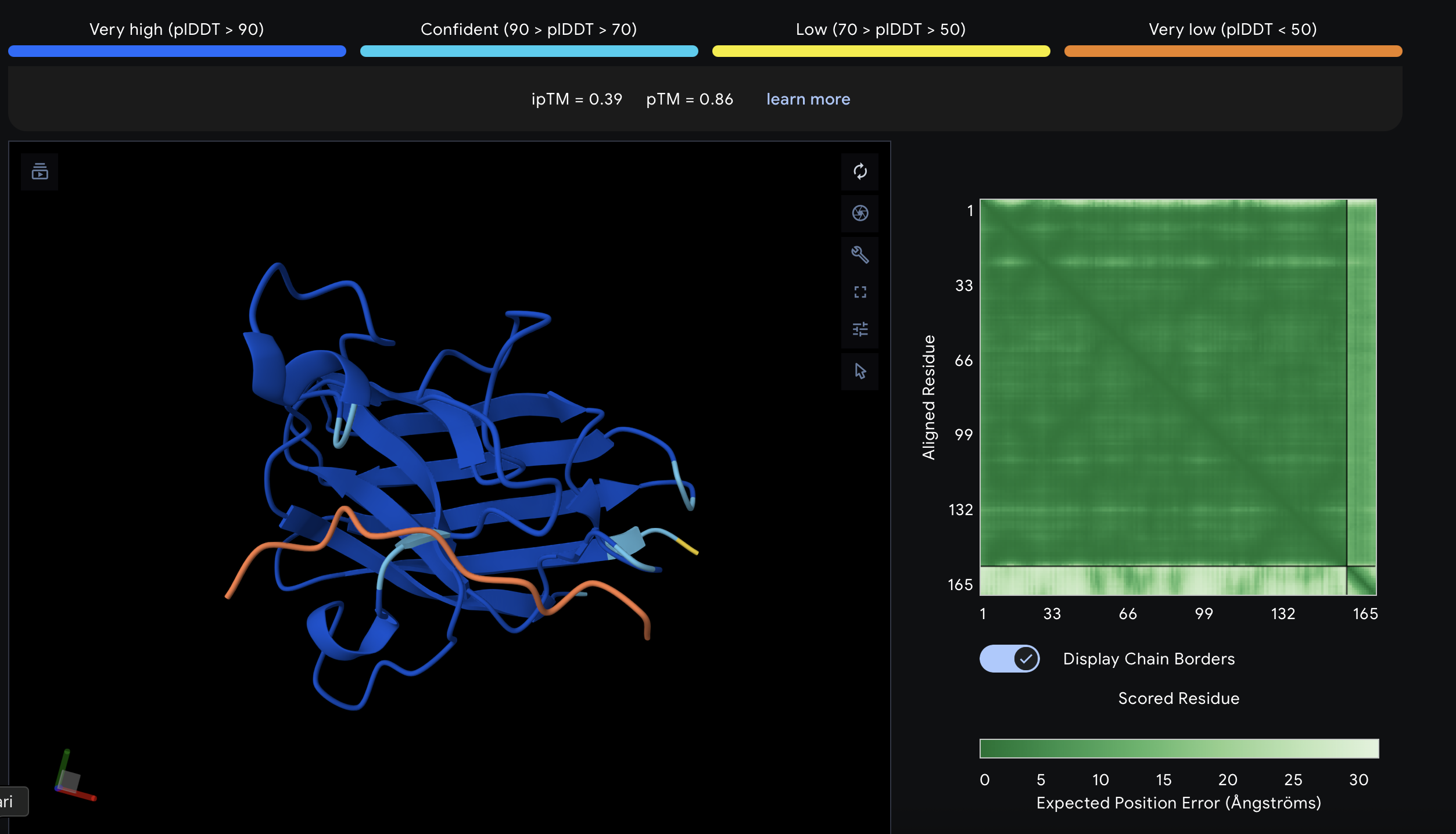
Known binder
The known binder surprisingly gave the weakest structural result in this AlphaFold screen, with a top-ranked ipTM of 0.33. In the chain-colored view, the peptide remains mostly separate from the protein and does not adopt a clear bound conformation. This does not necessarily mean it cannot bind experimentally, but in this prediction set it was less convincing than the best PepMLM-generated candidate.
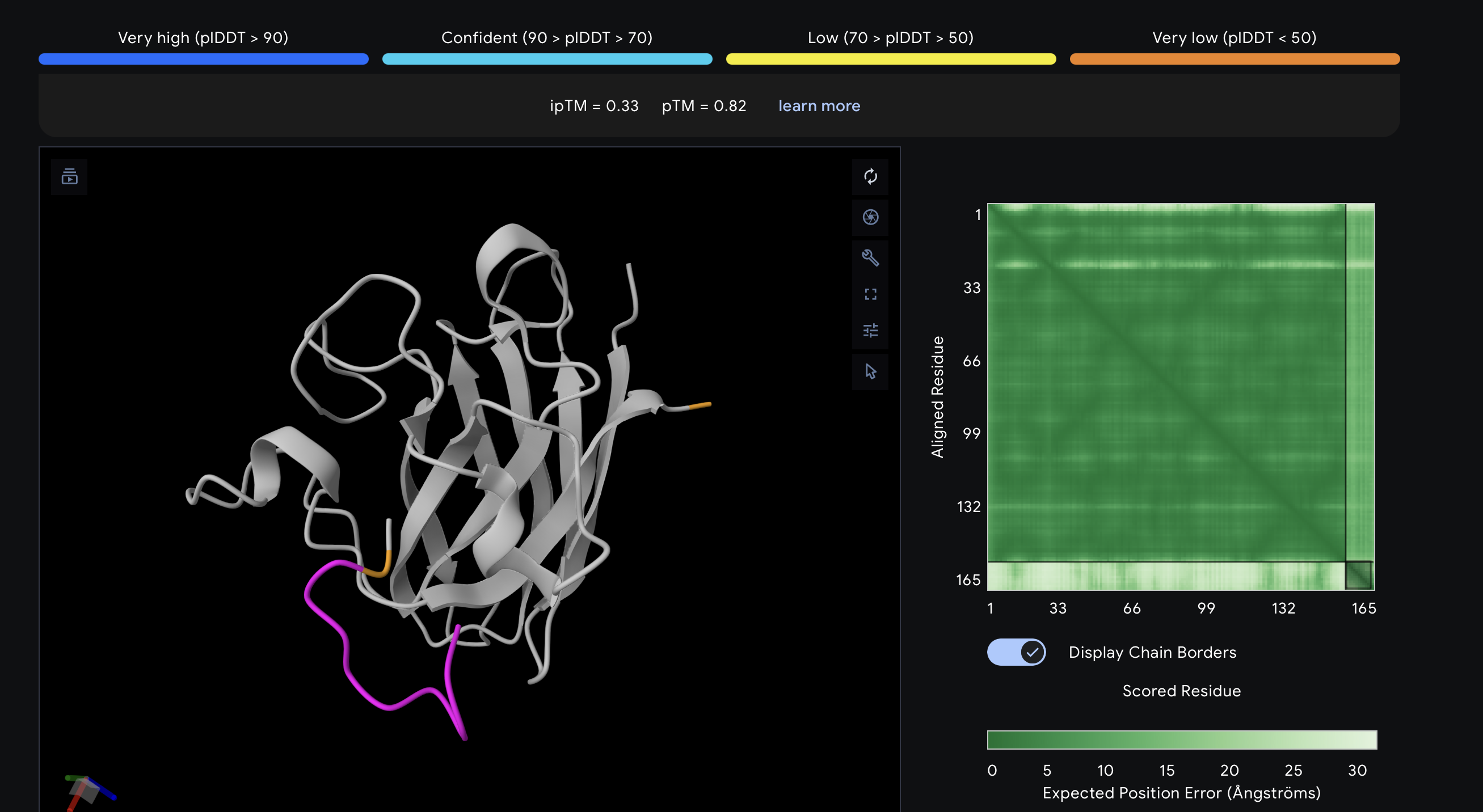
Interpretation
Overall, PepMLM-3 (WHSGPGAAAAAK) was the most promising peptide in the AlphaFold evaluation because it had the highest ipTM (0.64) and the most convincing surface-bound pose. PepMLM-1 was intermediate, while PepMLM-2, PepMLM-4, and the known binder all looked weaker in the structural screen.
An interesting result is that the peptide with the lowest PepMLM perplexity was PepMLM-2, but the peptide with the best AlphaFold complex prediction was PepMLM-3. This shows that sequence-level model confidence and structure-level interface confidence are related but not identical. In this case, I would prioritize PepMLM-3 for follow-up testing.
Another important observation is that none of the peptides clearly docked directly at the extreme N-terminal A4V mutation site itself. Instead, the predicted interactions were mostly distributed over broader exposed surfaces of SOD1. So the best candidate here appears to behave more like a surface-binding peptide than a mutation-site-specific binder.
Final ranking from Part 2
- PepMLM-3 — best overall AlphaFold interface
- PepMLM-1 — moderate but weaker than PepMLM-3
- PepMLM-2 — weaker structural support despite best PepMLM perplexity
- PepMLM-4 — poor interface
- Known binder — weakest in this AlphaFold screen
Part 3: Evaluate Properties of Generated Peptides in PeptiVerse
This part answers even if this peptide looks like the best binder, is it also a realistic peptide to pursue?
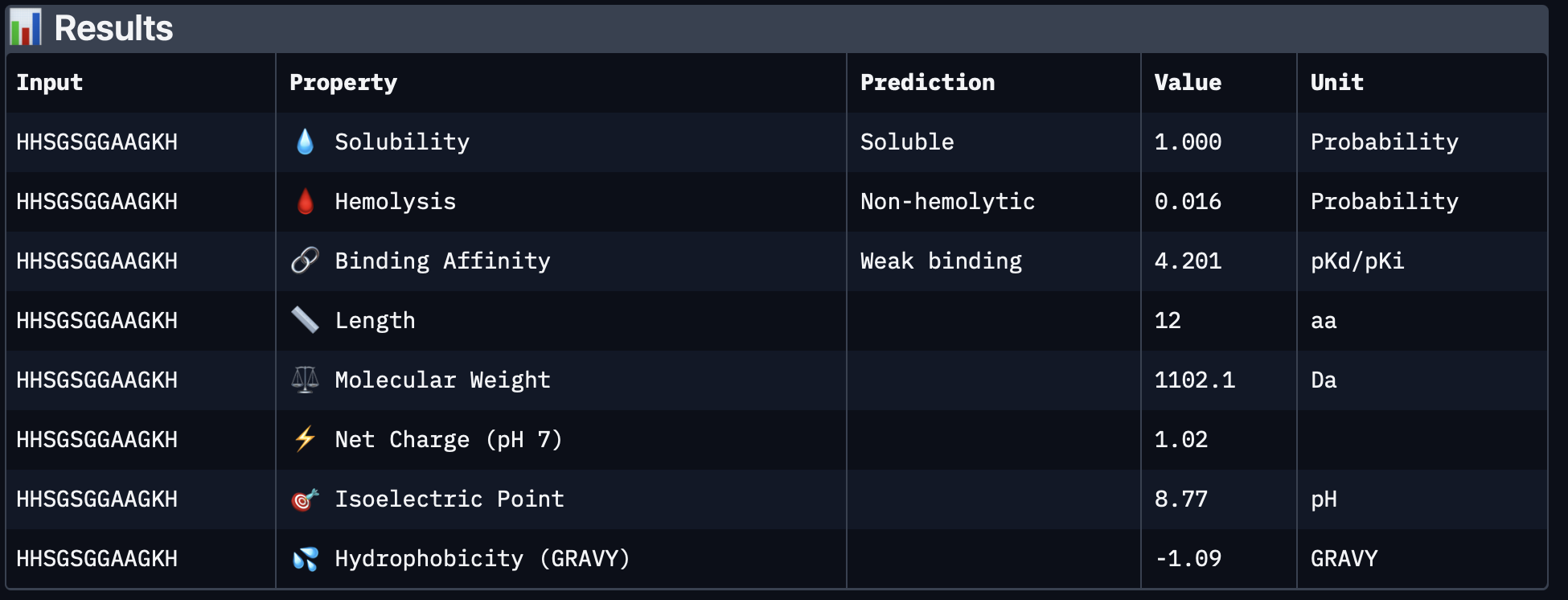
To further compare the PepMLM-generated peptides, I evaluated each one in PeptiVerse using the A4V mutant SOD1 sequence as the protein target. I recorded the required outputs from the homework prompt: predicted binding affinity, solubility, hemolysis probability, net charge (pH 7), and molecular weight.
why is predicted binding affinity, solubility, hemolysis probability, net charge (pH 7), and molecular weight important metrics and what do they acctually mean?
binding affinity A stronger binder usually means the peptide is more likely to stay attached long enough to have an effect. If binding is very weak, the peptide may just drift away and not do much.
solubility This is very important because most biological experiments happen in aqueous environments. If a peptide is poorly soluble, it may:
Hemolysis means breaking open red blood cells. So hemolysis probability is a prediction of whether the peptide might damage cell membranes strongly enough to lyse red blood cells. This matters because a peptide might bind a target but still be too toxic or membrane-disruptive to be a good therapeutic lead. low hemolysis probability = safer-looking peptide, high hemolysis probability = warning sign for toxicity
Net charge at pH 7 This is the peptide’s overall electrical charge around neutral pH. Some amino acids are positively charged, some negatively charged, and some neutral. When you add them up, you get the peptide’s net charge. This matters because charge affects: a) solubility b) how the peptide interacts with proteins c) how it interacts with membranes d) whether it tends to stick nonspecifically to other molecules
Molecular weight how heavy the peptide is,for a peptide, this is closely related to how many amino acids it contains and what those amino acids are.
Why all of these matter?:
- able to bind reasonably well
- soluble enough to test
- not obviously toxic
- have a reasonable charge
- have a manageable size
PeptiVerse results
| Peptide ID | Sequence | Binding affinity (pKd/pKi) | Solubility | Hemolysis probability | Net charge (pH 7) | Molecular weight (Da) |
|---|---|---|---|---|---|---|
| PepMLM-1 | WSDDAVVDAVHA | 5.632 | 1.000 | 0.065 | -3.15 | 1284.3 |
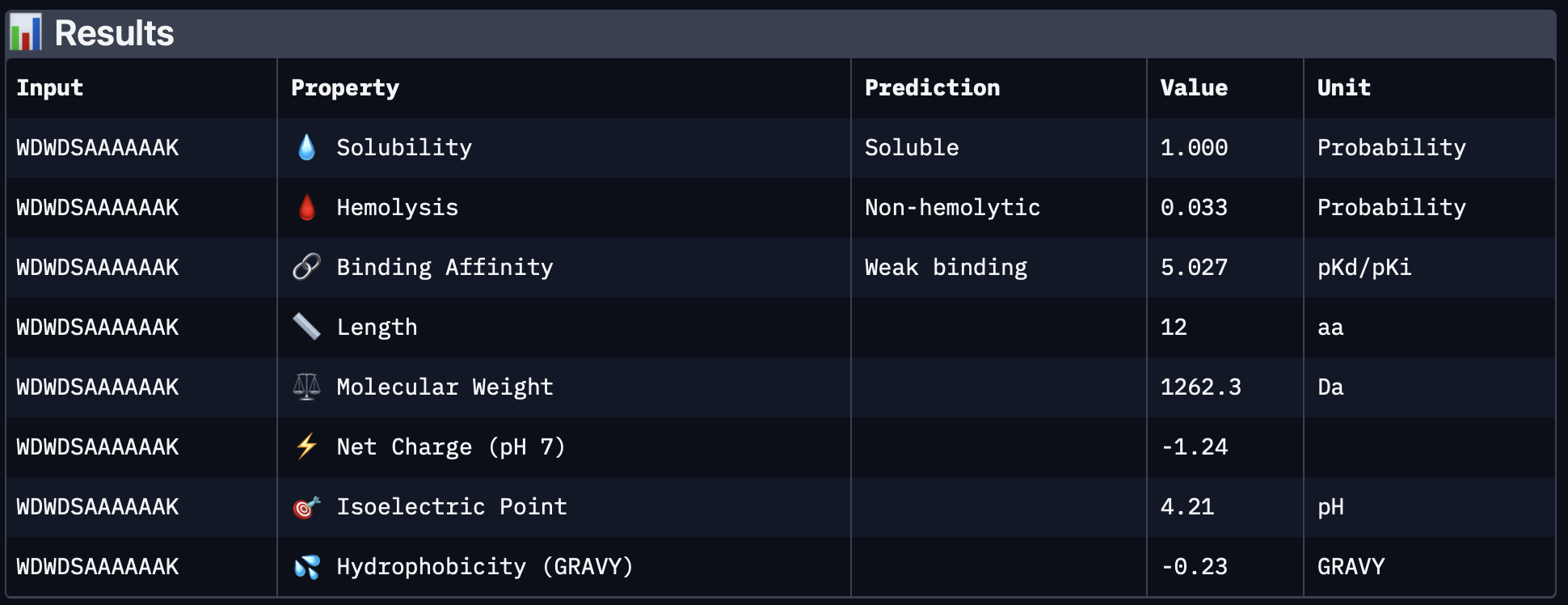
| PepMLM-2 | WDWDSAAAAAAK | 5.027 | 1.000 | 0.033 | -1.24 | 1262.3 |
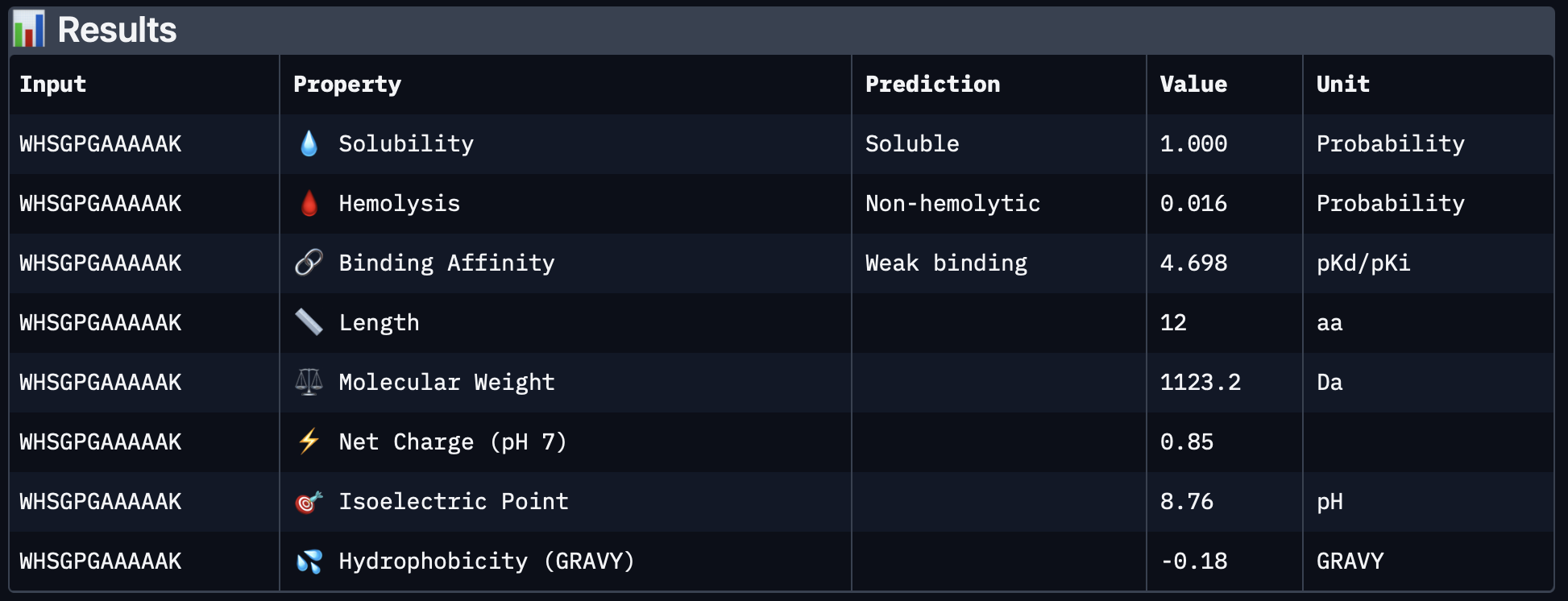
| PepMLM-3 | WHSGPGAAAAAK | 4.698 | 1.000 | 0.016 | 0.85 | 1123.2 |
| PepMLM-4 | HHSGSGGAAGKH | 4.201 | 1.000 | 0.016 | 1.02 | 1102.1 |
Individual PeptiVerse outputs
PepMLM-1
PeptiVerse predicted that PepMLM-1 is fully soluble and non-hemolytic, but it had the highest hemolysis probability of the four peptides and was also the most negatively charged. It showed the highest predicted binding affinity in PeptiVerse, although it was still classified as weak binding overall.
PepMLM-2
PepMLM-2 was also predicted to be fully soluble and non-hemolytic. Compared with PepMLM-1 it had a slightly lower predicted binding affinity, lower hemolysis probability, and a less negative charge. This makes it somewhat more balanced than PepMLM-1 from a developability perspective.
PepMLM-3
PepMLM-3 had full predicted solubility, very low hemolysis probability, and a slightly positive net charge, which could be favorable for interaction with exposed protein surfaces. Its predicted binding affinity was lower than PepMLM-1 and PepMLM-2 in PeptiVerse, but it still looked attractive overall because of its better safety/developability profile.
PepMLM-4
PepMLM-4 had the lowest predicted binding affinity of the four peptides, but it was also fully soluble, very low in hemolysis probability, and the lightest peptide by molecular weight. It looked like a safe and soluble candidate, but less promising from a binding perspective.
Interpretation
A clear pattern from PeptiVerse is that all four peptides were predicted to be soluble, and all four had low hemolysis probabilities, so none of them looked immediately problematic from a basic safety/solubility perspective. The differences were mainly in relative binding affinity, charge, and molecular weight.
If I rank the peptides by PeptiVerse predicted binding affinity alone, the order is:
- PepMLM-1 — 5.632
- PepMLM-2 — 5.027
- PepMLM-3 — 4.698
- PepMLM-4 — 4.201
However, PeptiVerse and AlphaFold did not rank the peptides in the same way. In Part 2, PepMLM-3 gave the best AlphaFold complex result with the highest ipTM and the most convincing surface-bound pose, while PepMLM-1 only showed a weaker and more ambiguous interface. This means that the peptide with the highest predicted affinity in PeptiVerse was not the same peptide that gave the strongest structural complex prediction.
Final decision
Based on the combined results from PepMLM, AlphaFold, and PeptiVerse, I would advance PepMLM-3 (WHSGPGAAAAAK).
My reasoning is:
- it had the strongest AlphaFold result from Part 2,
- it remained fully soluble in PeptiVerse,
- it had a very low hemolysis probability (0.016),
- it had a relatively low molecular weight (1123.2 Da),
- and its slightly positive net charge (0.85) may be more favorable than the strongly negative charge of PepMLM-1.
So even though PepMLM-1 had the highest PeptiVerse binding score, PepMLM-3 appears to offer the best overall balance between predicted binding geometry and peptide properties. For that reason, PepMLM-3 would be my lead candidate for follow-up testing.
Part 4: Generate Optimized Peptides with moPPIt
For the final design step, I used moPPIt to generate peptides that were explicitly guided toward a selected region of the target protein, rather than only sampling general binders from sequence context as in PepMLM. I used the A4V mutant SOD1 sequence as the target and chose a motif around the N-terminal region (residues 1–8) in order to bias the model toward the area surrounding the disease-associated A4V mutation.
Input settings used
- Target protein: A4V mutant SOD1
- Targeted motif / residue region: 1–8
- Peptide length: 12 aa
- Guidance enabled: Affinity + Motif
- Number of samples requested: 3
moPPIt-generated peptides
| Peptide ID | Sequence | Targeted motif | Notes |
|---|---|---|---|
| moPPIt-1 | RSKTKLCGEKQV | 1–8 | Positively charged / mixed-polar sequence, quite different from the PepMLM peptides |
| moPPIt-2 | GCGDLFTYYYYG | 1–8 | More aromatic and hydrophobic, with several tyrosines |
| moPPIt-3 | Not completed | 1–8 | Colab GPU limit interrupted the run before the third peptide finished |
Interpretation
Compared with the PepMLM peptides, the moPPIt peptides look quite different in sequence composition. The earlier PepMLM candidates were enriched in small and simple residues such as A, G, S, and D, while the moPPIt peptides contain more clearly designed features, including charged residues in moPPIt-1 and aromatic residues in moPPIt-2. This makes sense, because moPPIt was run with an explicit motif-targeting objective rather than only sequence-conditioned peptide generation.
The most important difference is conceptual:
- PepMLM generated peptides that behaved mostly like general surface binders
- moPPIt was used here to bias peptide design toward the N-terminal A4V-adjacent region
So even though I have not yet structurally validated these new peptides, they are more directly aligned with the biological goal of targeting the mutation-associated region of SOD1.
Limitation of this run
The moPPIt run was interrupted by Colab GPU usage limits before the third sample completed, so I only obtained two finished peptides in this session. I therefore treat this as a partial design round rather than a complete final screen.
Comparison to PepMLM peptides
In Parts 1–3, the best overall PepMLM candidate was PepMLM-3 (WHSGPGAAAAAK), because it showed the strongest AlphaFold interface while also maintaining good PeptiVerse properties. However, those PepMLM peptides did not clearly dock at the extreme A4V/N-terminal site. The moPPIt design step was therefore useful because it shifted the strategy from simply finding plausible binders to generating peptides that are more likely to engage the chosen mutation-adjacent motif.
How I would evaluate the moPPIt peptides before advancing them
Before considering these peptides as therapeutic leads, I would next:
- predict their complexes with AlphaFold to check whether they actually bind near residues 1–8 of SOD1,
- evaluate their binding affinity, solubility, hemolysis, charge, and molecular weight in PeptiVerse,
- compare them directly against PepMLM-3, which was the strongest candidate from the previous steps,
- test whether they show better site specificity for the mutant N-terminal region rather than general surface sticking.
After computational screening, the next stage would be experimental validation, including peptide synthesis, in vitro binding assays, comparison between wild-type and A4V mutant SOD1, and functional assays related to aggregation or stabilization.
Conclusion
Even with only two completed outputs, moPPIt was useful because it produced a new set of peptides specifically optimized toward the A4V-adjacent N-terminal motif of SOD1. The two peptides generated in this run were:
RSKTKLCGEKQVGCGDLFTYYYYG
These would be the next candidates I would test computationally against PepMLM-3 to see whether motif-guided design can produce a more mutation-focused binder than the original PepMLM approach.
Part C — Mutation Analysis with ESM
To explore how mutations may affect the stability and plausibility of my protein sequence, I used the ESM protein language model to perform a single-site mutational scan across the entire sequence. This analysis calculates a log-likelihood ratio (LLR) score for substituting each amino acid at each position in the protein.
The LLR score estimates how likely a mutation is according to the learned statistical patterns of natural proteins.
- Positive LLR values indicate that the mutation is plausible or tolerated.
- Negative LLR values suggest that the mutation may destabilize the protein or be less compatible with natural sequence patterns.
This approach allows us to identify positions that are mutation-tolerant and potentially useful for protein design.
Global Mutation Landscape
The heatmap below shows the predicted effects of all possible amino acid substitutions across the protein sequence.
- X-axis: position in the protein sequence
- Y-axis: substituted amino acid
- Color: predicted mutation effect (LLR score)
Brighter yellow regions represent mutations predicted to be more favorable, while darker blue/purple regions represent unfavorable substitutions.
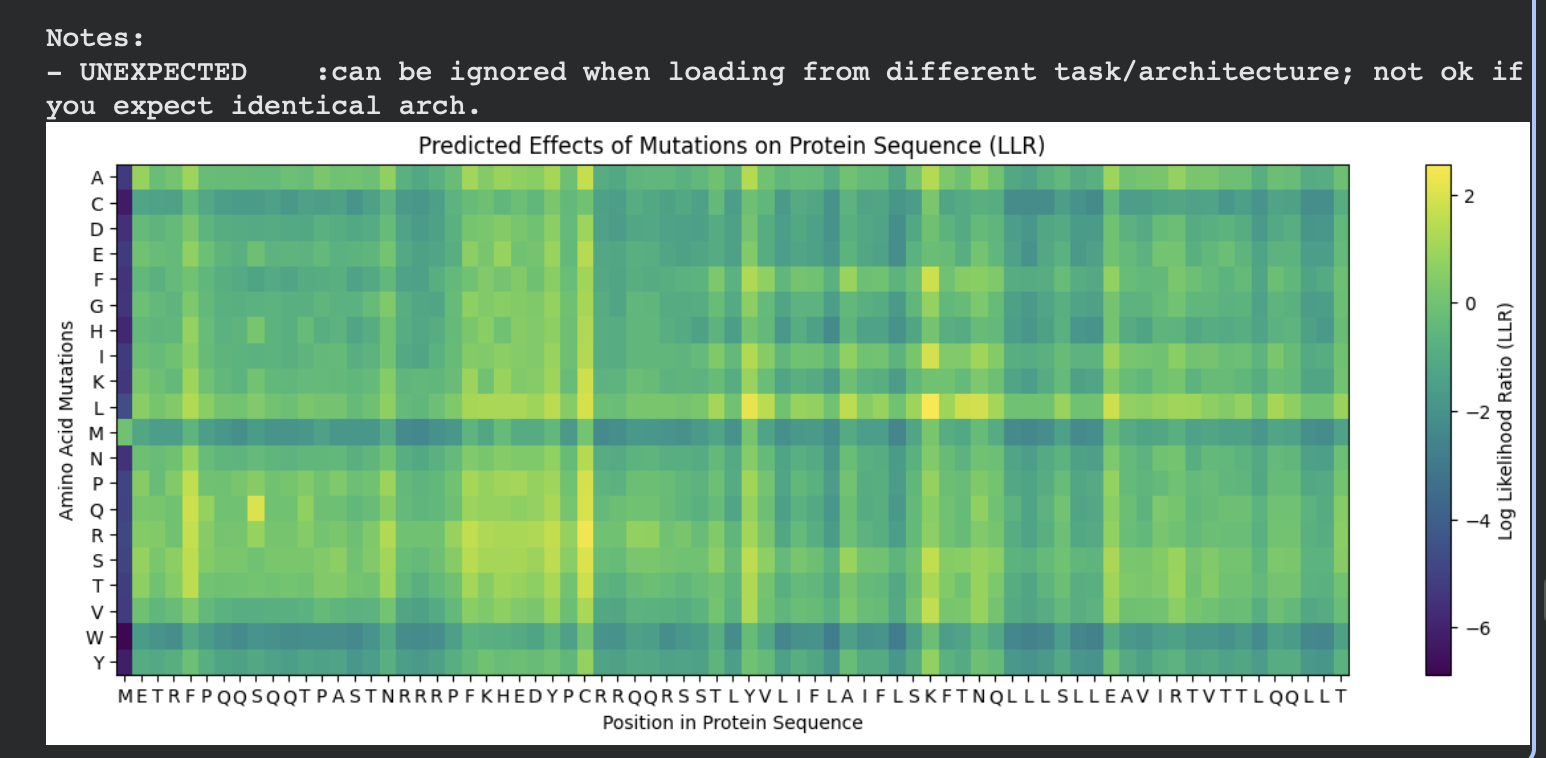
From this visualization we can see that:
- Some positions are highly constrained (mostly negative scores), suggesting that mutations there would likely disrupt the protein.
- Other positions show several neutral or positive substitutions, indicating that these sites may tolerate mutation.
- A few positions show strong positive signals for specific amino acids, suggesting potential candidates for protein engineering.
Detailed View of Mutation Effects
The following heatmap provides another view of the mutation landscape, confirming the overall pattern of mutation tolerance across the sequence.
In both visualizations, several residues show clusters of positive LLR values for specific substitutions, suggesting that these positions may accommodate changes without disrupting the protein fold.
Protein Representation Learned by ESM
The ESM model also generates a high-dimensional representation (embedding) of the protein sequence. These embeddings capture patterns such as evolutionary constraints and structural signals.
The visualization below shows the representation dimensions learned by the model across the sequence.
Although the representation values appear relatively uniform across most positions, subtle variations encode contextual information about each residue within the protein sequence.
Candidate Mutations
Based on the LLR mutation analysis, I selected several candidate mutations with relatively favorable scores. These mutations occur at positions where the model predicts that substitutions may be tolerated.
Example candidate mutations include:
- S9Q
- C29R
- Y39L
- K50L
- N53L
These mutations were chosen because they showed relatively high LLR scores compared to other substitutions at the same positions, suggesting that the protein language model considers them compatible with natural protein sequence patterns.
Residue 39 appeared particularly permissive to mutation, with multiple substitutions showing similar scores. This suggests that this region may tolerate amino-acid changes without strongly disrupting the protein structure.
Interpretation
The ESM mutational scan provides a data-driven way to identify potentially tolerable mutations in a protein sequence. While these predictions do not guarantee functional improvements, they highlight mutations that are consistent with evolutionary patterns learned by the model.
In protein engineering workflows, such predictions can be used to:
- prioritize mutations for experimental testing
- explore sequence space while maintaining structural plausibility
- identify flexible regions of the protein
Overall, the analysis suggests that several positions in this protein may tolerate mutation and could serve as starting points for further design or optimization.