I’m genuinely interested in neuroscience, research, and understanding how diseases work at a deeper level. I enjoy bench work and the process of experimenting, failing, and trying again.That’s where I feel most connected to science, not just reading about it.
I also love solo travel and discovering new places. Being on my own in different environments has shaped how I think and how I see the world, especially when it comes to people, systems, and inequalities. It’s made me more independent and more aware that science never exists in isolation.
My long-term goal is to contribute something meaningful through science, whether that’s helping develop better ways to understand or treat disease, improving how research is done, or being part of work that actually helps people. I know big impact takes time, learning, and persistence, and I’m committed to that journey.
Question 1: I propose a digital, governance-aware health data platform designed to support a population-level understanding of cancer and tumor prevalence in Iraq. At present, most medical records in Iraq are paper-based and fragmented across hospitals or retained by patients, making them vulnerable to loss and preventing the creation of a reliable national picture of cancer types, trends, and possible contributing factors. As a result, medical research, evidence-based policymaking, and long-term public health planning are severely limited. This proposed tool would not collect full patient records, enable diagnosis, or identify individuals. Instead, it would focus on aggregated, de-identified clinical and contextual data that can be used to understand broader cancer patterns while respecting patient privacy, consent, and cultural sensitivities. The primary goal of this platform is to address a critical infrastructure gap in Iraq’s health system by enabling ethical research and informed decision-making, while explicitly avoiding surveillance, stigmatization, or misuse of sensitive medical information. While neurological and psychological conditions represent equally serious challenges in Iraq, they are intentionally excluded from the initial scope of this design due to heightened ethical, privacy, and stigma-related risks.
Homework Questions from Professor Jacobson: According to the Lecture 2 slides, the intrinsic error rate of biological DNA polymerase is approximately 1 error per 10⁶ base pairs. The slides also indicate that the human genome is approximately 3.2 × 10⁹ base pairs in length. At this error rate, replication of the human genome would result in thousands of errors per replication cycle if no additional correction mechanisms existed. The slides explain that biology addresses this discrepancy through error-correcting mechanisms, including proofreading activity associated with DNA polymerase and post-replication mismatch repair systems, such as the MutS pathway. Together, these mechanisms reduce the effective mutation rate and allow large genomes to be stably maintained.
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications. Article: “Automation of protein crystallization scaleup via Opentrons-2 liquid handling”
Jacob B. DeRoo, Alec A. Jones, Caroline K. Slaughter, Tim W. Ahr, Sam M. Stroup, Grace B. Thompson, Christopher D. Snow, SLAS Technology, Volume 32, 2025, 100268, ISSN 2472-6303,
https://doi.org/10.1016/j.slast.2025.100268
General overview: Protein crystallization is a complex and time-consuming process that is essential for determining protein structures in structural biology. Producing well-formed protein crystals requires careful optimization of multiple conditions, including protein concentration, precipitant composition, and mixing accuracy. Because these parameters cannot be predicted in advance, crystallization is largely a trial-and-error process that demands repeated setup of crystallization plates. Traditionally, this process is performed manually, making it labor-intensive and susceptible to human error and variability. In addition, viscous protein solutions are difficult to handle consistently, which further complicates crystallization experiments.
Part A: Conceptual Questions For an average amino acid, the molecular weight is about 100 Daltons, which is equivalent to 100 g/mol. If I assume meat is about 20% protein, then 500 g of meat contains roughly 100 g of protein. The relationship is: number of moles = mass (g) / molar mass (g/mol) So, 100 g ÷ 100 g/mol ≈ 1 mole of amino acids. One mole corresponds to approximately 6×1023molecules. Therefore, consuming 500 g of meat corresponds to on the order of 1023amino acid molecules.
Image source: Genetic Lifehacks. https://www.geneticlifehacks.com/sod1-gene-polymorphisms/
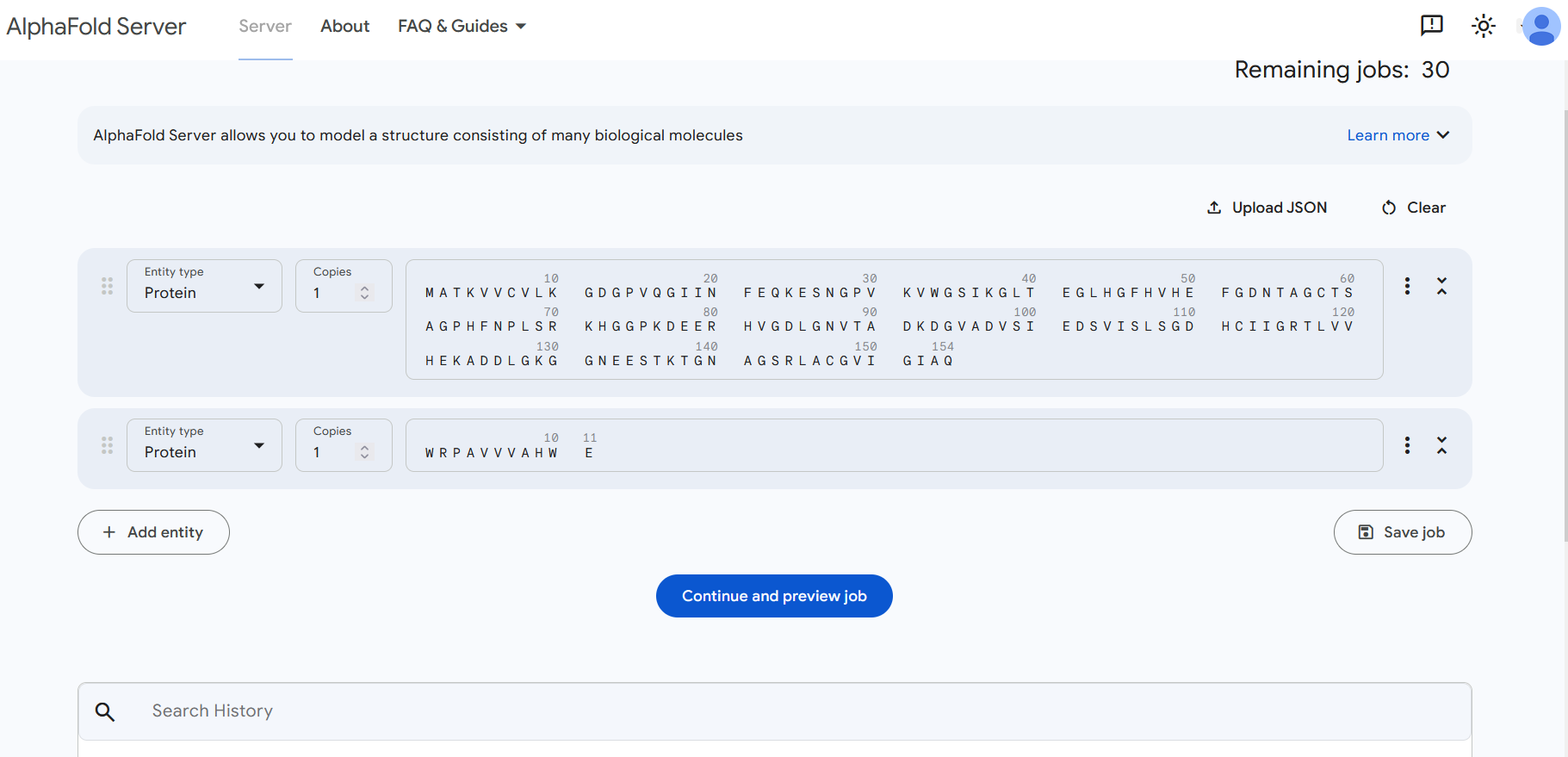
Part 1: SOD1 Binder Peptide Design “MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ”
This is the SOD1 original sequence
The mutation A4V means:
Alanine (A) at position 4 → Valine (V)
But UniProt includes the starting M, so the alanine that changes is the 5th residue in the sequence.
1 M 2 A 3 T 4 K 5 A ← this is the one that changes 6 V We change from this MATKAV to this MATKVV
DNA Assembly Phusion High-Fidelity PCR Master Mix contains DNA polymerase, dNTPs, buffer, and Mg²⁺. The polymerase has proofreading activity (3′→5′ exonuclease), which reduces errors during DNA amplification. The dNTPs act as building blocks, while the buffer and Mg²⁺ maintain optimal conditions for enzyme activity. This is important in cloning because even small mutations can affect protein function. https://www.neb.com/en/products/e0553-phusion-high-fidelity-pcr-kit?srsltid=AfmBOooI-JWWTJ01XuZL3foWSnvq5kqQol7r8q61xRo95a6S7amAGeiH
Part 1: Intracellular Artificial Neural Networks Q.1. Traditional genetic circuits mostly behave like Boolean logic, meaning everything is either ON or OFF. That works for simple designs like AND or OR gates, but it doesn’t really match what actually happens inside cells.
In real biology, nothing is strictly binary. Gene expression can be low, medium, or high, and signals are noisy and constantly changing. So forcing everything into ON/OFF makes the system too limited.
Part A: General and Lecturer-Specific Questions Q.1. One of the main advantages of cell-free protein synthesis over traditional in vivo methods is the flexibility and level of control over the system. In normal in vivo systems, cells are still trying to survive, grow, divide, regulate metabolism, and maintain their own functions. Because of that, researchers are working with the cell’s own priorities and limitations.
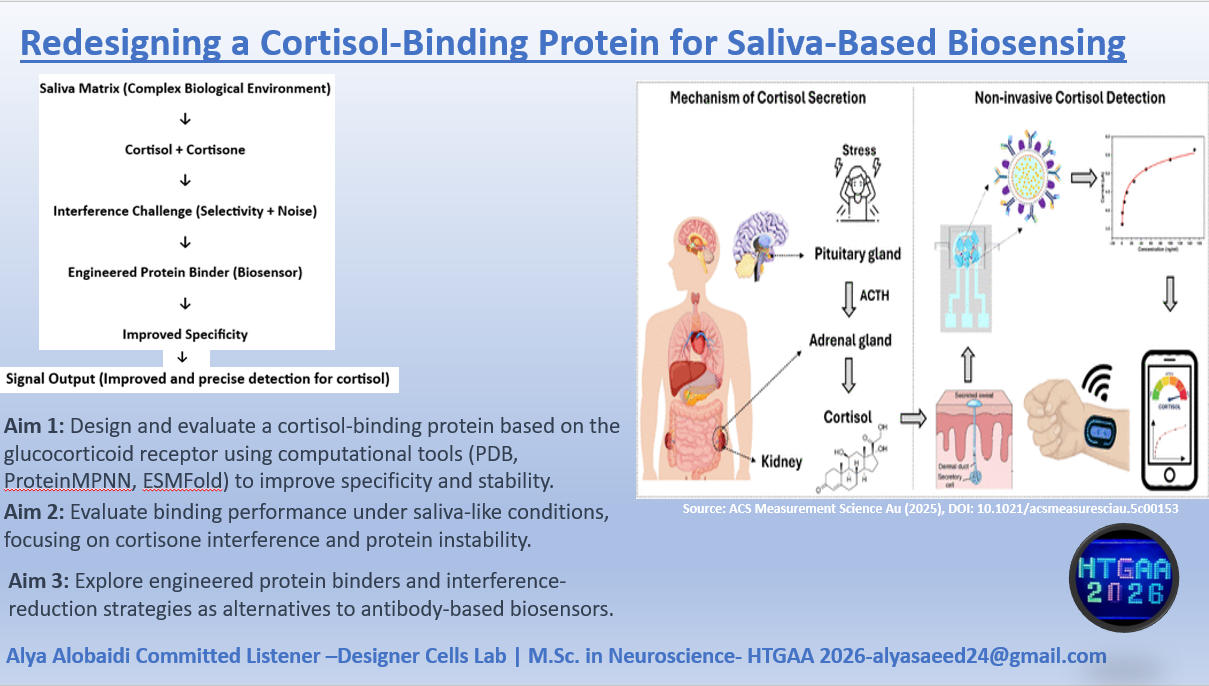
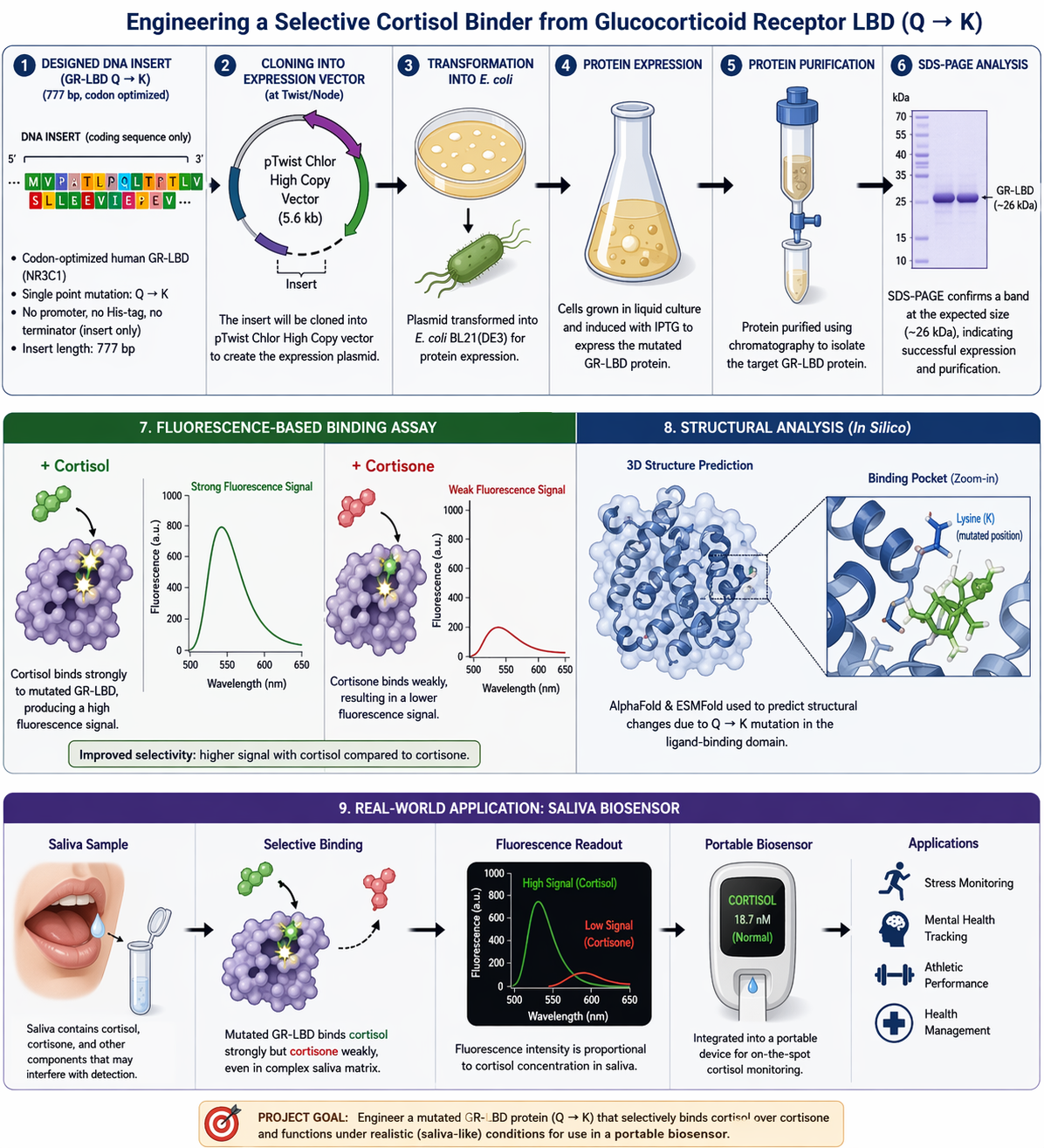
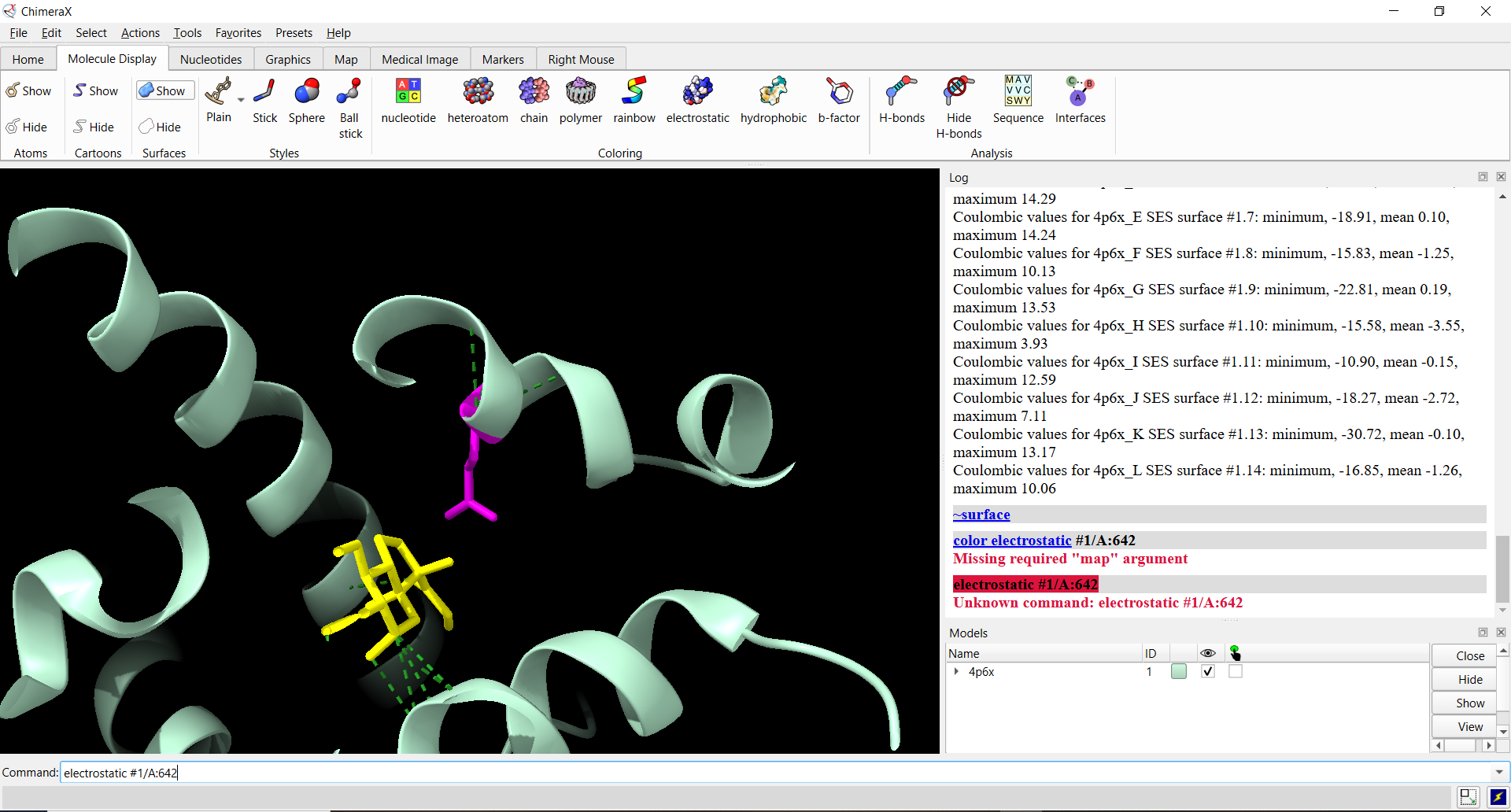
Homework: Final Project I would mainly want to see whether the redesigned GR-LBD protein is successfully produced and whether introducing the Q642K mutation improves cortisol selectivity compared with cortisone. Since the DNA construct was designed and ordered through Twist Bioscience, I would first verify the received construct sequence before moving into expression studies.
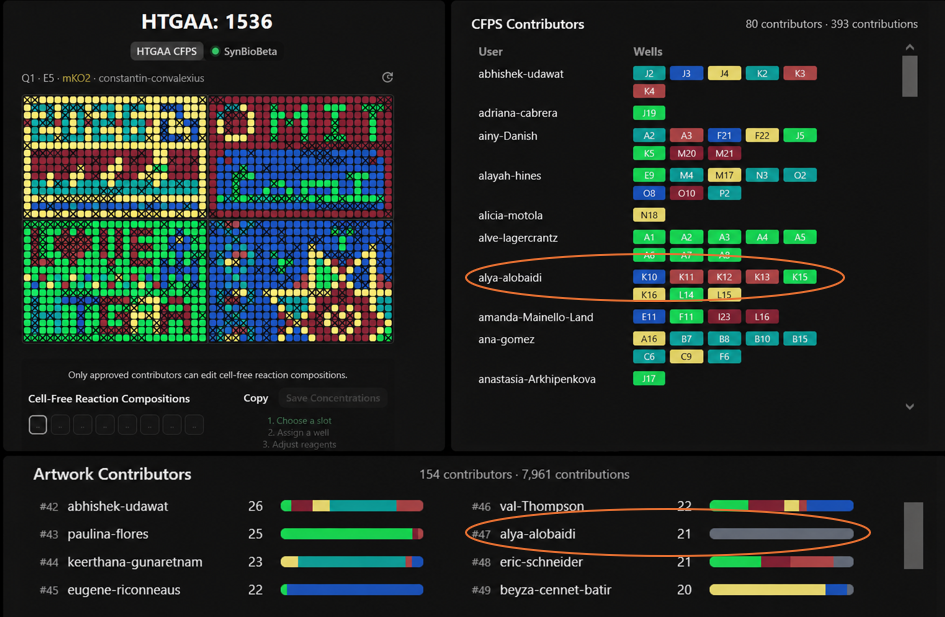
For the collaborative bioart project, I contributed to several wells including K10, K11, K12, K13, K15, K16, L14, and L15. Most of my contributions appeared in the right-side region of the artwork.
What I liked most about this project was seeing how many people from different backgrounds worked together to create one large scientific artwork. It honestly felt chaotic at first, but super cool once the final pattern started forming. I also liked how science and art were combined together in a very interactive way. One thing that could make the project even more fun next year is adding a little more guidance or visualization for first-time participants to better follow how the final image develops collectively over time. I really enjoyed the project overall, but at the beginning it took me some time to fully understand how everything was coming together. I wish I have participated more and on time!
I propose a digital, governance-aware health data platform designed to support a population-level understanding of cancer and tumor prevalence in Iraq.
At present, most medical records in Iraq are paper-based and fragmented across hospitals or retained by patients, making them vulnerable to loss and preventing the creation of a reliable national picture of cancer types, trends, and possible contributing factors. As a result, medical research, evidence-based policymaking, and long-term public health planning are severely limited.
This proposed tool would not collect full patient records, enable diagnosis, or identify individuals. Instead, it would focus on aggregated, de-identified clinical and contextual data that can be used to understand broader cancer patterns while respecting patient privacy, consent, and cultural sensitivities.
The primary goal of this platform is to address a critical infrastructure gap in Iraq’s health system by enabling ethical research and informed decision-making, while explicitly avoiding surveillance, stigmatization, or misuse of sensitive medical information.
While neurological and psychological conditions represent equally serious challenges in Iraq, they are intentionally excluded from the initial scope of this design due to heightened ethical, privacy, and stigma-related risks.
Question 2:
Goal 1: Protect patient dignity, privacy, and trust
The primary governance goal of this project is to protect patient dignity and privacy in a context where cancer remains highly stigmatized and medical ethics are inconsistently enforced. Given the absence of robust digital infrastructure and uneven adherence to confidentiality standards, there is a significant risk that sensitive health information could be misused, disclosed without consent, or lead to social harm.
This goal emphasizes minimizing data collection, enforcing de-identification by design, and ensuring that patients and communities can trust that participation will not expose them to discrimination, blame, or loss of dignity.
Goal 2: Enable ethical, feasible research under limited resources
A second key goal is to enable responsible cancer research in Iraq without creating governance barriers that make research impossible in practice. While strong safeguards are necessary, overly restrictive rules, lack of funding, limited governmental support, and dependence on expensive foreign technologies could unintentionally suppress research and innovation.
This goal therefore prioritizes governance structures that are realistic for a low-resource setting, support researcher autonomy within ethical boundaries, and allow gradual capacity building rather than imposing idealized systems that cannot be sustained locally.
Question 3:
Governance Action 1: Privacy-by-design transition from paper to aggregated digital reporting
(Technical + institutional action | Led by academic researchers and hospitals)
Purpose: Currently, cancer-related data in Iraq is largely paper-based, fragmented, and vulnerable to loss or unauthorized access. This governance action proposes a transition from individual paper records to aggregated, de-identified digital reporting, enabling population-level insight while minimizing privacy risks.
Design
• Hospitals and clinics report summary-level cancer data (e.g., tumor type, age range, region, and high-level risk factors where available).
• No personal identifiers such as names, national IDs, or addresses are collected.
• Data entry tools are designed to be low-cost, simple, and compatible with limited digital infrastructure.
• Training emphasizes what data should not be collected, reinforcing privacy-by-design principles.
Assumptions
• Aggregated data is sufficient to identify national cancer trends.
• Healthcare staff can be trained to follow simplified digital reporting protocols.
Risks of Failure & “Success”
• Failure risk: Limited technical capacity or staff resistance could result in incomplete or inconsistent data reporting.
• Success risk: Even aggregated data could be misinterpreted or misused if governance oversight is weak.
Governance Action 2: Strengthened ethics enforcement and hospital-level conduct standards
(Institutional rule | Led by hospitals, universities, and health authorities)
Purpose: Although ethical guidelines exist, they are not consistently enforced. This action aims to strengthen ethical conduct within hospitals, particularly around patient privacy, infection control, and respect for patient dignity.
Design
• Establish clear, enforceable standards for:
o patient confidentiality
o limits on hospital visitors for immuno-compromised cancer patients
o basic sterilization and infection-control practices
• Ethics training is integrated into routine hospital operations rather than optional workshops.
• Accountability mechanisms focus on institutional responsibility rather than individual blame.
Assumptions
• Institutional enforcement is more effective than relying solely on individual compliance.
• Hospitals have the authority to implement and monitor conduct standards.
Risks of Failure & “Success”
• Failure risk: Standards may exist only on paper without consistent enforcement.
• Success risk: Strict enforcement could be perceived as culturally insensitive if not accompanied by clear communication.
Governance Action 3: Community-centered cancer education and engagement strategy
(Social and educational action | Led by hospitals, NGOs, and public health educators)
Purpose: In many rural and underserved areas, cancer and tumors are misunderstood, sometimes viewed as contagious or caused by moral failure. This action treats education as a governance tool to reduce stigma, misinformation, and resistance to ethical data sharing.
Design
• Community education initiatives explaining:
o what cancer and tumors are and are not
o common risk factors (genetic factors, environmental pollution, viral causes, lifestyle and diet)
o The importance of limiting hospital visits to protect patient immunity
• Education delivered by trusted local healthcare workers and community figures.
• No requirement for digital literacy or individual data submission.
Assumptions
• Trust in local messengers increases cooperation and understanding.
• Education can reduce stigma and harmful practices.
Risks of Failure & “Success”
• Failure risk: Misinformation may spread faster than educational efforts.
• Success risk: Communities may expect direct medical treatment or financial support beyond the scope of the project.
Question 4:
Option 1: Privacy-by-design aggregated digital reporting
Option 2: Ethics enforcement and hospital-level conduct standards
Option 3: Community-centered cancer education and engagement
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
n/a
• By helping respond
1
2
3
Foster Lab Safety
• By preventing incident
2
1
n/a
• By helping respond
2
1
n/a
Protect the environment
• By preventing incidents
n/a
n/a
n/a
• By helping respond
n/a
n/a
n/a
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
2
3
1
• Not impede research
1
2
1
• Promote constructive applications
1
2
1
Question 5:
I prioritize a sequenced combination of governance actions, rather than treating all options as equally urgent.
Governance Action 1 (privacy-by-design aggregated digital reporting) is the primary priority. It directly addresses the central problem identified in this proposal—the absence of reliable, population-level cancer data due to fragmented, paper-based records—while also providing the strongest protection for patient privacy and dignity. Without this ethical data foundation, other governance efforts would lack a practical and legitimate basis.
Governance Action 3 (community-centered cancer education and engagement) is prioritized as a supporting and parallel action. Public understanding and trust are essential for any data-related initiative to be ethically acceptable and practically feasible in the Iraqi context, particularly in rural and underserved communities. Education helps reduce stigma, misinformation, and harmful practices, and supports voluntary participation without coercion.
Governance Action 2 (ethics enforcement and hospital-level conduct standards) is recognized as critically important but is treated as a longer-term priority. Although it has strong potential to improve patient safety and ethical compliance, it depends on sustained institutional capacity, enforcement mechanisms, and governmental support, which remain uncertain in the short term.
This prioritization reflects key trade-offs between ethical protection, feasibility under limited resources, and institutional readiness. It also acknowledges uncertainty regarding long-term enforcement and funding. These recommendations are intended for local hospitals, universities, and public health institutions in Iraq, as well as international academic and public-health collaborators supporting ethical research and capacity building.
Reflecting on this week’s class and assignment, one thing that really stood out to me was how easily health data initiatives can cause harm—even when the intentions are good—if governance is treated as something secondary rather than built in from the start. Before this week, I mostly thought of ethical risk as something tied to deliberate misuse. Working through this assignment made it clear to me that harm can also come from structural issues, such as paper-based systems, unclear responsibility, and weak enforcement, even when no one is acting maliciously.
I was also struck by the tension between protecting patient dignity and making research actually possible in low-resource settings. While strong safeguards are clearly necessary, overly idealized governance frameworks can unintentionally exclude researchers and communities that don’t have the funding or infrastructure to meet those standards. This reinforced for me the importance of privacy-by-design approaches, community engagement, and realistic, step-by-step governance strategies that build trust over time, rather than relying only on top-down rules.
AI Use Statement:
I used ChatGPT as a support tool to help clarify assignment instructions, organize my thinking, and refine the wording of my responses. All ideas, decisions, and final interpretations reflect my own understanding.
Week 2 HW: DNA read, write, and edit
Homework Questions from Professor Jacobson:
According to the Lecture 2 slides, the intrinsic error rate of biological DNA polymerase is approximately 1 error per 10⁶ base pairs. The slides also indicate that the human genome is approximately 3.2 × 10⁹ base pairs in length. At this error rate, replication of the human genome would result in thousands of errors per replication cycle if no additional correction mechanisms existed.
The slides explain that biology addresses this discrepancy through error-correcting mechanisms, including proofreading activity associated with DNA polymerase and post-replication mismatch repair systems, such as the MutS pathway. Together, these mechanisms reduce the effective mutation rate and allow large genomes to be stably maintained.
The Lecture 2 slides indicate that an average human protein is approximately 1036 base pairs in length. Because DNA consists of four possible nucleotides, the total number of possible nucleotide sequences of this length is 4¹⁰³⁶, which follows directly from basic combinatorics (four choices at each position).
The slides further show that, in practice, only a small subset of these sequences are usable. Constraints illustrated in the slides include GC content effects, secondary structure formation, and sequence-dependent synthesis limitations, all of which can interfere with DNA synthesis, transcription, or downstream use. As a result, most theoretically possible sequences are not viable in biological or synthetic contexts.
Homework Questions from Dr. LeProust:
According to the Lecture 2 slides, the most commonly used method for oligonucleotide synthesis is solid-phase phosphoramidite chemical synthesis. The slides describe this as a stepwise process in which nucleotides are added sequentially to a growing DNA strand attached to a solid support through repeated chemical cycles.
The historical overview in the slides also notes that the phosphoramidite method, developed by Caruthers (1981), remains the foundation of modern DNA synthesis technologies.
The Lecture 2 slides explain that direct oligonucleotide synthesis becomes difficult beyond approximately 200 nucleotides because errors accumulate with each synthesis cycle. Each nucleotide addition has a finite probability of failure, and as the number of synthesis steps increases, the yield of full-length, correct oligos decreases sharply.
The slides also show that longer oligos suffer from truncation products, base incorporation errors, and sequence-dependent effects, including high GC content and secondary structure formation. These factors reduce both yield and purity, making long oligos impractical to synthesize reliably in a single continuous process.
As shown in the Lecture 2 slides, a 2000 base-pair gene cannot be synthesized directly because the cumulative error rate of chemical synthesis over thousands of nucleotide additions would result in an extremely low fraction of correct, full-length molecules.
Instead, the slides describe classical gene synthesis, in which long genes are assembled from many shorter oligos using enzymatic methods such as PCR-based assembly and ligation. This hierarchical approach allows errors to be managed and corrected during assembly, making long gene construction feasible.
Homework Question from George Church:
Animals require a conserved set of essential amino acids that must be obtained through diet. These include histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine.
Prof. Church’s Lecture 2 slide #4 highlights the genetic code as a fixed mapping between DNA codons and amino acids. In this context, the Lysine Contingency reflects a fundamental biological constraint: lysine is essential and encoded by the genetic code, yet animals cannot synthesize it. This suggests that biological systems are historically and chemically constrained, and that changing or replacing core amino acids such as lysine would require large-scale re-engineering of both metabolism and the genetic code.
References & Use of Tools:
The primary sources for all homework answers are the:
Jacobson, J.
HTGAA Lecture 2: Gene Synthesis (MIT, 2026).
Used for questions on DNA polymerase error rates, genome scale, protein length estimates, combinatorics of DNA sequences, GC content effects, secondary structure, and biological error correction.
LeProust, E.
HTGAA Lecture 2: Oligonucleotide and Gene Synthesis (MIT, 2026).
Used for questions on phosphoramidite oligonucleotide synthesis, limitations of long oligo synthesis, error accumulation, and classical gene assembly strategies.
Church, G.
HTGAA Lecture 2: Reading & Writing Life (MIT, 2026), Slide #4.
Used for conceptual framing of the genetic code (DNA → mRNA → amino acids) and interpretation of the “Lysine Contingency.”
All interpretations derived from lecture material are explicitly tied to the concepts presented in these slides.
(Used as background confirmation that lysine is classified as an essential amino acid.)
No external references were used for the Jacobson or LeProust questions beyond the lecture slides.
An AI-based writing tool (ChatGPT) was used solely to assist with wording, organization, and clarity. All factual content was derived from the cited lectures and explicitly listed external sources.
Part 0: Basics of Gel Electrophoresis
Gel electrophoresis is a technique used to separate DNA fragments based on their size by applying an electric field. I have performed gel electrophoresis multiple times during molecular biology laboratory work in Iraq, as well as during my internship at DGIST University in South Korea in the Molecular Neuroscience Department. Through these experiences, I became familiar with the practical workflow, while the HTGAA lectures helped reinforce the underlying concepts.
In this technique, DNA migrates through an agarose gel toward the positive electrode because DNA is negatively charged. The gel is prepared using agarose, poured into a casting tray, and fitted with a comb to form wells. DNA samples are mixed with a loading dye and carefully loaded into the wells along with a DNA ladder for size reference. After applying an electric current, DNA fragments separate within the gel and are visualized using a gel imaging system to capture and document the results.
In my previous lab work, gel electrophoresis was mainly used for genotyping and for verifying DNA samples prior to sequencing, helping confirm fragment size and sample quality before downstream analysis. Revisiting this technique in the context of HTGAA helped me better articulate how gel electrophoresis fits into broader molecular biology and sequencing workflows.
In many workflows, DNA samples are PCR-amplified prior to gel electrophoresis to ensure sufficient DNA quantity and to analyze specific target fragments, particularly in genotyping and sequencing preparation.
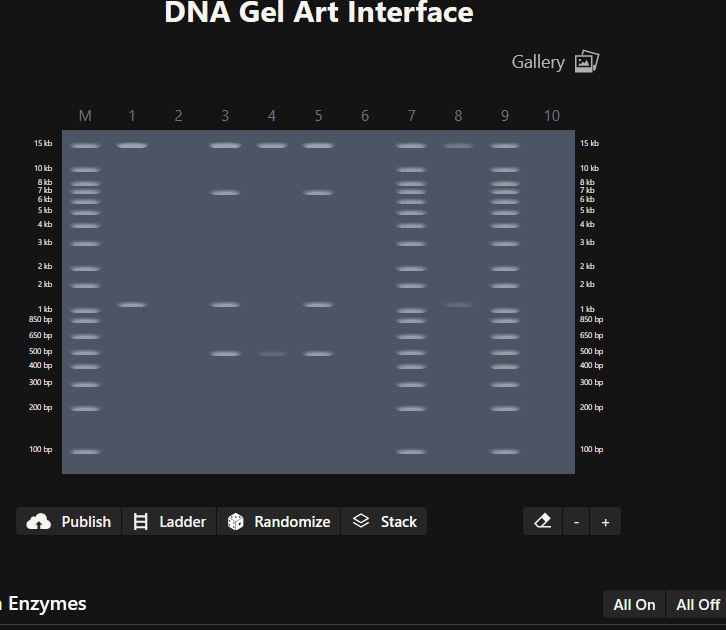
Part 1: Benchling & In-silico Gel Art
Figure 1:
Figure 2:
Part 3: DNA Design Challenge
3.1. Choose your protein
Amyloid Beta (Aβ), specifically the Aβ(1–42) peptide derived from the human amyloid precursor protein (APP).
I chose Amyloid-β because I want to understand how a normal peptide becomes harmful when it misfolds and aggregates, and how that process can disrupt brain function, memory, and overall body function in Alzheimer’s disease. Alzheimer’s affects many older adults and can gradually remove their ability to access their memories and daily independence, so I’m personally motivated to understand the molecular pathway that leads to these changes. In addition, Aβ is strongly linked to the classic Alzheimer’s pathology of extracellular plaques, which makes it a clear and widely studied starting point for connecting sequence → structure → disease mechanism.
Amyloid-beta is not encoded as an independent gene. Instead, it is generated by proteolytic cleavage of the amyloid-beta precursor protein (APP). Therefore, the APP protein sequence was used as the source sequence, with specific focus on the region that produces the Aβ peptide.
I used an online reverse translation tool (bioinformatics.org SMS2) to convert the amino acid sequence of my chosen protein (human APP; UniProt P05067) into a corresponding DNA coding sequence. Reverse translation is not unique because the genetic code is degenerate, meaning multiple codons can encode the same amino acid. Therefore, the sequence below represents one valid nucleotide sequence that could encode the same protein.
Reverse-translated DNA sequence (coding DNA; A/T/G/C only):
reverse translation of sp|P05067|A4_HUMAN Amyloid-beta precursor protein OS=Homo sapiens OX=9606 GN=APP PE=1 SV=3 to a 2310 base sequence of most likely codons.
atgctgccgggcctggcgctgctgctgctggcggcgtggaccgcgcgcgcgctggaagtg
ccgaccgatggcaacgcgggcctgctggcggaaccgcagattgcgatgttttgcggccgc
ctgaacatgcatatgaacgtgcagaacggcaaatgggatagcgatccgagcggcaccaaa
acctgcattgataccaaagaaggcattctgcagtattgccaggaagtgtatccggaactg
cagattaccaacgtggtggaagcgaaccagccggtgaccattcagaactggtgcaaacgc
ggccgcaaacagtgcaaaacccatccgcattttgtgattccgtatcgctgcctggtgggc
gaatttgtgagcgatgcgctgctggtgccggataaatgcaaatttctgcatcaggaacgc
atggatgtgtgcgaaacccatctgcattggcataccgtggcgaaagaaacctgcagcgaa
aaaagcaccaacctgcatgattatggcatgctgctgccgtgcggcattgataaatttcgc
ggcgtggaatttgtgtgctgcccgctggcggaagaaagcgataacgtggatagcgcggat
gcggaagaagatgatagcgatgtgtggtggggcggcgcggataccgattatgcggatggc
agcgaagataaagtggtggaagtggcggaagaagaagaagtggcggaagtggaagaagaa
gaagcggatgatgatgaagatgatgaagatggcgatgaagtggaagaagaagcggaagaa
ccgtatgaagaagcgaccgaacgcaccaccagcattgcgaccaccaccaccaccaccacc
gaaagcgtggaagaagtggtgcgcgaagtgtgcagcgaacaggcggaaaccggcccgtgc
cgcgcgatgattagccgctggtattttgatgtgaccgaaggcaaatgcgcgccgtttttt
tatggcggctgcggcggcaaccgcaacaactttgataccgaagaatattgcatggcggtg
tgcggcagcgcgatgagccagagcctgctgaaaaccacccaggaaccgctggcgcgcgat
ccggtgaaactgccgaccaccgcggcgagcaccccggatgcggtggataaatatctggaa
accccgggcgatgaaaacgaacatgcgcattttcagaaagcgaaagaacgcctggaagcg
aaacatcgcgaacgcatgagccaggtgatgcgcgaatgggaagaagcggaacgccaggcg
aaaaacctgccgaaagcggataaaaaagcggtgattcagcattttcaggaaaaagtggaa
agcctggaacaggaagcggcgaacgaacgccagcagctggtggaaacccatatggcgcgc
gtggaagcgatgctgaacgatcgccgccgcctggcgctggaaaactatattaccgcgctg
caggcggtgccgccgcgcccgcgccatgtgtttaacatgctgaaaaaatatgtgcgcgcg
gaacagaaagatcgccagcataccctgaaacattttgaacatgtgcgcatggtggatccg
aaaaaagcggcgcagattcgcagccaggtgatgacccatctgcgcgtgatttatgaacgc
atgaaccagagcctgagcctgctgtataacgtgccggcggtggcggaagaaattcaggat
gaagtggatgaactgctgcagaaagaacagaactatagcgatgatgtgctggcgaacatg
attagcgaaccgcgcattagctatggcaacgatgcgctgatgccgagcctgaccgaaacc
aaaaccaccgtggaactgctgccggtgaacggcgaatttagcctggatgatctgcagccg
tggcatagctttggcgcggatagcgtgccggcgaacaccgaaaacgaagtggaaccggtg
gatgcgcgcccggcggcggatcgcggcctgaccacccgcccgggcagcggcctgaccaac
attaaaaccgaagaaattagcgaagtgaaaatggatgcggaatttcgccatgatagcggc
tatgaagtgcatcatcagaaactggtgttttttgcggaagatgtgggcagcaacaaaggc
gcgattattggcctgatggtgggcggcgtggtgattgcgaccgtgattgtgattaccctg
gtgatgctgaaaaaaaaacagtataccagcattcatcatggcgtggtggaagtggatgcg
gcggtgaccccggaagaacgccatctgagcaaaatgcagcagaacggctatgaaaacccg
acctataaattttttgaacagatgcagaac
3.3 Codon optimization
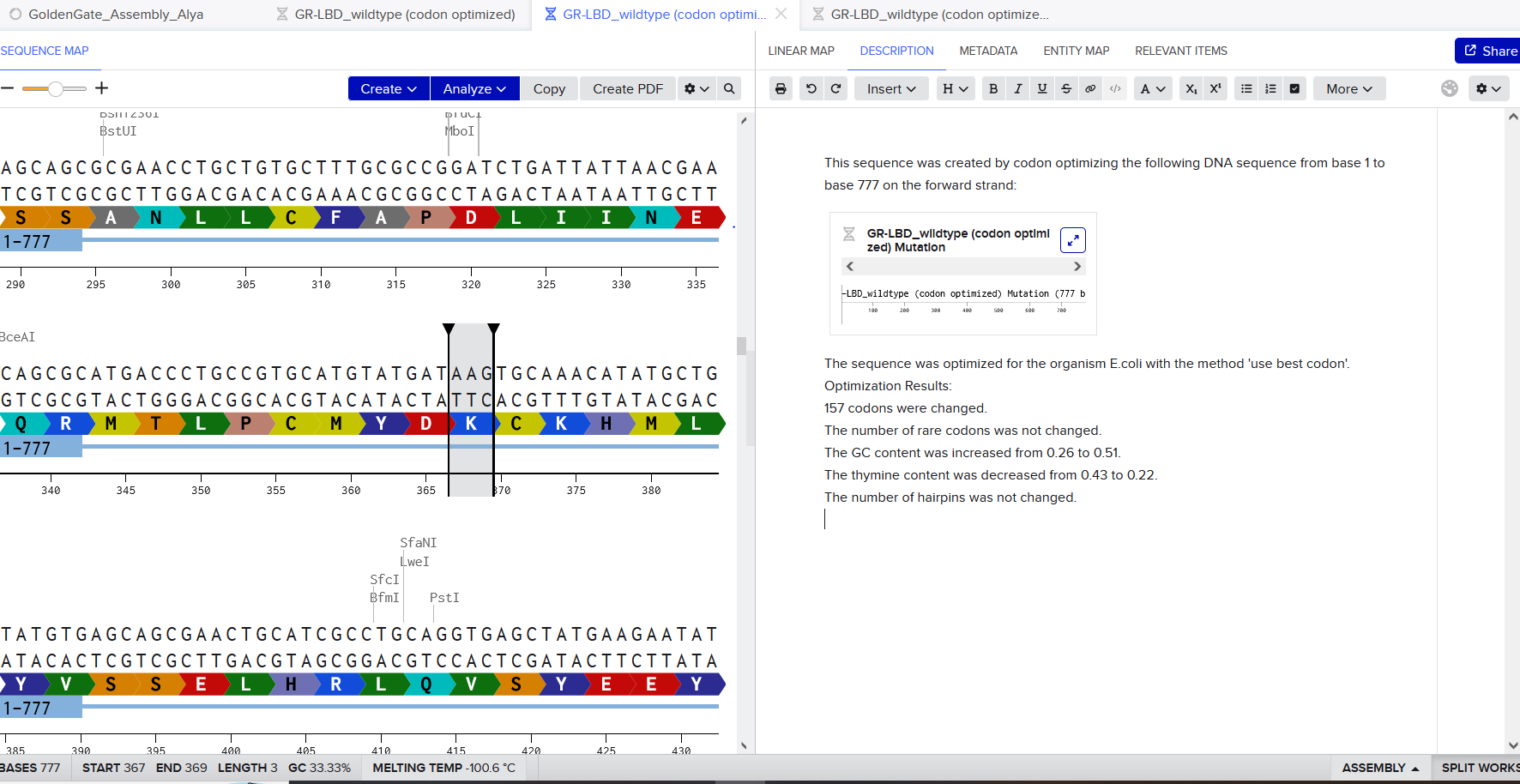
To optimize the codon usage of the nucleotide sequence obtained in the previous step, I used the VectorBuilder online codon optimization tool. The reverse-translated DNA sequence was entered into the tool and Mus musculus (mouse) was selected as the target organism.
Codon optimization is necessary because, although the genetic code is universal, different organisms preferentially use specific codons due to differences in tRNA abundance and translation efficiency. If a gene contains codons that are rarely used in the host organism, translation can be inefficient and protein expression levels may be reduced. Codon optimization replaces rare codons with synonymous codons that are more frequently used by the host, while preserving the amino acid sequence of the protein.
After optimization, improvements were observed in sequence quality metrics. The GC content increased slightly from 55.37% to 57.62%, remaining within an optimal range for stability and transcription. In addition, the Codon Adaptation Index (CAI) increased from 0.74 to 0.92, indicating a substantially improved match between the codon usage of the sequence and the translational machinery of the mouse host. These changes suggest a higher likelihood of efficient translation and protein expression in mouse-based experimental systems.
Mouse was chosen as the target organism because Alzheimer’s disease research is commonly conducted using mouse models, including APP-related transgenic and knock-in models. Optimizing the codon usage for mouse therefore increases the biological relevance of the designed sequence.
Codon-optimized DNA sequence:
Improved DNA[1]: GC=57.62%, CAI=0.92
ATGCTGCCAGGCCTGGCCCTGCTGCTGCTCGCCGCCTGGACAGCCCGGGCCCTGGAAGTGCCAACCGACGGCAACGCTGGACTGCTGGCTGAGCCTCAGATCGCCATGTTTTGTGGGCGGCTGAATATGCACATGAATGTGCAGAACGGAAAGTGGGACTCTGACCCCTCCGGCACCAAAACCTGTATCGATACAAAGGAAGGCATTCTGCAGTACTGTCAGGAGGTGTATCCCGAGCTGCAGATCACCAACGTGGTGGAGGCCAACCAGCCTGTGACCATCCAAAATTGGTGCAAAAGGGGTAGAAAGCAGTGTAAGACACACCCACACTTTGTGATCCCATATAGATGTCTGGTGGGGGAGTTCGTGTCCGACGCCCTGCTGGTGCCCGACAAGTGCAAGTTTCTGCACCAGGAGAGAATGGACGTGTGCGAGACACACCTGCACTGGCACACAGTGGCTAAGGAGACCTGTAGTGAGAAGAGCACCAACCTGCACGACTACGGGATGCTGCTGCCCTGCGGTATCGACAAGTTTAGAGGTGTGGAATTCGTGTGCTGTCCTCTGGCCGAGGAGTCCGACAATGTGGATAGCGCCGACGCCGAGGAGGACGACAGCGACGTGTGGTGGGGCGGCGCCGATACAGACTACGCCGATGGCTCCGAAGACAAGGTGGTGGAGGTGGCCGAGGAAGAGGAAGTGGCCGAGGTGGAGGAGGAGGAGGCTGACGACGACGAGGACGATGAGGACGGCGATGAGGTTGAGGAGGAGGCCGAGGAGCCTTACGAGGAAGCCACCGAGCGGACTACTTCCATTGCTACCACCACCACCACCACTACCGAGAGCGTGGAGGAGGTGGTGAGAGAGGTGTGCAGCGAGCAGGCCGAGACCGGCCCTTGTAGAGCCATGATCTCCCGGTGGTATTTCGATGTGACCGAGGGAAAGTGCGCCCCTTTCTTCTACGGAGGCTGTGGAGGCAACAGGAACAATTTTGACACTGAGGAGTACTGTATGGCCGTGTGTGGCTCCGCCATGAGCCAGTCCCTGCTGAAGACCACTCAGGAGCCCCTGGCACGGGACCCTGTGAAGCTGCCCACCACCGCCGCTAGCACACCCGACGCCGTGGACAAGTATTTGGAGACCCCAGGAGACGAGAATGAGCACGCACACTTTCAGAAGGCTAAGGAGCGCCTGGAGGCTAAGCACCGAGAAAGGATGTCTCAGGTGATGCGCGAGTGGGAGGAAGCCGAGAGGCAGGCTAAGAACCTGCCTAAAGCTGACAAAAAAGCCGTGATCCAGCATTTCCAGGAGAAGGTGGAGAGCCTGGAACAGGAGGCTGCCAACGAGAGACAGCAGCTGGTGGAGACTCACATGGCTCGAGTGGAGGCCATGCTGAACGACAGGAGGAGGCTGGCCCTGGAGAACTACATCACCGCTCTGCAGGCCGTGCCTCCCAGGCCAAGGCATGTGTTTAACATGCTGAAGAAGTACGTGAGGGCAGAACAGAAGGACCGGCAACACACCCTGAAACACTTCGAGCACGTTAGAATGGTGGATCCTAAGAAAGCCGCTCAGATTAGAAGCCAGGTGATGACCCACCTGAGAGTGATTTACGAGAGAATGAACCAAAGCCTGTCTCTGCTGTATAATGTGCCCGCCGTCGCCGAGGAGATCCAGGACGAGGTGGACGAACTGCTGCAGAAGGAGCAAAATTACTCAGATGACGTGCTGGCAAACATGATCAGCGAACCACGCATCTCCTACGGCAACGACGCCCTGATGCCTTCCCTGACCGAAACTAAGACCACTGTGGAGCTGCTCCCAGTGAACGGCGAATTCTCCCTCGACGACCTGCAGCCTTGGCACAGCTTCGGGGCCGACTCCGTGCCTGCAAACACTGAAAACGAGGTGGAGCCTGTGGACGCAAGACCTGCCGCCGATAGAGGACTGACAACAAGACCTGGCAGCGGACTGACCAACATCAAGACCGAGGAGATTAGTGAGGTGAAGATGGATGCCGAGTTCAGGCACGATAGCGGGTACGAGGTACACCACCAGAAGCTGGTGTTCTTCGCTGAGGATGTGGGCAGCAATAAAGGAGCCATTATCGGCCTGATGGTGGGAGGGGTGGTGATCGCCACAGTGATCGTTATCACCCTGGTGATGCTGAAGAAGAAGCAGTACACCTCCATTCACCATGGGGTCGTCGAAGTGGATGCCGCCGTGACTCCAGAGGAGAGACACCTGAGCAAGATGCAGCAGAACGGGTATGAGAACCCAACCTATAAGTTCTTCGAGCAGATGCAGAAC
Once a codon-optimized DNA sequence is obtained, the protein can be produced using either cell-dependent or cell-free expression technologies.
Cell-dependent expression
In a cell-dependent system, the codon-optimized DNA sequence is first inserted into an expression vector that contains a promoter and other regulatory elements required for transcription. This vector is then introduced into host cells, such as mouse or mammalian cells. Inside the cell, the DNA sequence is transcribed into messenger RNA (mRNA) by RNA polymerase. The mRNA is subsequently translated by ribosomes, which read the nucleotide codons and use transfer RNAs (tRNAs) to assemble the corresponding amino acids into the protein. This approach allows protein production in a biologically relevant cellular environment and is commonly used in disease-related research.
Cell-free expression
Alternatively, the DNA sequence (or the corresponding mRNA) can be used in a cell-free expression system. These systems contain purified ribosomes, enzymes, and translation factors, allowing transcription and translation to occur in vitro without living cells. Cell-free expression enables rapid protein production and precise experimental control, although it may not fully replicate cellular processes such as protein trafficking or degradation.
3.5 (Optional) How does it work in nature / biological systems?
How a single gene can code for multiple proteins
In biological systems, a single gene can give rise to multiple proteins through transcriptional and post-transcriptional mechanisms. One major mechanism is alternative splicing, where different combinations of exons are joined from the same pre-mRNA to produce multiple mRNA transcripts. Additional diversity can arise from alternative transcription start sites or alternative polyadenylation. In the case of the amyloid precursor protein (APP), different processing pathways generate distinct fragments, including amyloid-beta peptides, demonstrating how one gene can produce multiple biologically relevant protein products.
DNA → RNA → Protein alignment
To visualize the flow of genetic information, the DNA sequence was translated using the ExPASy Translate tool, which displays all possible reading frames. The biologically relevant open reading frame was identified in the 5’→3’ Frame 1, which begins with a start codon (ATG) and produces a continuous amino acid sequence.
Week 3 HW: lab-automation
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Article: “Automation of protein crystallization scaleup via Opentrons-2 liquid handling”
Jacob B. DeRoo, Alec A. Jones, Caroline K. Slaughter, Tim W. Ahr, Sam M. Stroup, Grace B. Thompson, Christopher D. Snow,
SLAS Technology,
Volume 32,
2025,
100268,
ISSN 2472-6303,
General overview: Protein crystallization is a complex and time-consuming process that is essential for determining protein structures in structural biology. Producing well-formed protein crystals requires careful optimization of multiple conditions, including protein concentration, precipitant composition, and mixing accuracy. Because these parameters cannot be predicted in advance, crystallization is largely a trial-and-error process that demands repeated setup of crystallization plates. Traditionally, this process is performed manually, making it labor-intensive and susceptible to human error and variability. In addition, viscous protein solutions are difficult to handle consistently, which further complicates crystallization experiments.
In this study, the authors demonstrate how an Opentrons OT-2 liquid-handling robot can be adapted to automate protein crystallization plate setup. The robot was programmed using Python scripts, allowing precise control over aspirating, dispensing, and positioning steps. The researchers used Hampton Research Cryschem 24-well plates, which are larger than standard microplates and not directly compatible with the OT-2 deck. To address this limitation, the team designed a custom 3D-printed adapter made from polylactic acid (PLA) that securely clips into two deck slots and holds the crystallization plate in place. This setup enabled accurate and reproducible preparation of sitting-drop crystallization experiments using an affordable, open-source automation platform.
Findings: The authors validated the automated workflow using multiple experimental approaches. First, food dyes (red, blue, and yellow) were dispensed into colorless water to visually confirm accurate gradient formation across the crystallization plate, showing no significant difference between automated and manual pipetting. The system was then tested using hen egg white lysozyme (HEWL), a protein known to crystallize reliably under suitable conditions. During testing, the authors identified that the GEN1 P10 pipette had difficulty consistently dispensing very small volumes (2 µL) onto the sitting-drop pedestal. To overcome this limitation, they increased the total drop volume to 4 µL, which improved consistency and reliability. Finally, the automated protocol was used to reproduce crystallization of a protein previously studied by the authors, demonstrating that the Opentrons-based workflow could successfully replicate known crystallization outcomes with reduced manual effort.
Figure 1: Crystallization results from OT-2–prepared Cryschem 24-well sitting-drop experiments.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
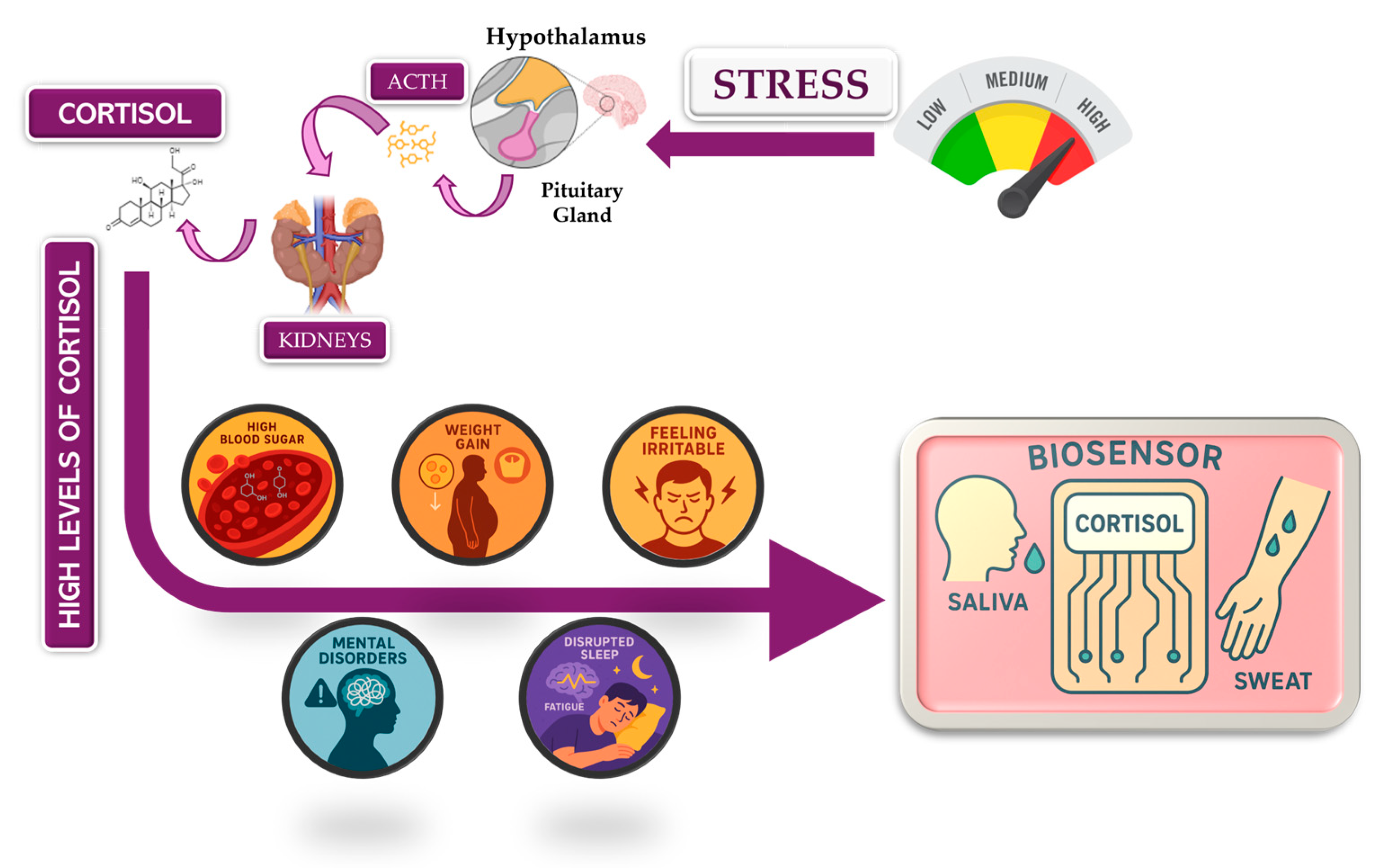
For my final project, I want to use lab automation tools to explore biological stress responses, with a focus on the hormone cortisol and its long-term effects on mental health. My motivation comes from the Iraqi context, where years of war, instability, environmental stress, and constant exposure to technology have contributed to high levels of anxiety, attention deficits, and stress-related disorders across the population.
According to the review article “https://pmc.ncbi.nlm.nih.gov/articles/PMC5619133/" chronic elevation of cortisol disrupts normal physiological balance and keeps the body in a prolonged fight-or-flight state. Long-term cortisol exposure affects the brain, particularly regions involved in attention, emotional regulation, and cognitive control, and is strongly associated with anxiety, impaired focus, and declining mental health. The article explains how sustained stress alters hypothalamic–pituitary–adrenal (HPA) axis regulation, leading to maladaptive stress responses rather than short-term protective ones.
In this project, I aim to simulate stress-related conditions in a controlled and automated way, rather than measuring stress directly in humans. Using automation tools such as the Opentrons liquid-handling robot, I would design workflows that represent different stress states (for example: baseline, moderate stress, chronic stress) through reproducible experimental conditions. Automation allows precise control of timing, volumes, and repetition, which is essential when modeling biological stress responses.
I would document the workflow using Python scripts or pseudocode, similar to what we learned in recitation, even if the protocol is not yet tested on the robot. Automation is critical here because stress biology depends on consistency and repetition, which manual handling cannot guarantee.
By replicating stress-associated conditions in vitro through automated workflows, this project aims to better understand how chronic stress environments such as those experienced by many Iraqi individuals may contribute to long-term cognitive and emotional effects. Understanding these mechanisms is an important step toward improved diagnosis, prevention, and treatment of stress-related disorders.
For now this is my pesudocode that will be developed more before the end of this course.
for condition in stress_conditions:
dispense_reagents(condition)
incubate_for_defined_time(condition)
prepare_samples_for_analysis()
Week 04 – Protein Design Part I
Part A: Conceptual Questions
For an average amino acid, the molecular weight is about 100 Daltons, which is equivalent to 100 g/mol. If I assume meat is about 20% protein, then 500 g of meat contains roughly 100 g of protein. The relationship is:
number of moles = mass (g) / molar mass (g/mol)
So, 100 g ÷ 100 g/mol ≈ 1 mole of amino acids. One mole corresponds to approximately 6×1023molecules. Therefore, consuming 500 g of meat corresponds to on the order of 1023amino acid molecules.
Proteins from food are first digested in the gastrointestinal tract into smaller peptides and eventually into individual amino acids. These amino acids are absorbed into the bloodstream and become part of the body’s amino acid pool. Human cells then reuse these amino acids to synthesize new proteins according to human gene expression and the cellular molecular machinery. Because each organism has its own genes and regulatory systems, the same amino acids can be assembled into completely different proteins. This is why eating fish or chicken does not make us become those organisms.The amino acids are simply recycled by our own cells to build human proteins.
According to the article from FEBS Press, the standard set of 20 amino acids was selected early in evolution because it provides a near-ideal mix of charge, size, and hydrophobicity that enables proteins to fold into soluble, stable, close-packed structures with functional binding pockets. This selection happened after the RNA World had cofactors that already performed most catalysis, so amino acids were chosen mainly to support folding and structural stability, not just chemistry. While other amino acids are chemically possible, these 20 were favored because they collectively cover the chemical properties needed for diverse protein structures.
(FEBS Press – Andrew J. Doig, 2016)
Yes, it is possible to design non-natural amino acids. All amino acids share the same backbone (amino + carboxyl), and the side chain (R group) is what varies. By changing or substituting the R group, many new amino acids can be created.
Non-natural amino acids are used in synthetic biology and drug design to explore chemistry beyond the 20 natural ones, but they are not part of the standard genetic code.
(Supported by general protein chemistry and synthetic biology literature)
The astrobiology article explains that amino acids could form abiotically through chemical reactions in the early Solar System and on early Earth. Processes like Strecker synthesis under prebiotic conditions, reactions in hydrothermal vents, and organic chemistry on planetesimals delivered by meteorites could produce amino acids before life existed. These amino acids do not require enzymes; they form through random but plausible chemical reactions in environments with water, heat, and simple carbon compounds.
In biology, proteins are made of L-amino acids, and these form right-handed α-helices because of the stereochemistry of the backbone. If the same sequence were built entirely from D-amino acids, the mirror image geometry would favor left-handed α-helices instead.
Yes. In addition to the common α-helix, proteins also contain other helical structures such as 3₁₀ helices and π-helices that are observed in real proteins. These alternative helices differ in hydrogen bonding patterns and geometry. More exotic or engineered helices could also be possible if non-natural amino acids or non-canonical backbones are used, expanding the structural possibilities.
Most protein helices are right-handed because proteins use L-amino acids almost exclusively in nature. The stereochemistry of L-amino acids puts constraints on backbone torsion angles that favor right-handed helices energetically and sterically. Left-handed helices are possible in principle but are unstable with L-amino acids due to steric clashes and unfavorable geometry.
β-sheets are secondary structures formed by extended polypeptide chains connected by backbone hydrogen bonds. The edges of β-sheets expose hydrogen-bond donors and acceptors. If these edges are not satisfied internally, they tend to pair with similar edges on other β-strands or sheets, leading to aggregation.
The main driving forces behind β-sheet aggregation are:
A. Backbone hydrogen bonding between β-strands in different molecules.
B. Hydrophobic interactions between nonpolar side chains, which reduce exposure to water.
C. Entropy gain from releasing ordered water as hydrophobic surfaces come together and as hydrogen bonds form.
This explains why β-sheet-rich structures such as amyloids and fibrils tend to form in aggregation-prone conditions.
10 and 11 are skipped.
Part B: Protein Analysis and Visualization
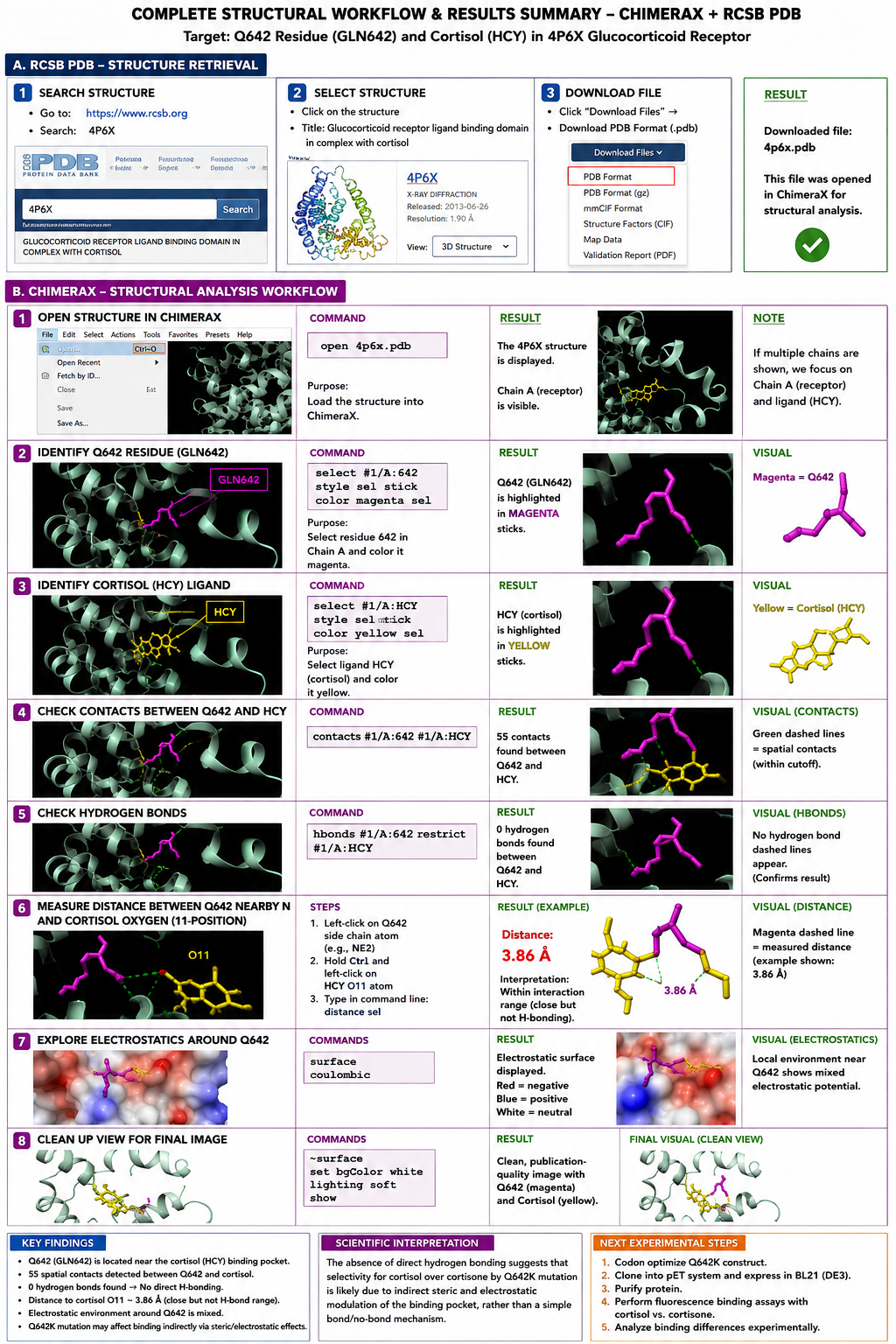
I chose the human glucocorticoid receptor because it is directly involved in sensing cortisol, which is one of the main stress hormones in the body. I was drawn to this protein because, in my own life and work in Iraq, I have seen how prolonged stress and anxiety can affect people deeply, especially students, through poor focus, memory problems, sleep disturbance, digestive complaints, and other health issues. This made me interested in a protein that helps the body detect and respond to stress at the molecular level. I also chose it because it connects neuroscience, hormones, and real-life health challenges in a very meaningful way.
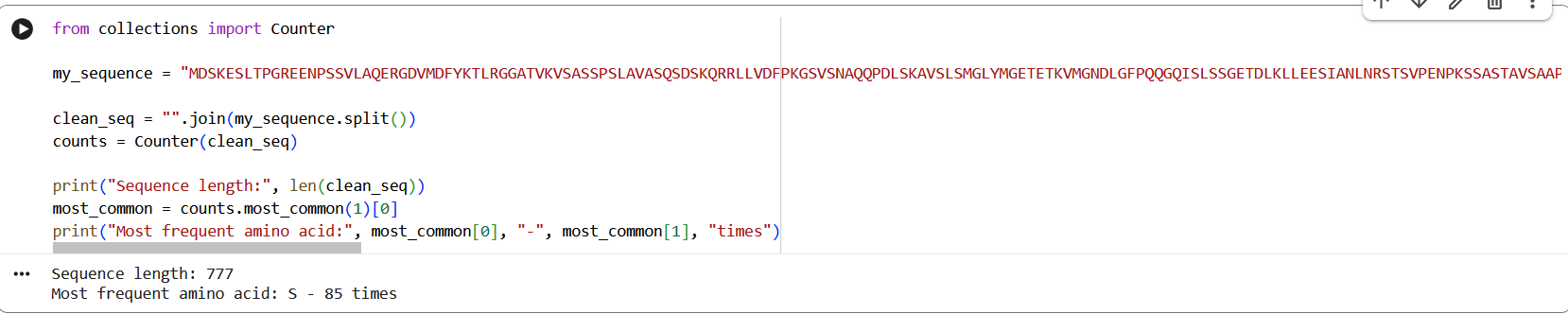
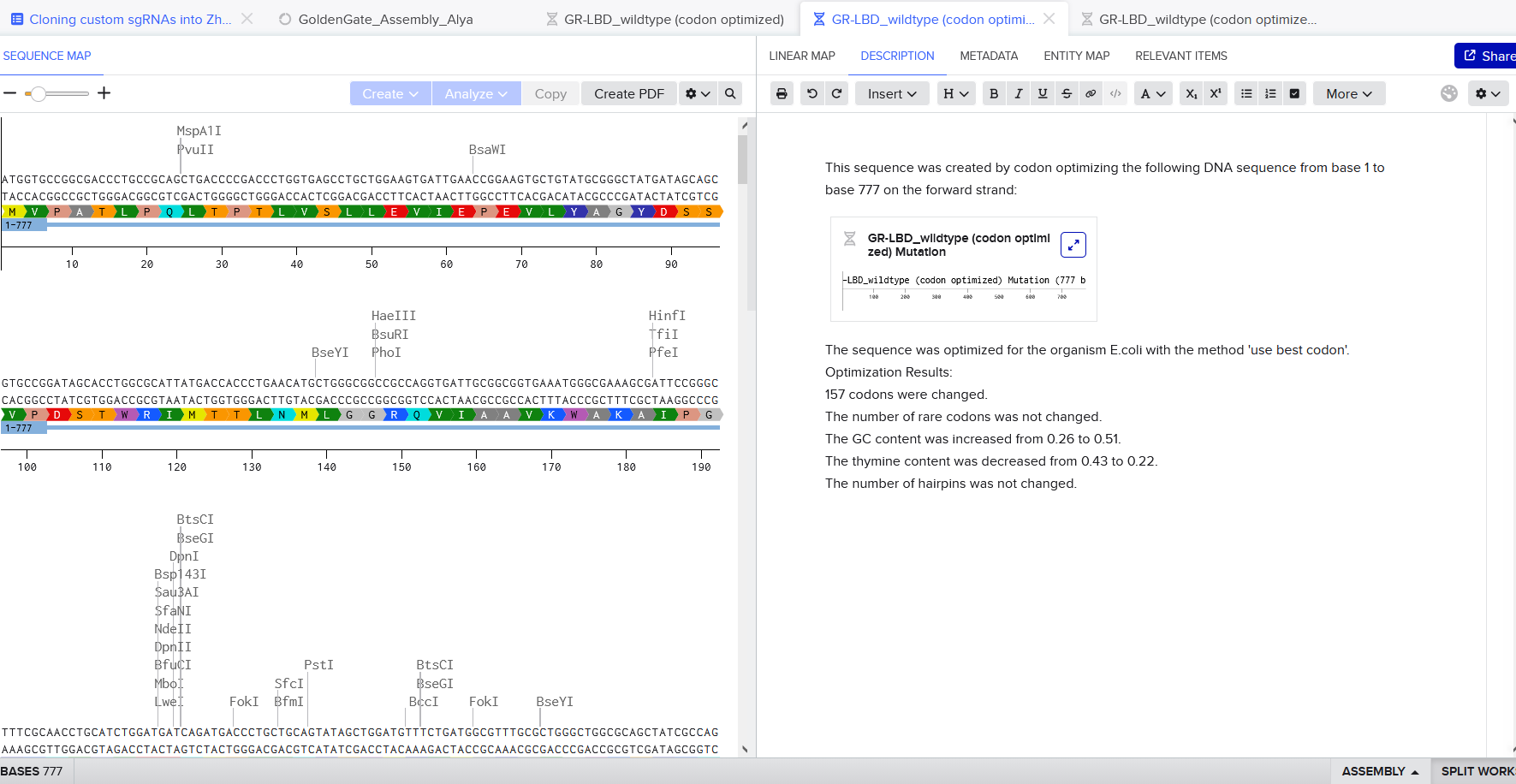
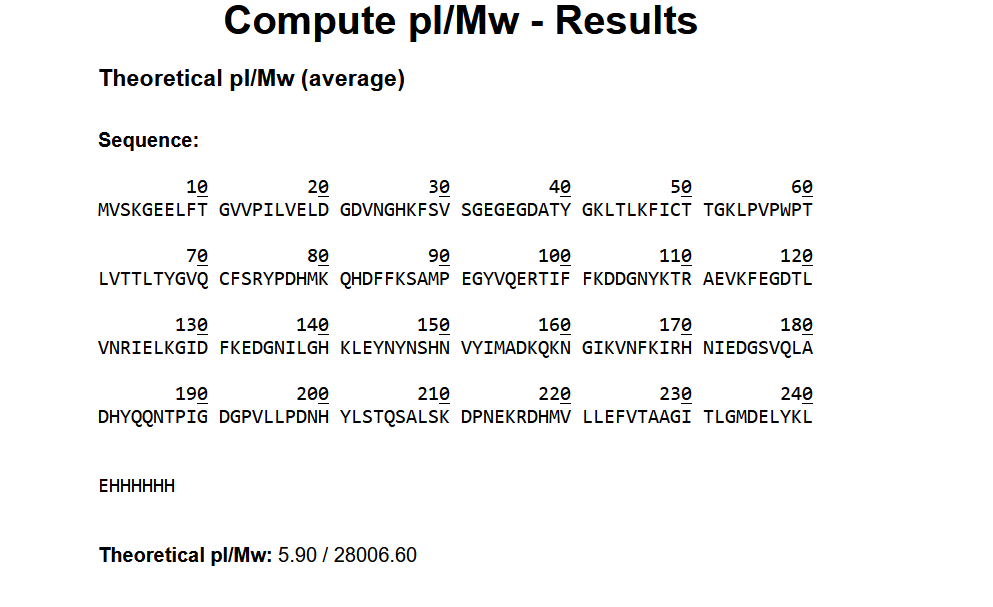
The amino acid sequence of the human glucocorticoid receptor (NR3C1) was obtained from the UniProt database (P04150).
Figure 1: NR3C1 human glucocorticoid receptor amino acid sequence
The protein consists of 777 amino acids. Using the Google Colab amino acid counting notebook, the most frequent amino acid in the sequence is serine (S), which appears 85 times.
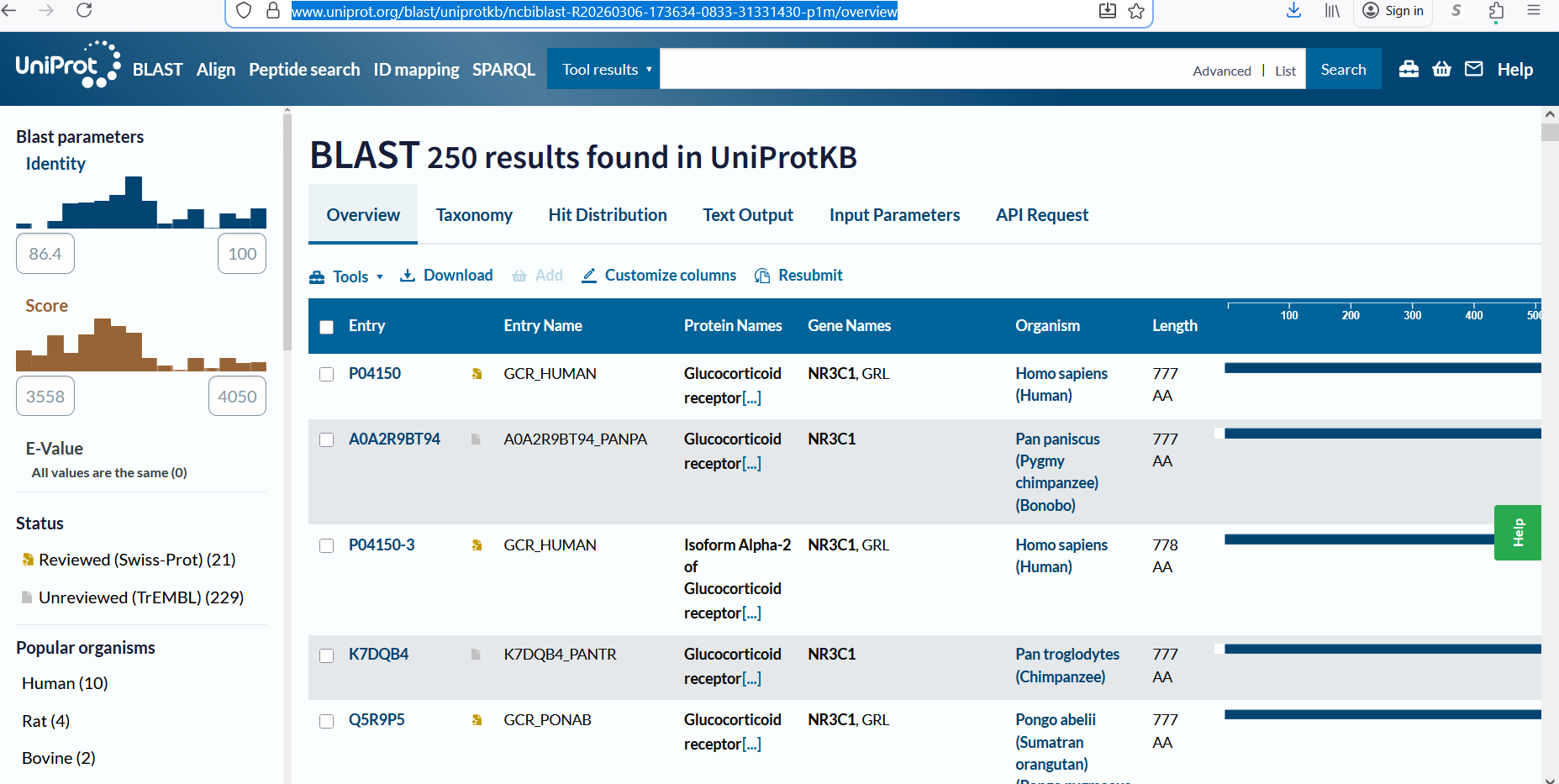
A UniProt BLAST search for the glucocorticoid receptor returned 250 homologous protein sequences in UniProtKB. These homologs are found across several vertebrate species, including humans, chimpanzees, bonobos, and orangutans, indicating that the protein is evolutionarily conserved.
Figure 2: Protein sequence homologs.
The glucocorticoid receptor belongs to the nuclear receptor protein family, specifically the steroid hormone receptor family, which functions as ligand-activated transcription factors that regulate gene expression.
Figure 3: Protein family
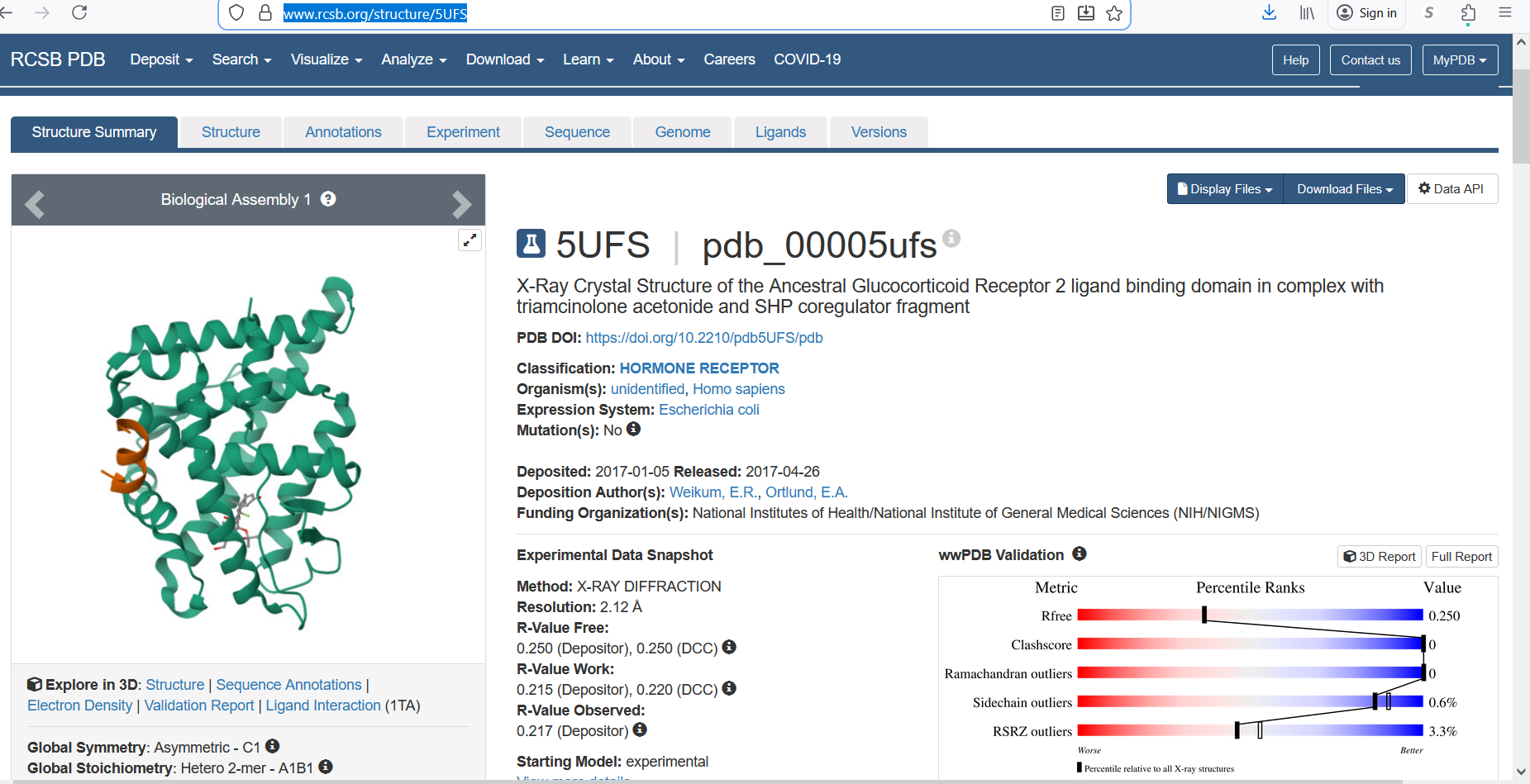
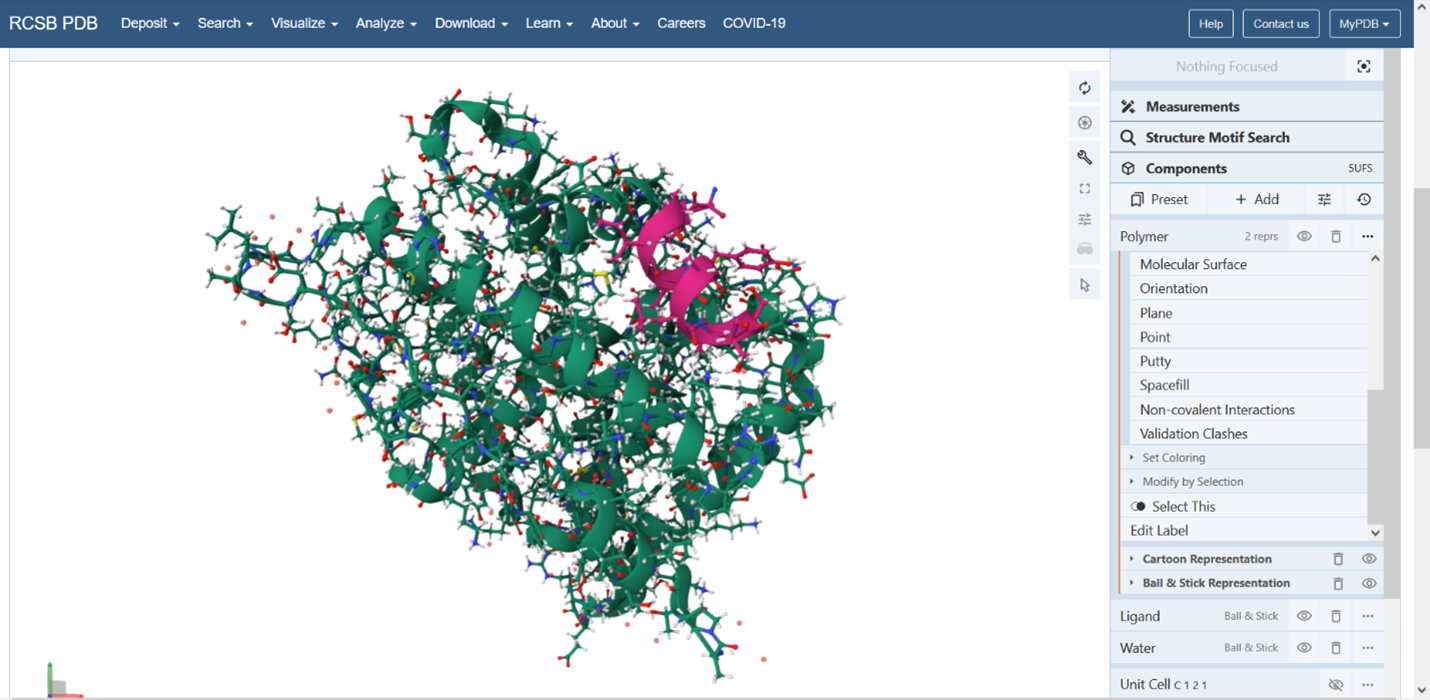
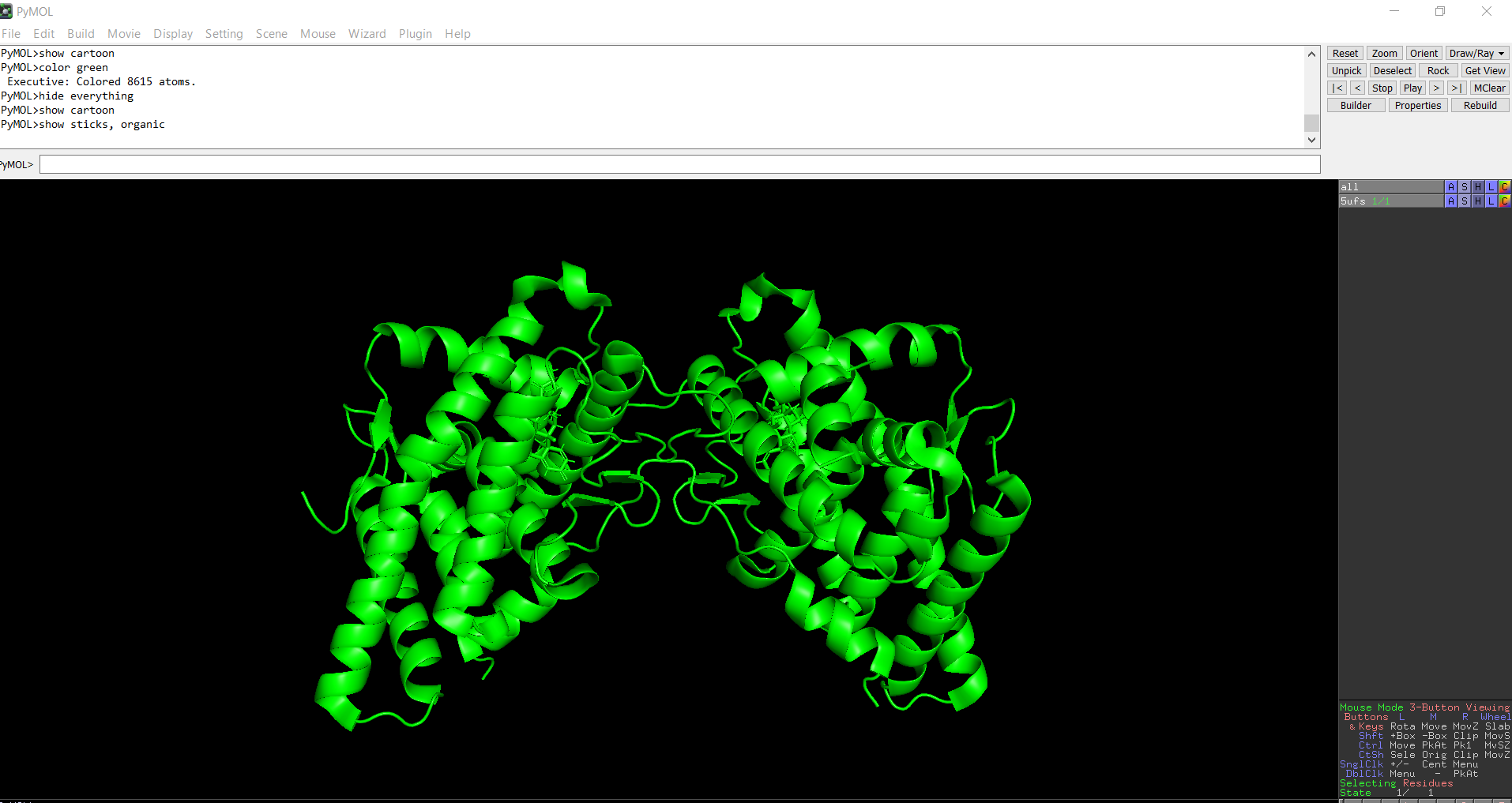
The protein was solved in 2017, and the structure was determined using X-ray diffraction. As well as the structure has a resolution of 2.12 Å, which indicates a high-quality crystal structure. The structure contains additional molecules besides the protein. These include triamcinolone acetonide, which is a glucocorticoid ligand bound in the receptor’s binding pocket, and a coregulator peptide fragment (SHP) that interacts with the receptor.
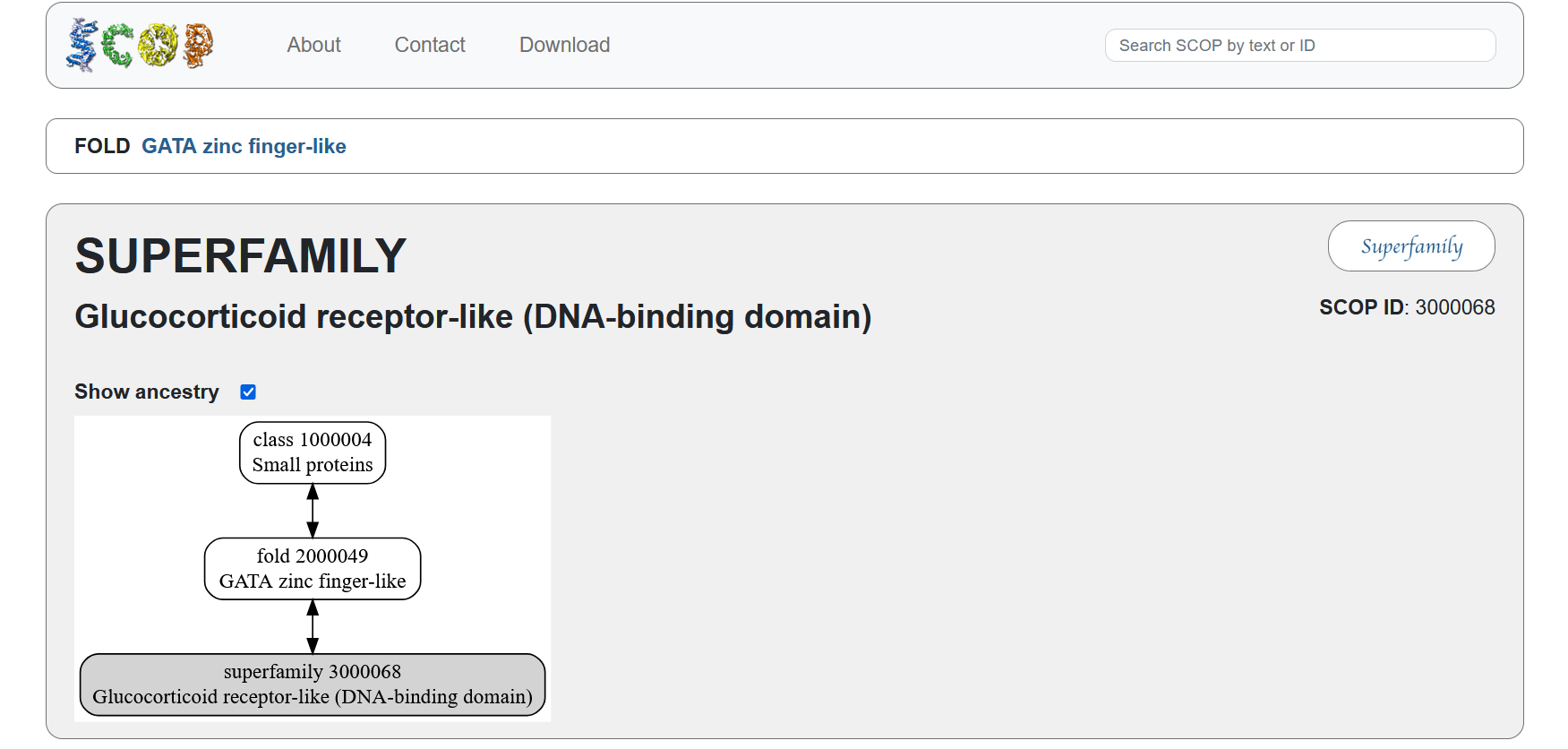
According to the SCOP structural classification database, the glucocorticoid receptor DNA-binding domain belongs to the glucocorticoid receptor-like DNA-binding domain superfamily. This superfamily adopts a GATA zinc-finger-like fold, which enables the receptor to bind specific DNA sequences and regulate gene transcription.
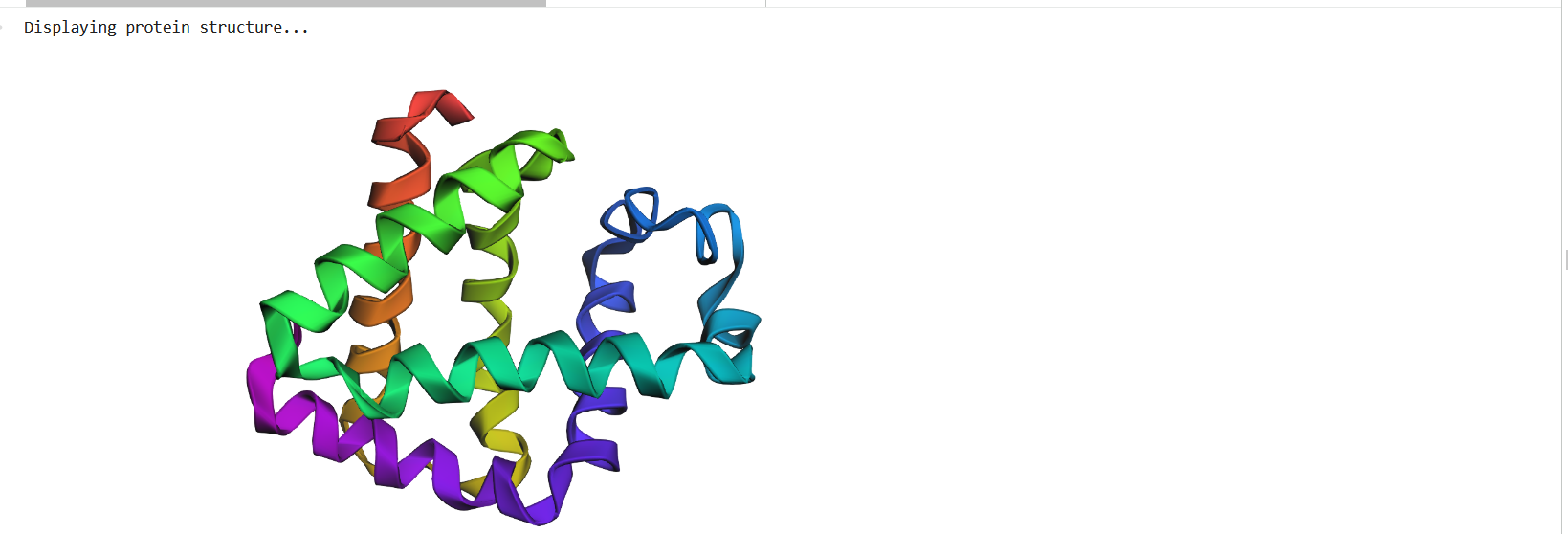
Figure 4: Protein Structure
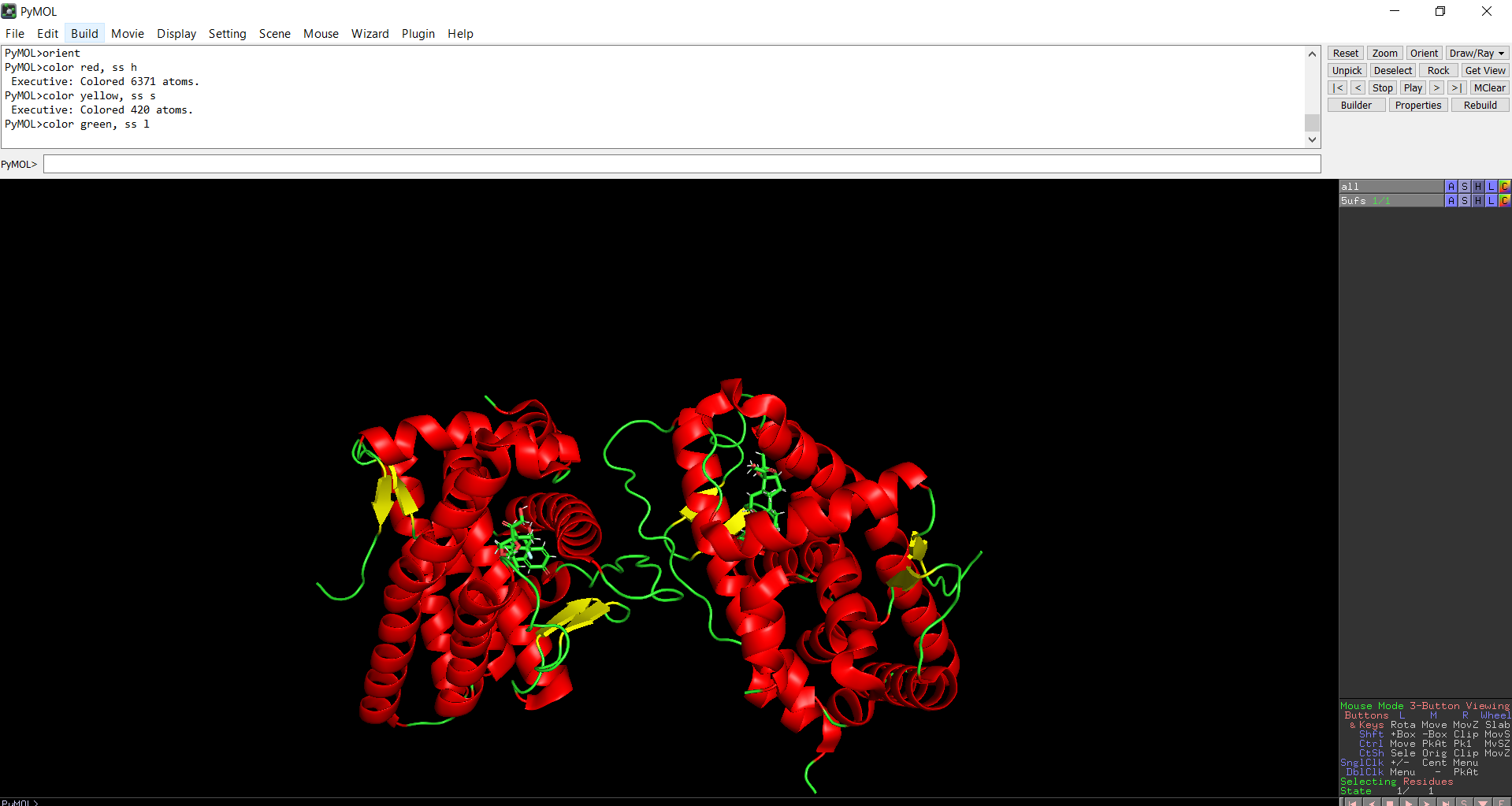
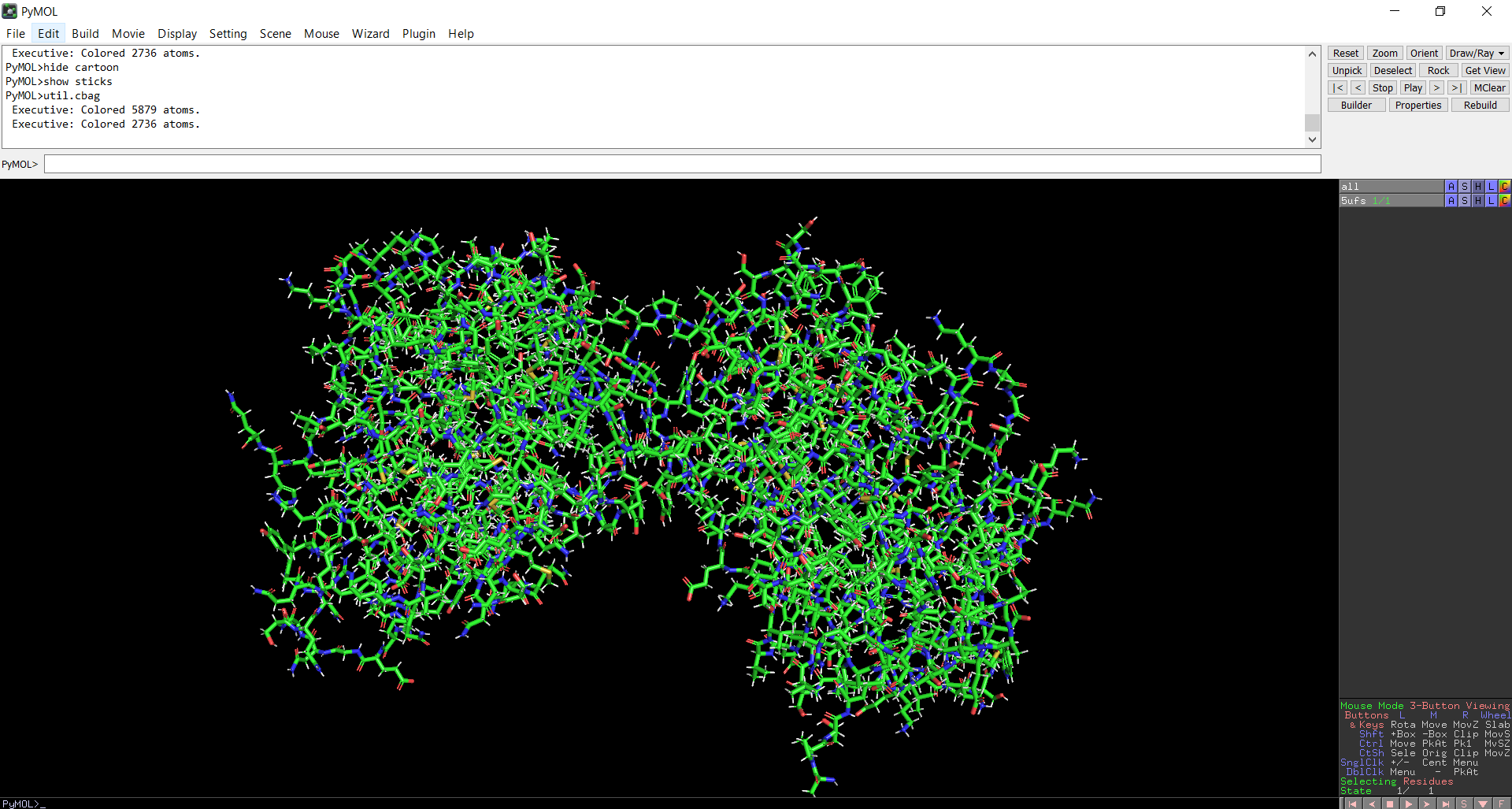
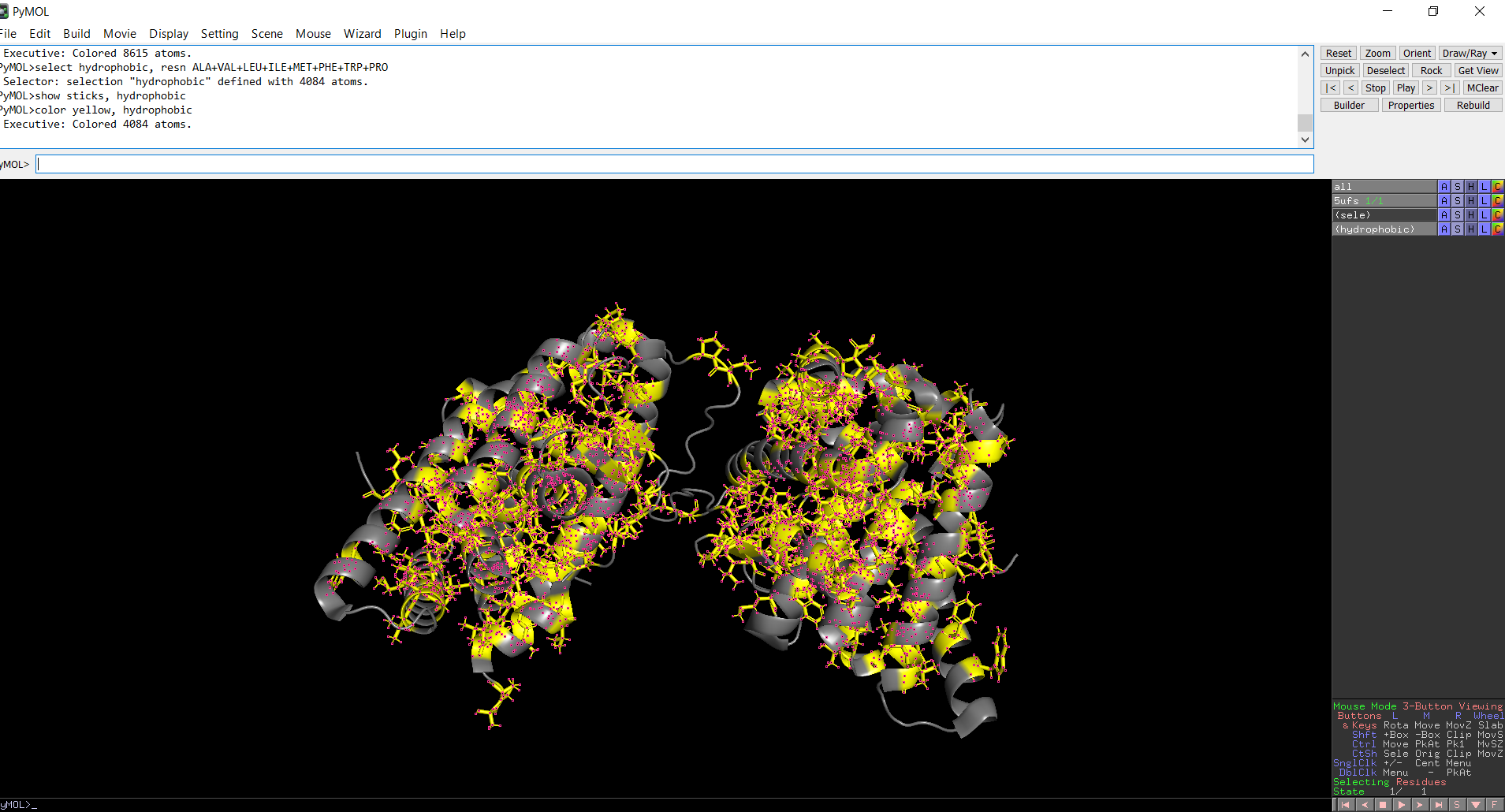
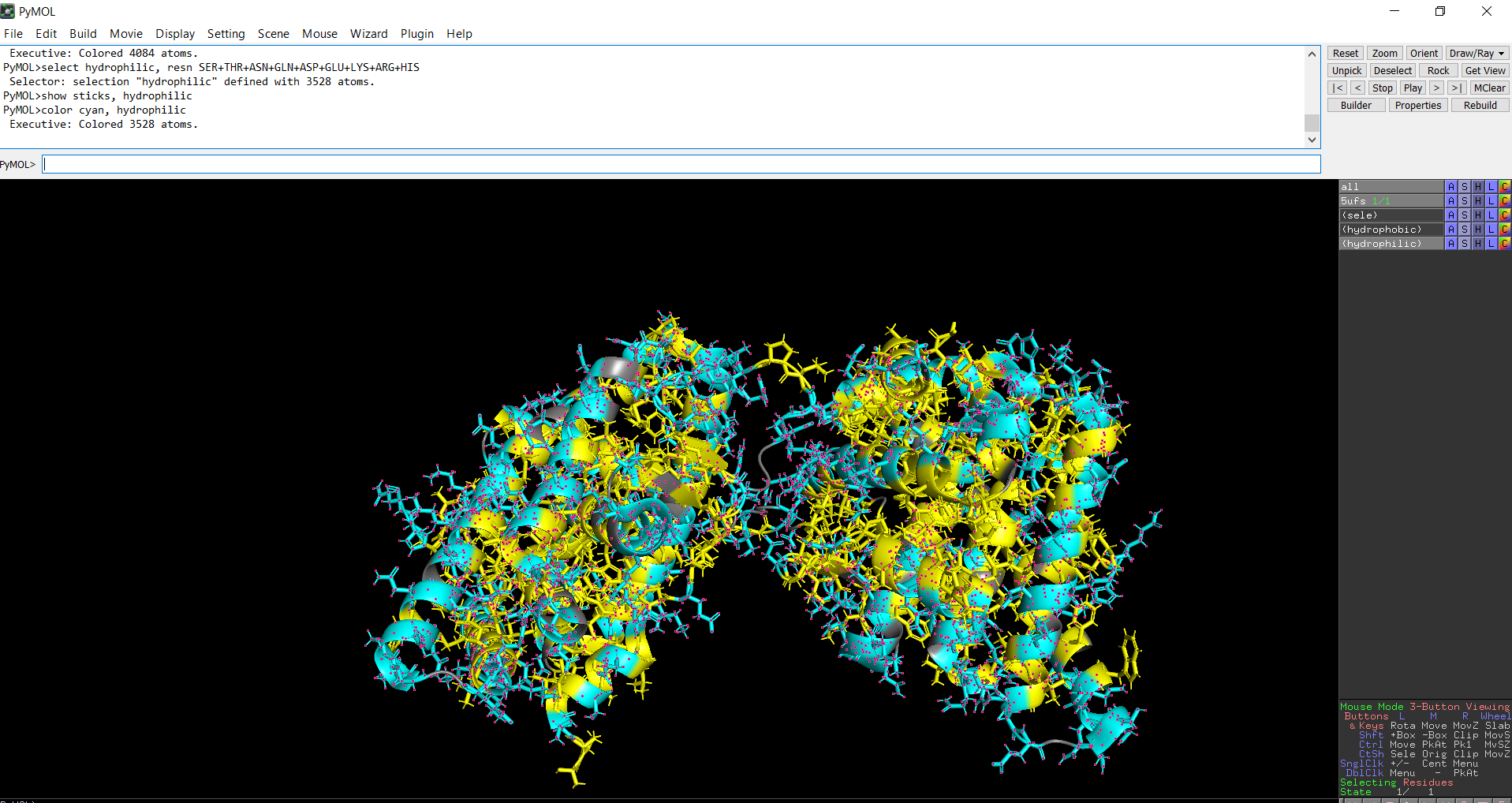
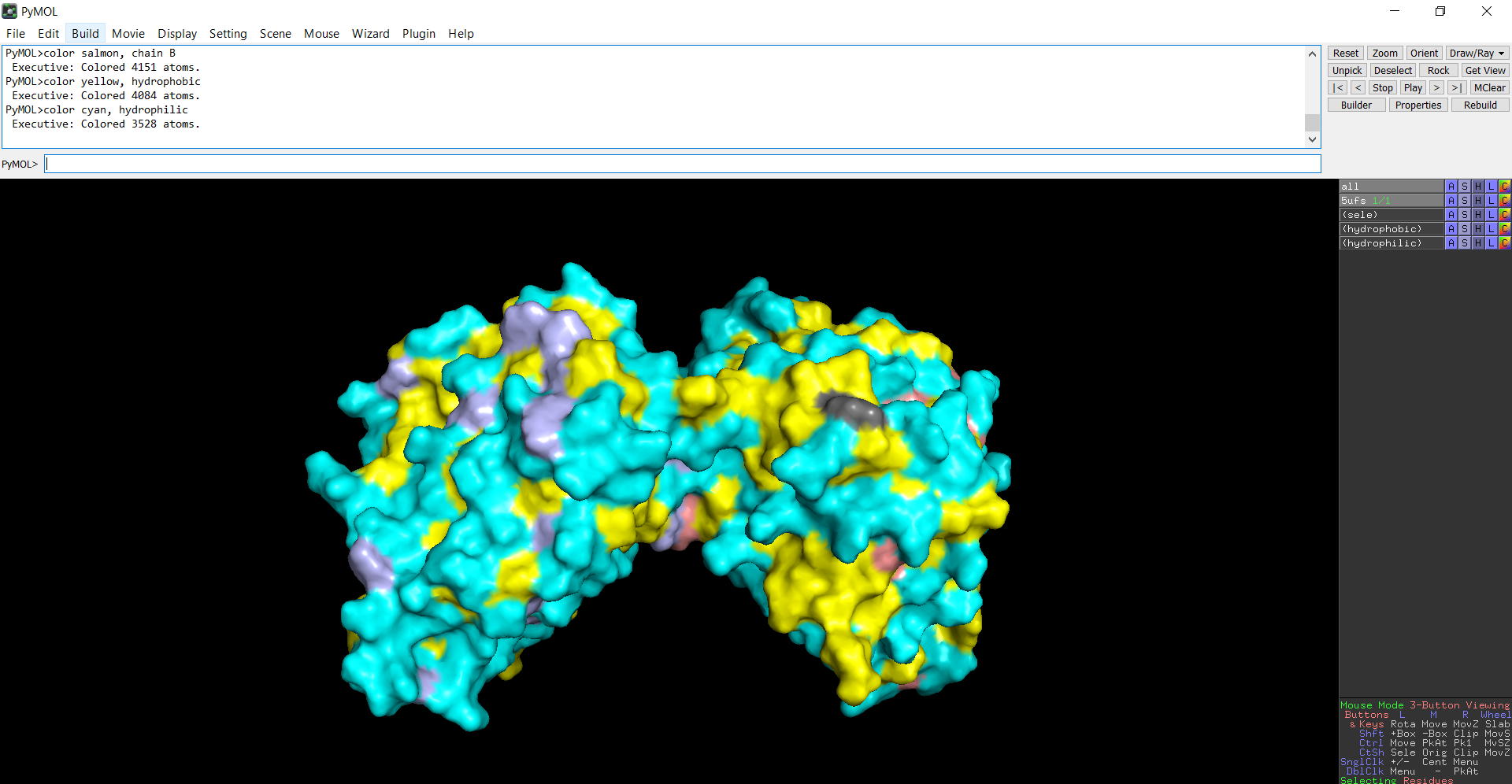
The protein structure was visualized in PyMOL and RBCS using different representations, including cartoon, ribbon, and ball-and-stick models, as shown in Figures 5 and 6. The secondary structure of the protein was examined by coloring the structure according to helices and sheets, which is presented in Figure 9. In addition, the distribution of hydrophobic and hydrophilic residues across the protein is illustrated in Figures 10, 11 and 12. Finally, the surface representation of the protein was generated to observe possible cavities or binding pockets, as shown in Figure 13.
Figure 5: Cartoon representation of the protein structure using RCSB
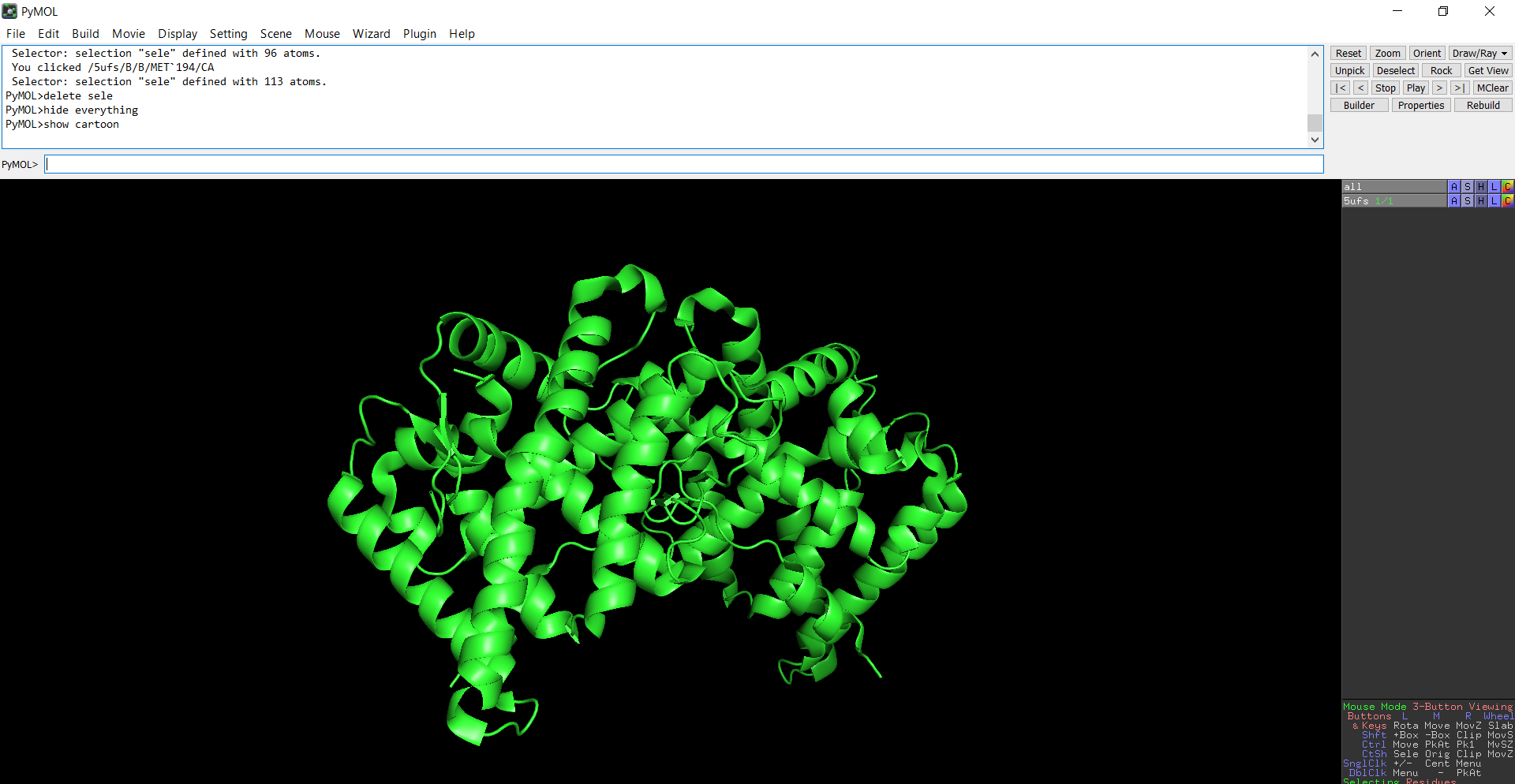
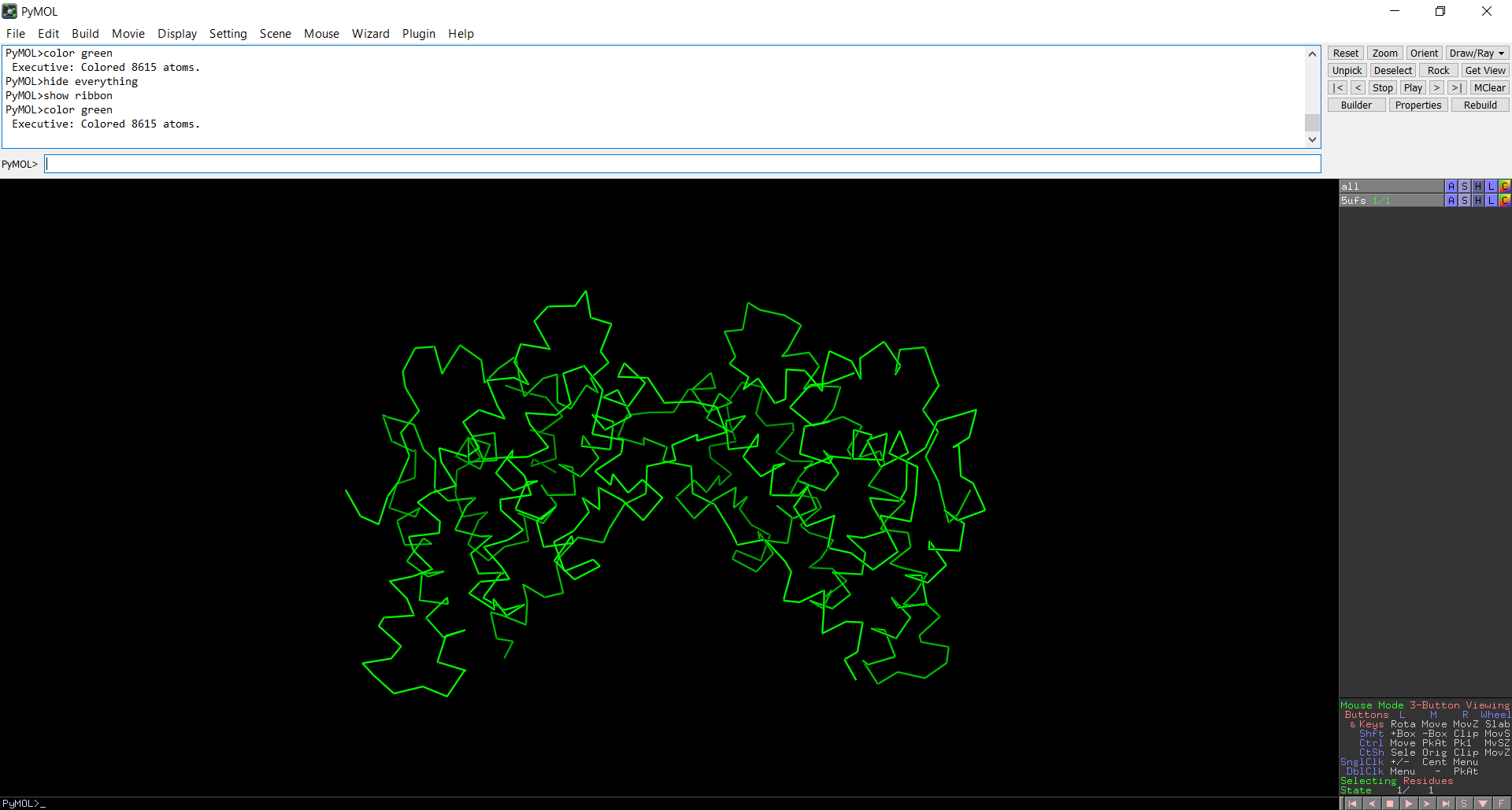
Figure 6: Cartoon representation of the protein structure using PyMol
Figure 7: Ribbon representation of the protein structure using PyMol
Figure 8: Ball-and-stick models representation of the protein structure using PyMol
The protein does contain more alpha helices than beta pleated sheets.
When the protein surface is colored by residue type, hydrophilic residues are primarily distributed on the outer surface of the protein where they can interact with water. Hydrophobic residues appear in patches and are more commonly buried within the protein interior or located in pockets, helping stabilize the protein structure and potentially forming ligand-binding regions.
Figure 9: Secondary structure of the protein was colored using PyMol
Figure 10: Distribution of hydrophobic and hydrophilic residues across the protein
Figure 11: Distribution of hydrophobic residues across the protein
Figure 12: Distribution of hydrophilic residues across the protein
The surface representation reveals several cavities and grooves on the protein surface. One noticeable pocket appears near the center between the two domains, which could function as a ligand-binding site.
Figure 13: The surface representation of the protein
Part C: Using ML-Based Protein Design Tools
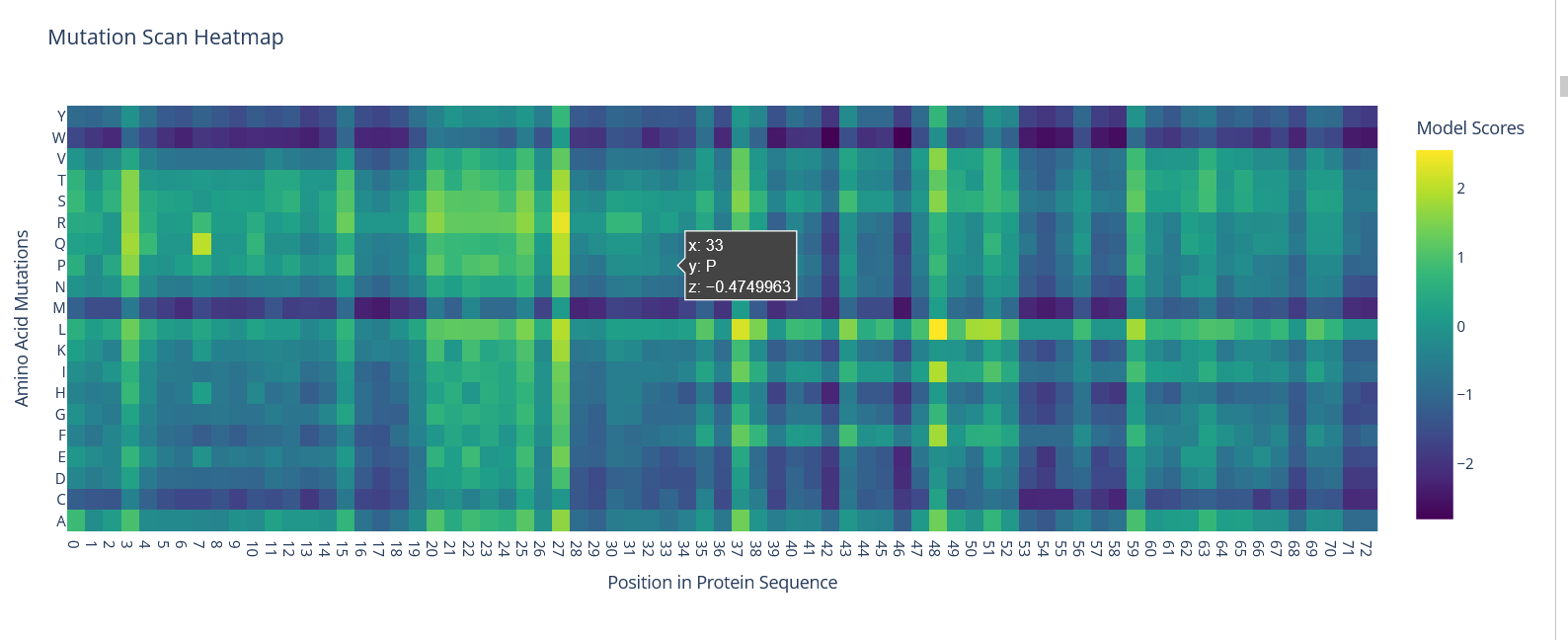
When looking at the mutation heatmap I noticed that position 58 shows a strong negative score for some substitutions. For example, mutating that residue to tryptophan (W) gives a score of about −2.62, which is one of the darkest values in the heatmap. This suggests that the original amino acid at that position is important for the protein structure and that introducing a bulky residue like tryptophan is not tolerated.
Figure 14: Mutation Scan HeatMap
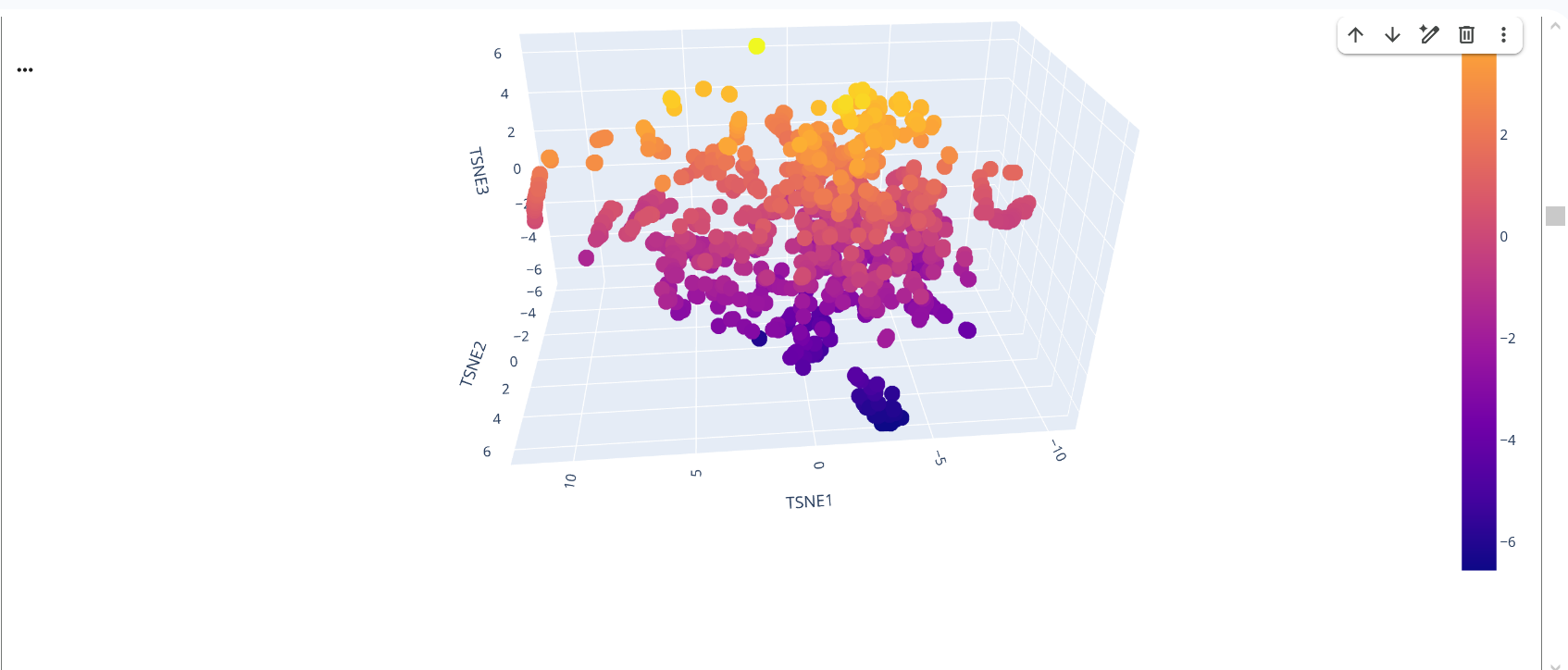
In the latent space visualization, proteins that appear closer together in the t-SNE space share more similar sequence embeddings generated by the ESM model. The clustering pattern suggests that proteins with related sequence features are grouped together. The color scale corresponds to the value of the third t-SNE dimension and mainly helps visualize the structure of the embedding space.
Figure 15: Protein Folding by ESM
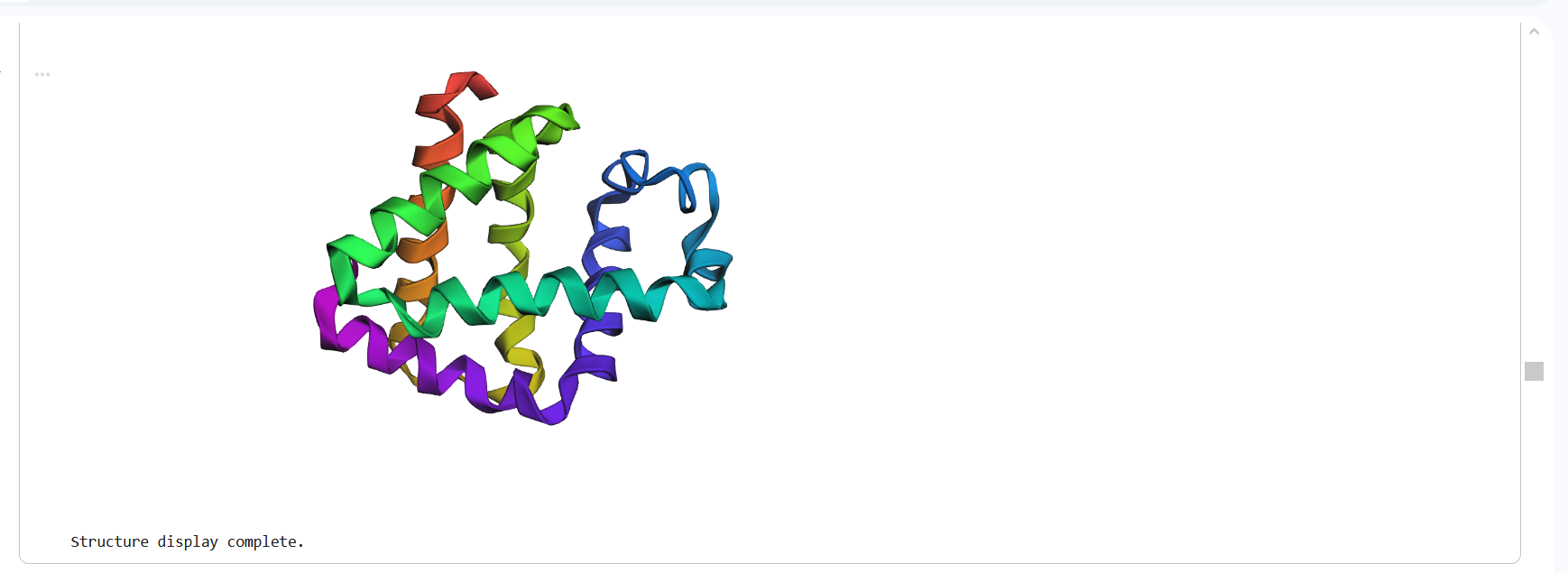
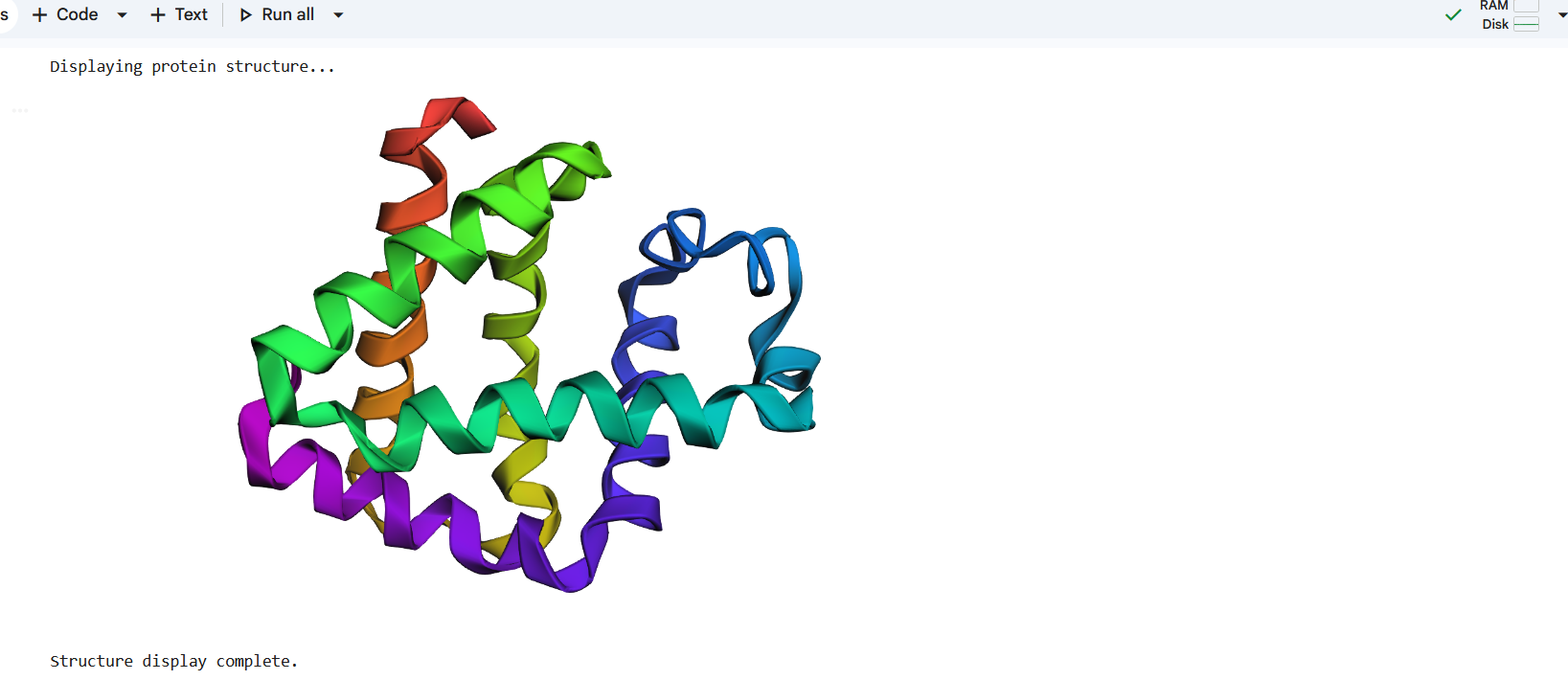
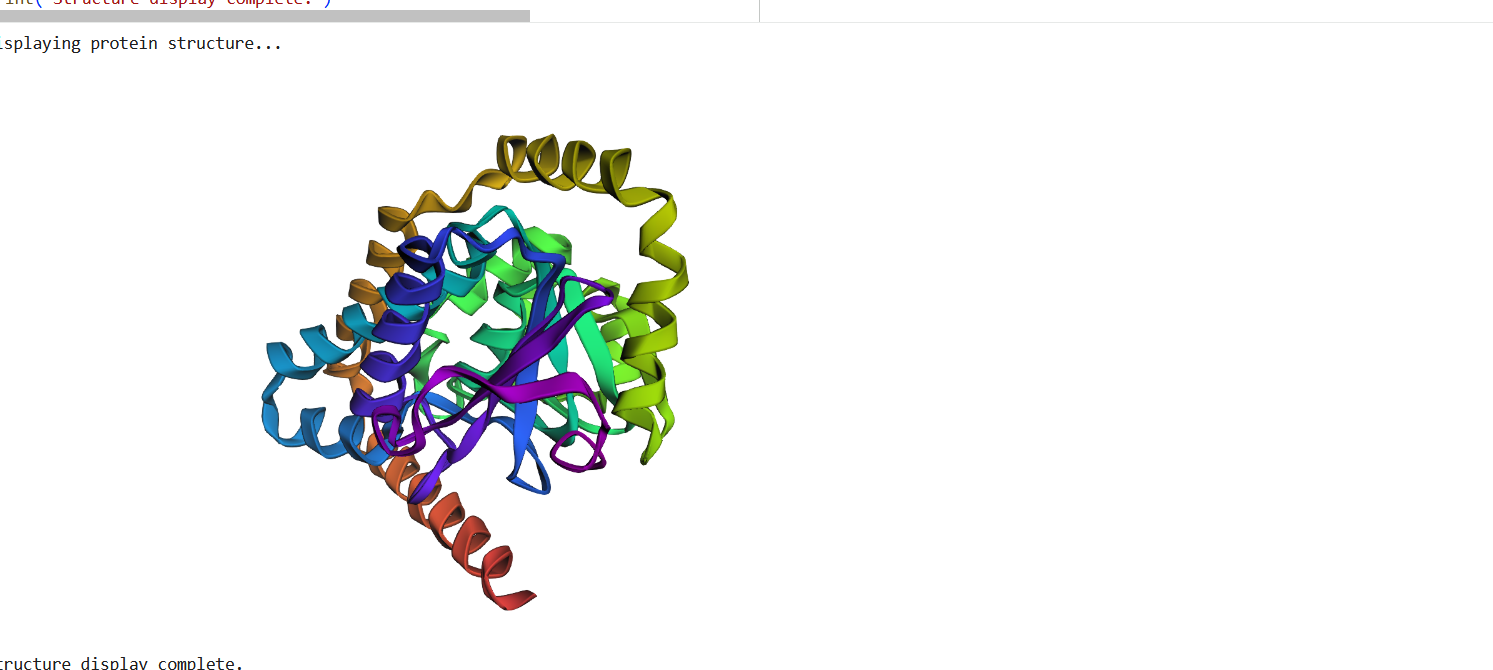
The structure predicted by ESMFold shows a similar overall fold compared to the experimentally determined PDB structure. Both structures contain multiple α-helices arranged in a compact configuration. However, the structures are not identical. The PDB structure represents a multimeric assembly with multiple chains, while the ESMFold model predicts a single-chain structure. Additionally, experimental structures often contain ligands or small molecules that are not predicted by ESMFold. Despite these differences, the general secondary structure arrangement appears consistent between the two models.
Figure 16: Protein Structure Predicted by ESMFold
I first introduced a single mutation by changing one amino acid in the sequence and then folded the modified sequence using ESMFold. The predicted structure remained very similar to the original structure, with the alpha-helical regions still preserved, as shown in Figure (17 ).
I then introduced larger mutations by replacing several residues with alanine (AAAAAA) and also tested additional mutations such as proline substitutions. Despite these modifications, the predicted structures remained largely similar to the original model, with only minor local differences, as shown in Figure (18 ). Overall, these results suggest that the protein fold is relatively resilient to moderate sequence mutations.
Figure 17: Making a mutation by changing one letter from P to A in the amino acid sequence.
Figure 18: Making a mutation by changing a large segment of the amino acid sequence.
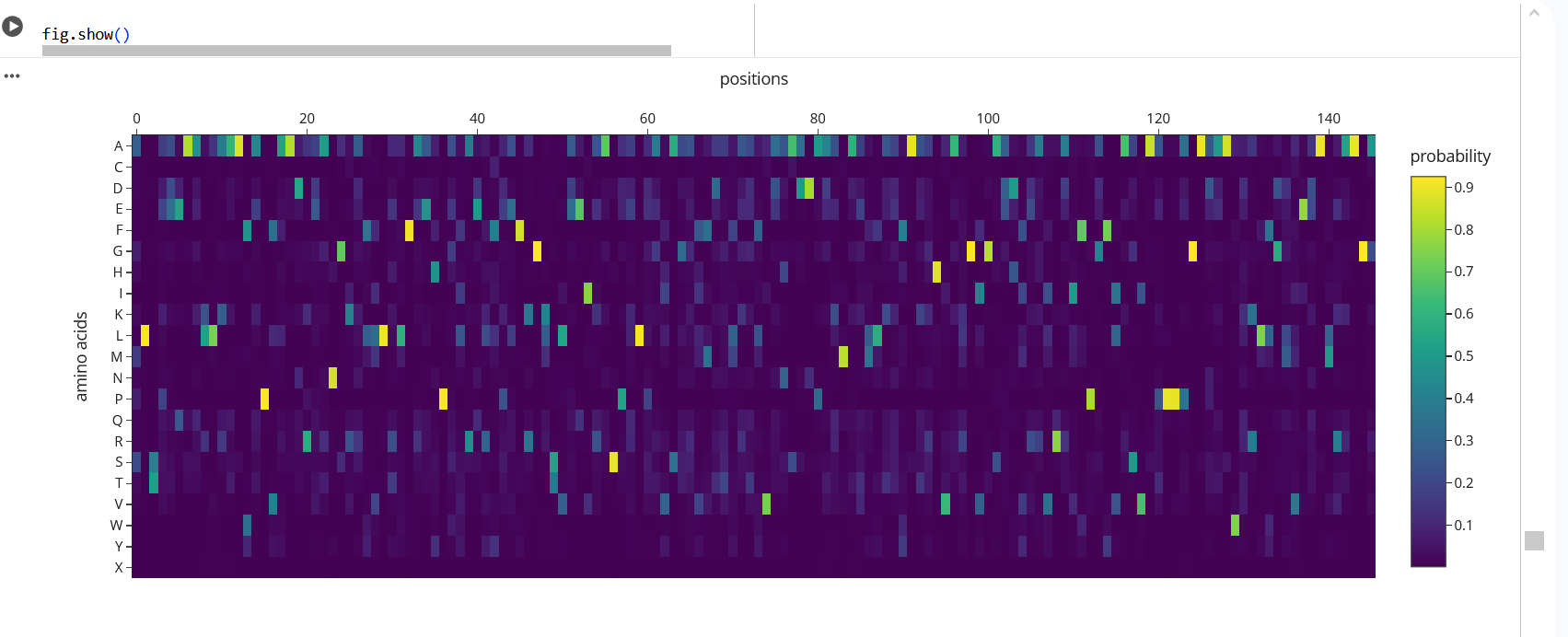
The heatmap shows the probability of different amino acids at each position in the protein. The yellow regions indicate higher probabilities, meaning the model strongly prefers those residues at those positions. For example, at position 58 leucine (L) has a probability of about 0.91, and at position 36 proline (P) has a probability of about 0.92. In contrast, the green regions show lower probabilities, meaning those positions are more flexible and can tolerate different amino acids. This suggests that some residues are more important for maintaining the protein structure while others are less constrained.
Figure 19: The heatmap shows the probability of different amino acids at each position in the protein.
The sequence generated by ProteinMPNN was folded using ESMFold and compared to the original structure. The predicted structure shows a very similar overall fold, with helices appearing in similar positions. Although there are small differences in some regions, the general shape of the protein remains the same. This suggests that the designed sequence is compatible with the same backbone structure.
Figure 20: The Protein sequence generated by ProteinMPNN
But UniProt includes the starting M, so the alanine that changes is the 5th residue in the sequence.
1 M
2 A
3 T
4 K
5 A ← this is the one that changes
6 V
We change from this MATKAV to this MATKVV
The new sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Figure 1: The original length of amino acids.
Figure 2: The length 12 amino acids conditioned on the mutant SOD1 sequence.
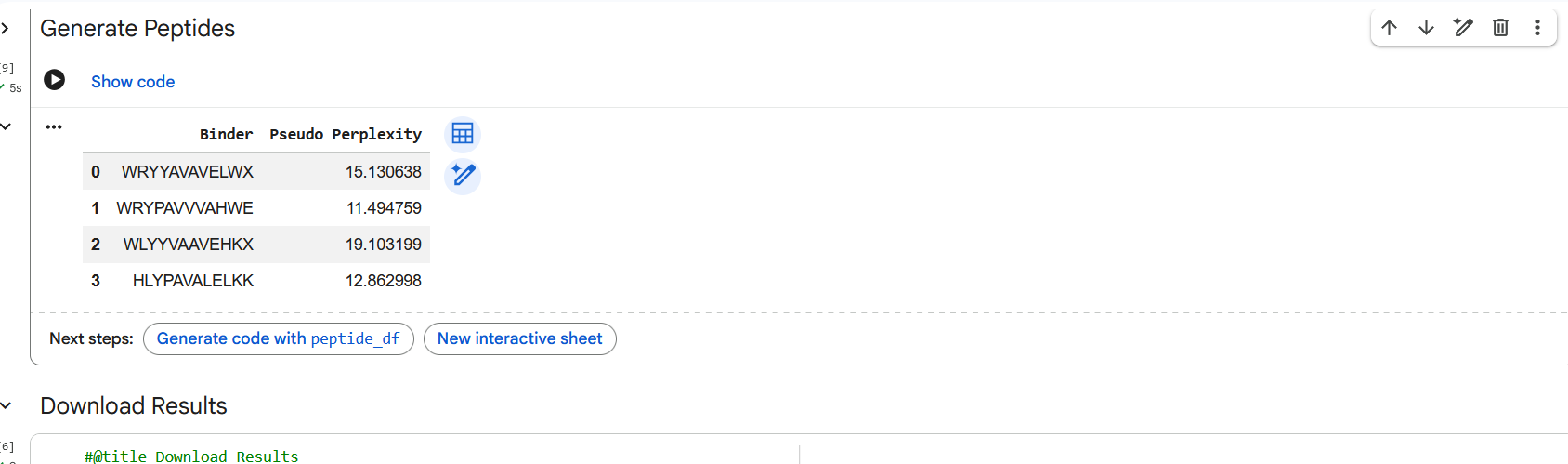
Figure 3: Generation of four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
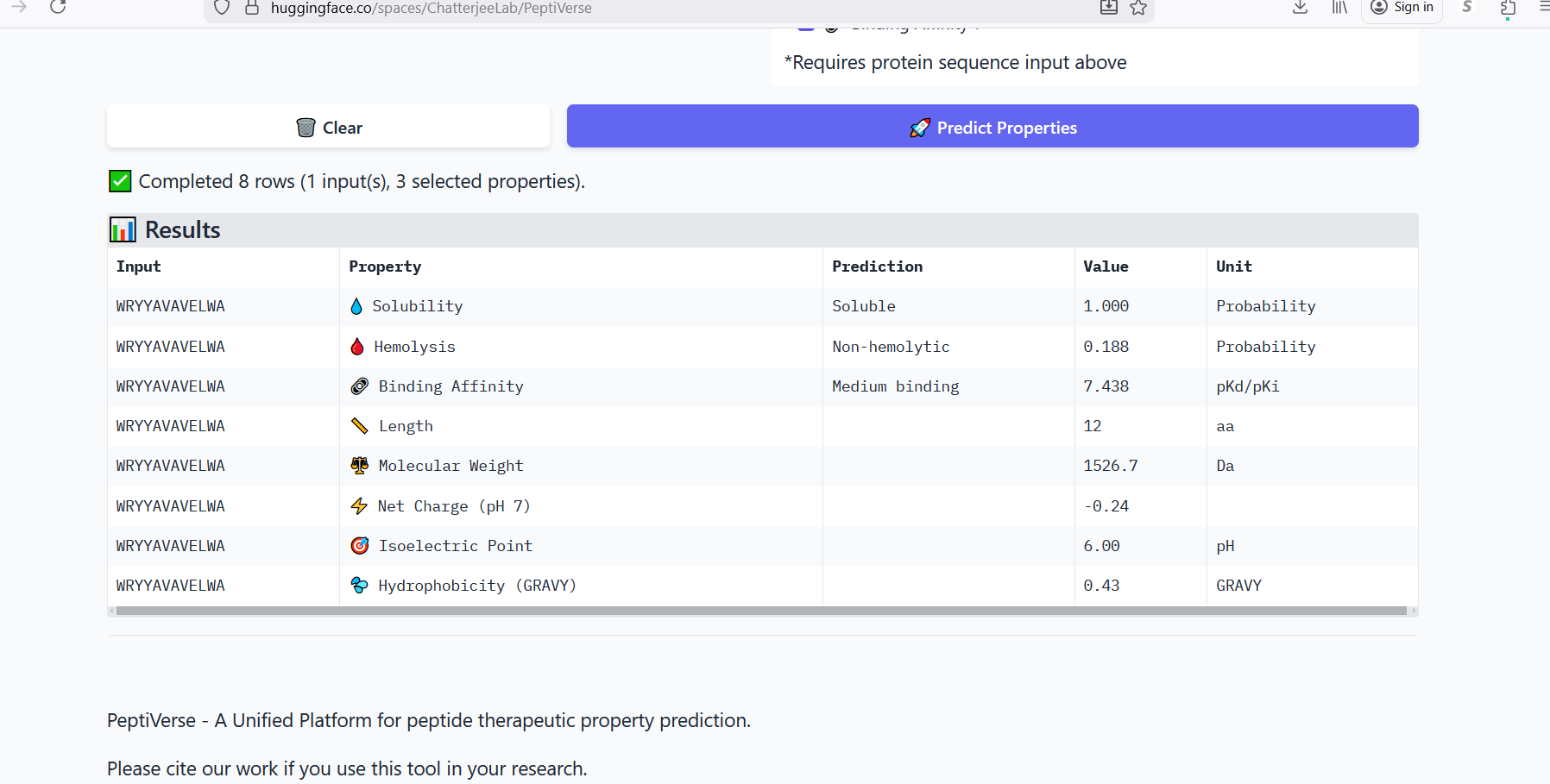
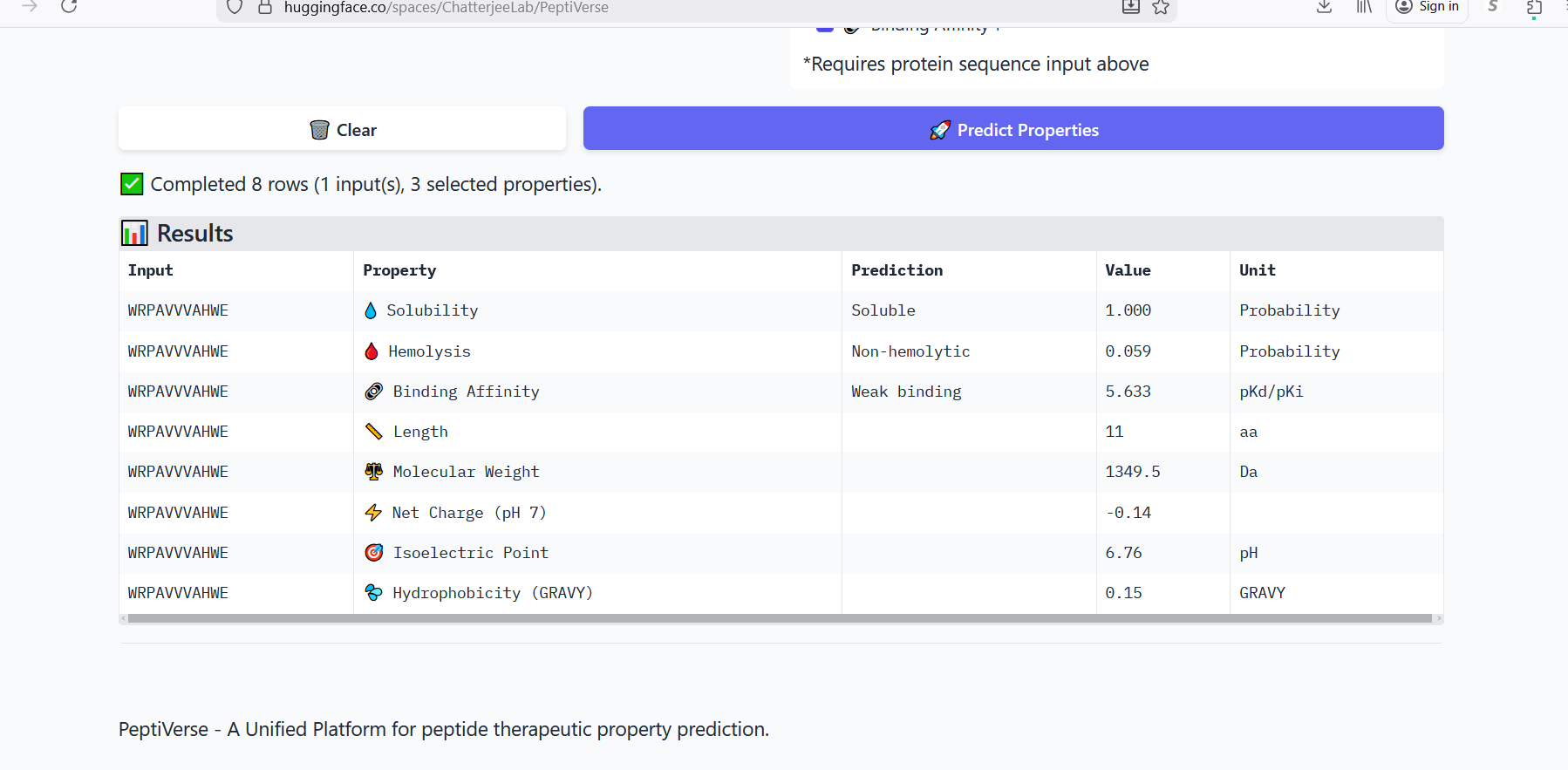
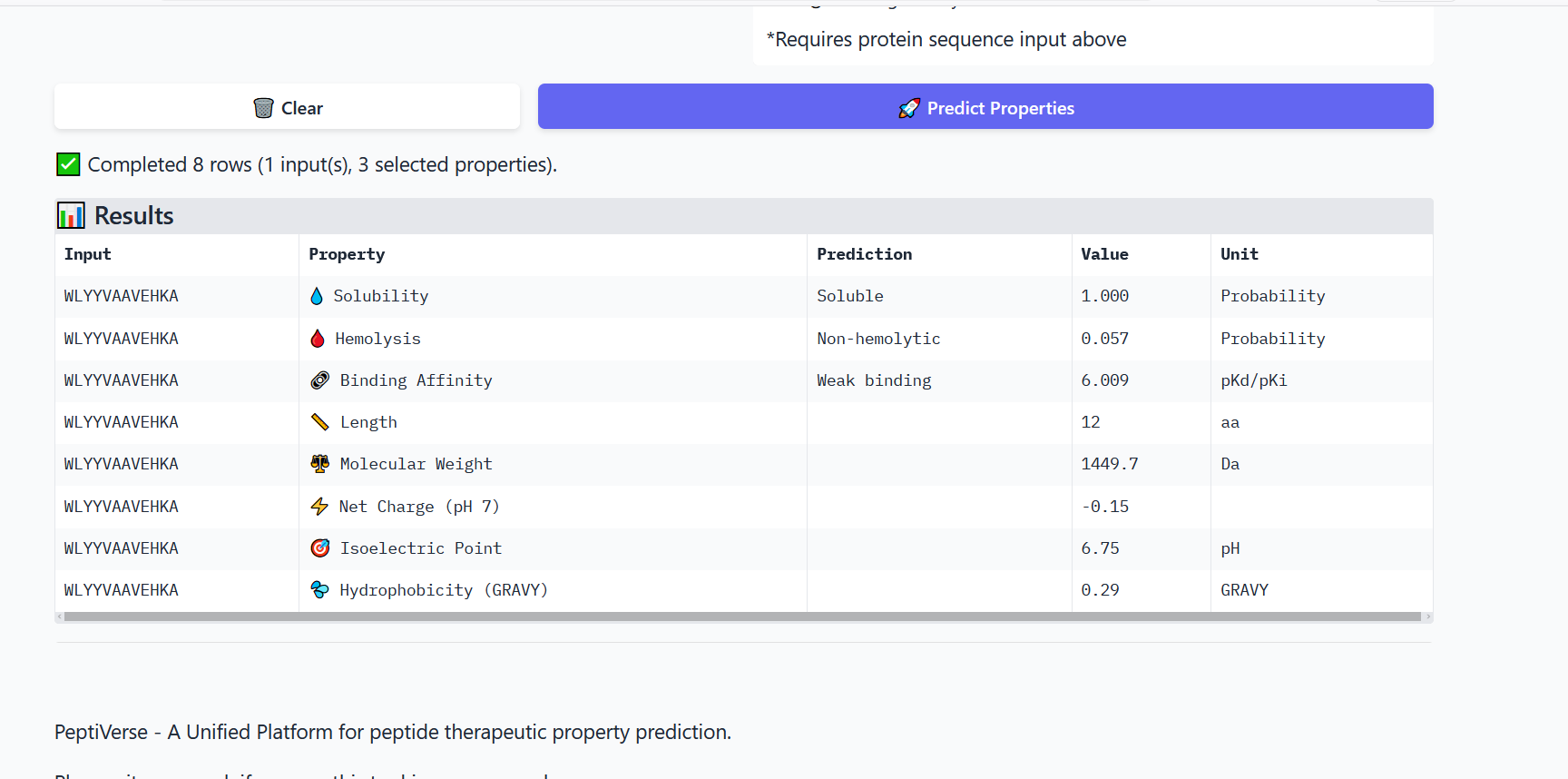
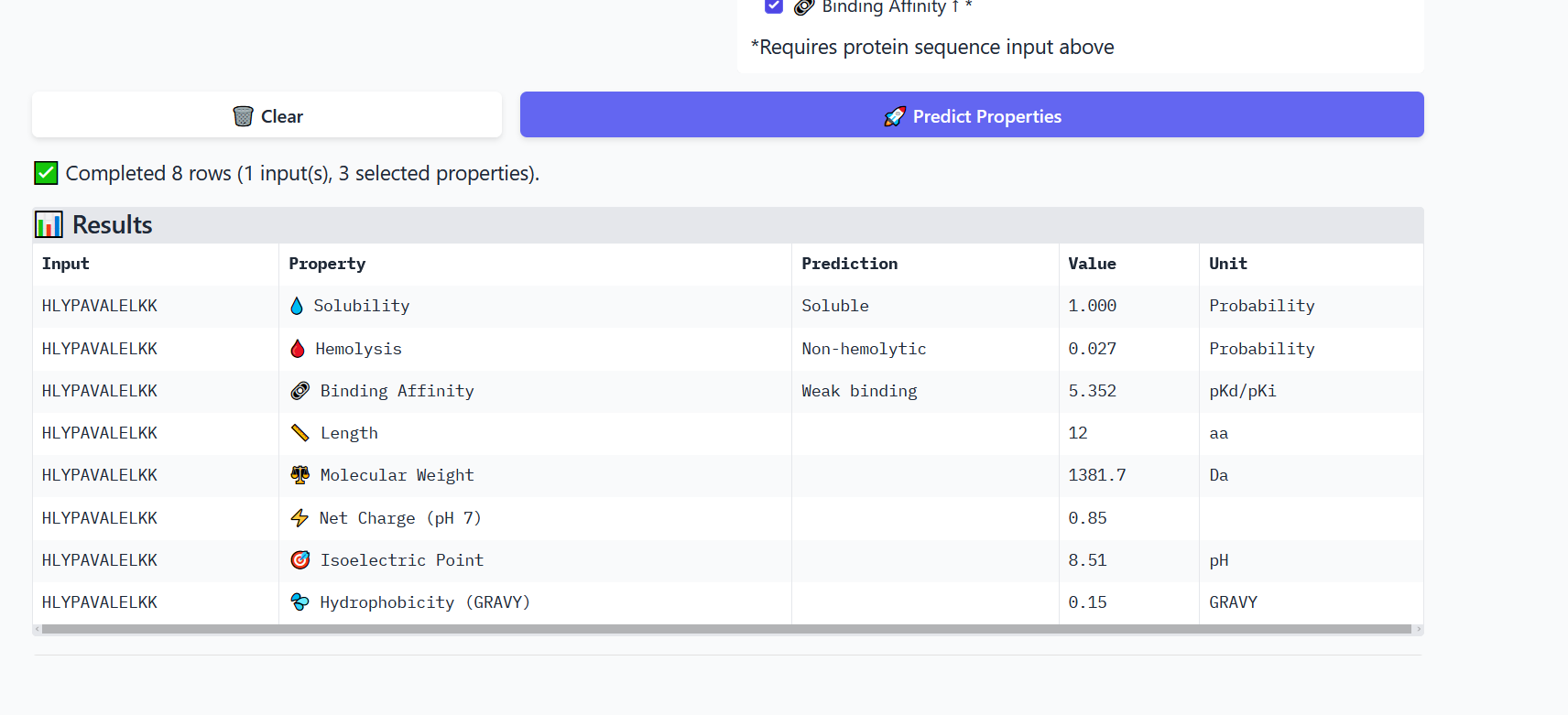
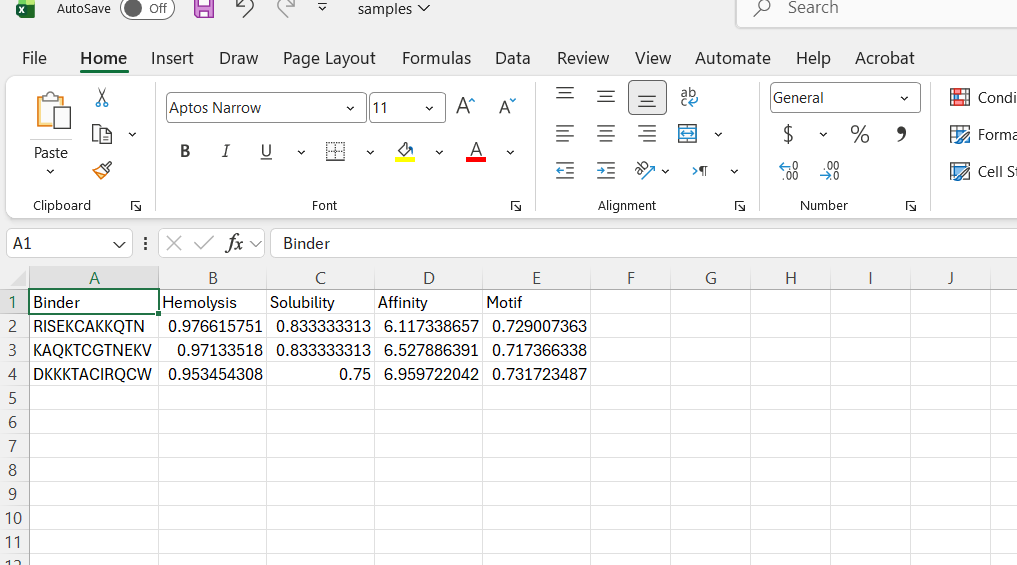
Perplexity reflects the model’s confidence in the generated peptide sequences. Lower perplexity values indicate that the sequence is more probable according to the language model. Among the generated candidates, WRPAVVVAHWE had the lowest perplexity (11.49), suggesting that it is the most confident peptide predicted by PepMLM.
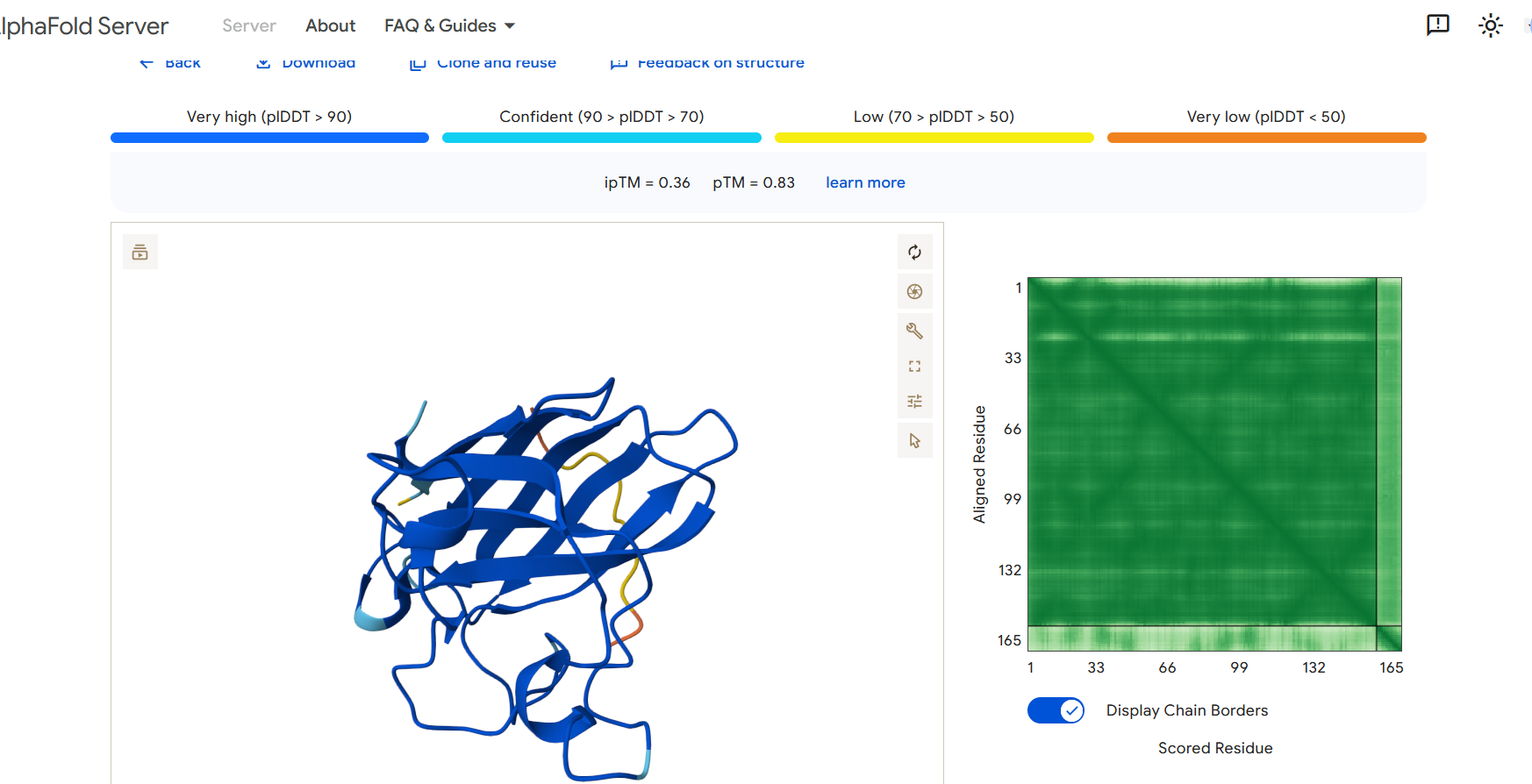
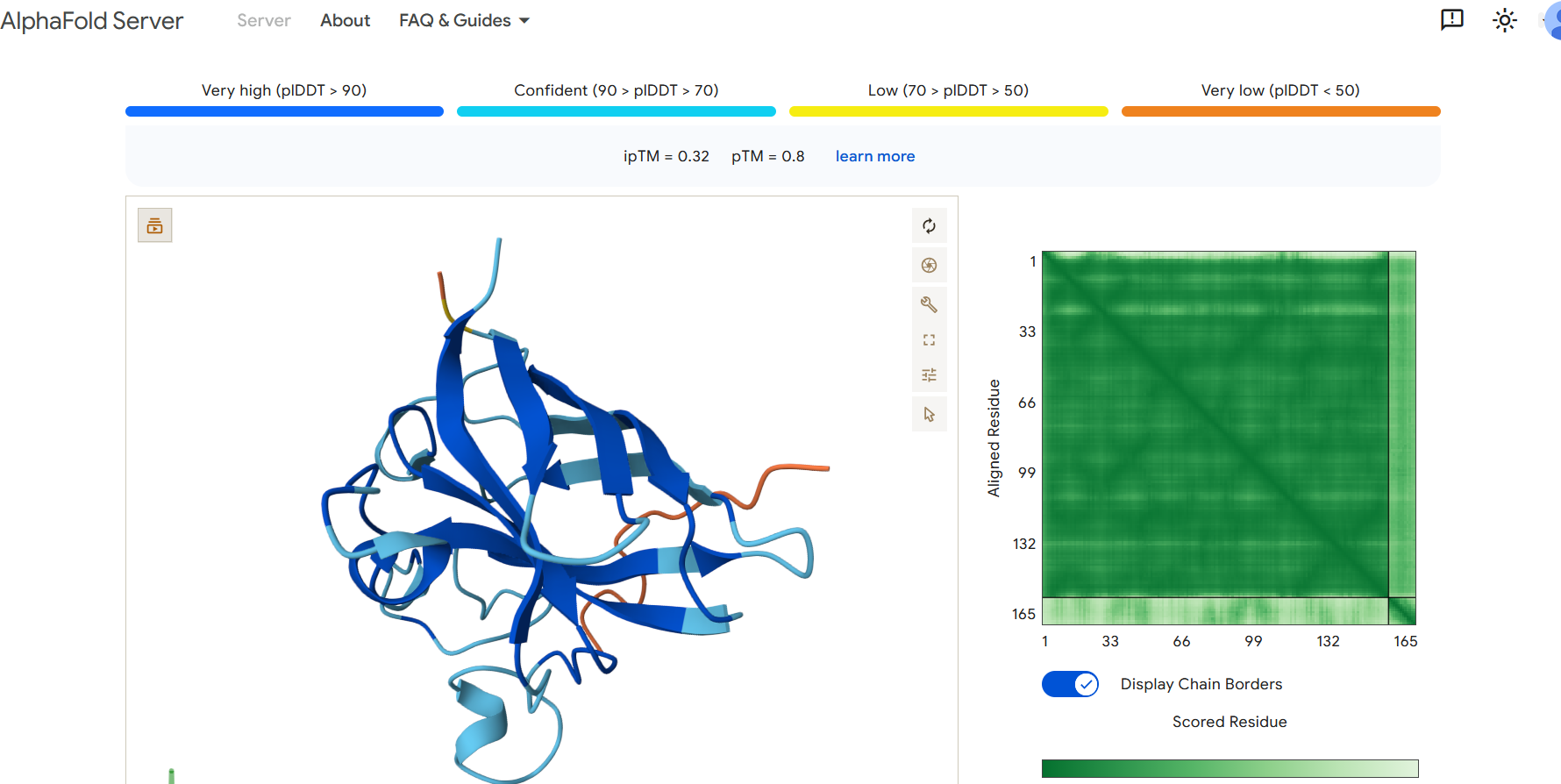
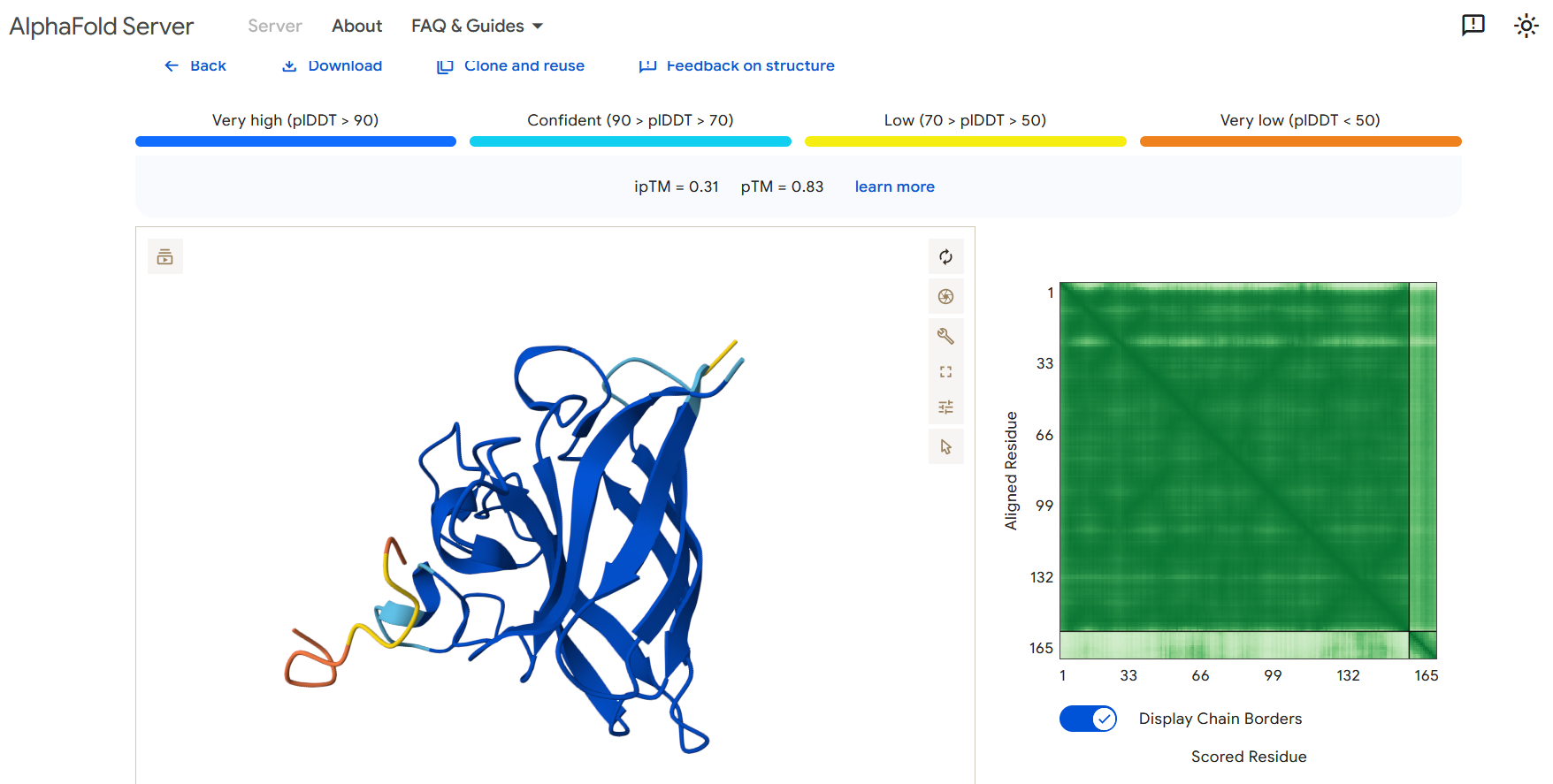
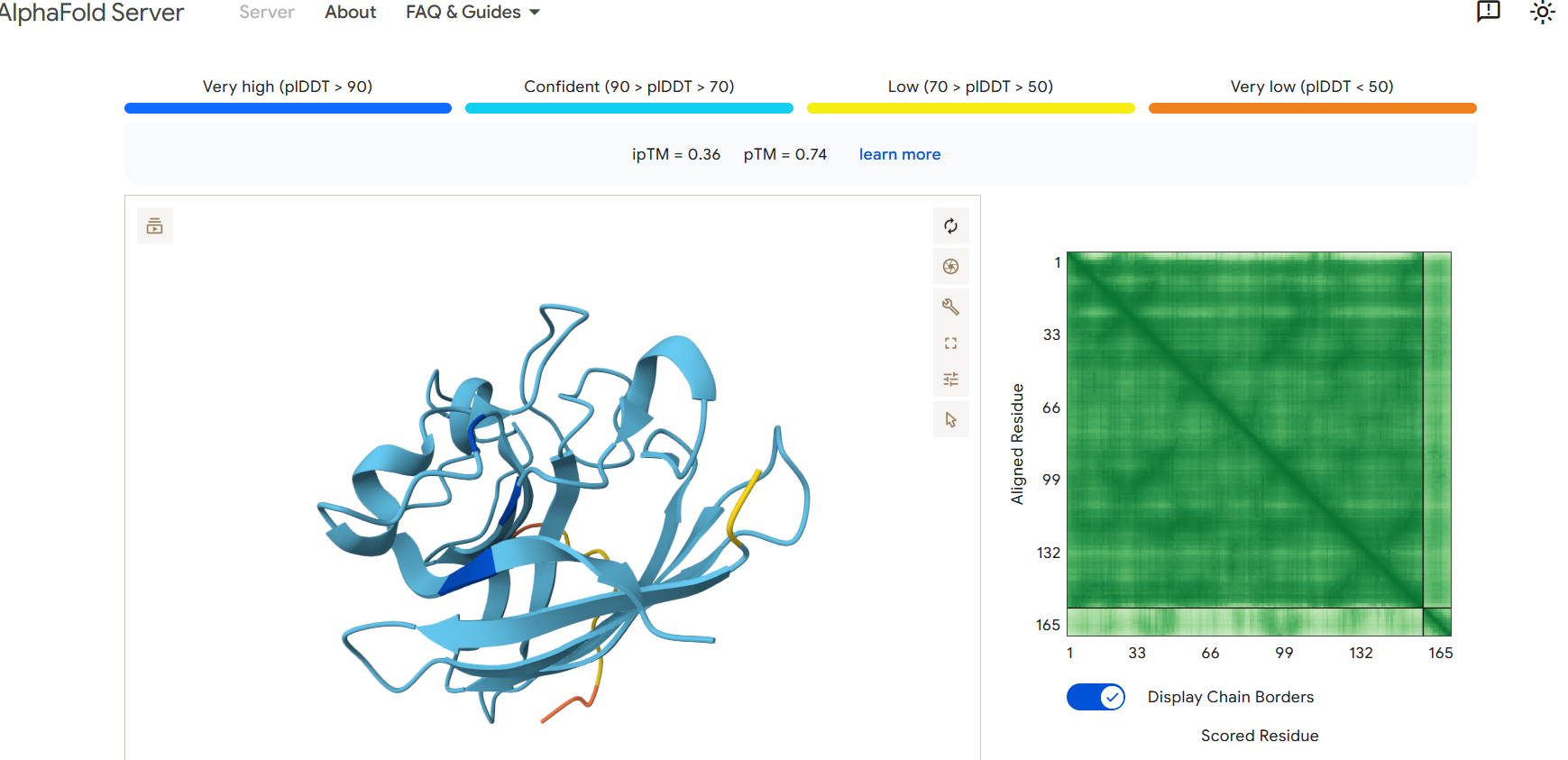
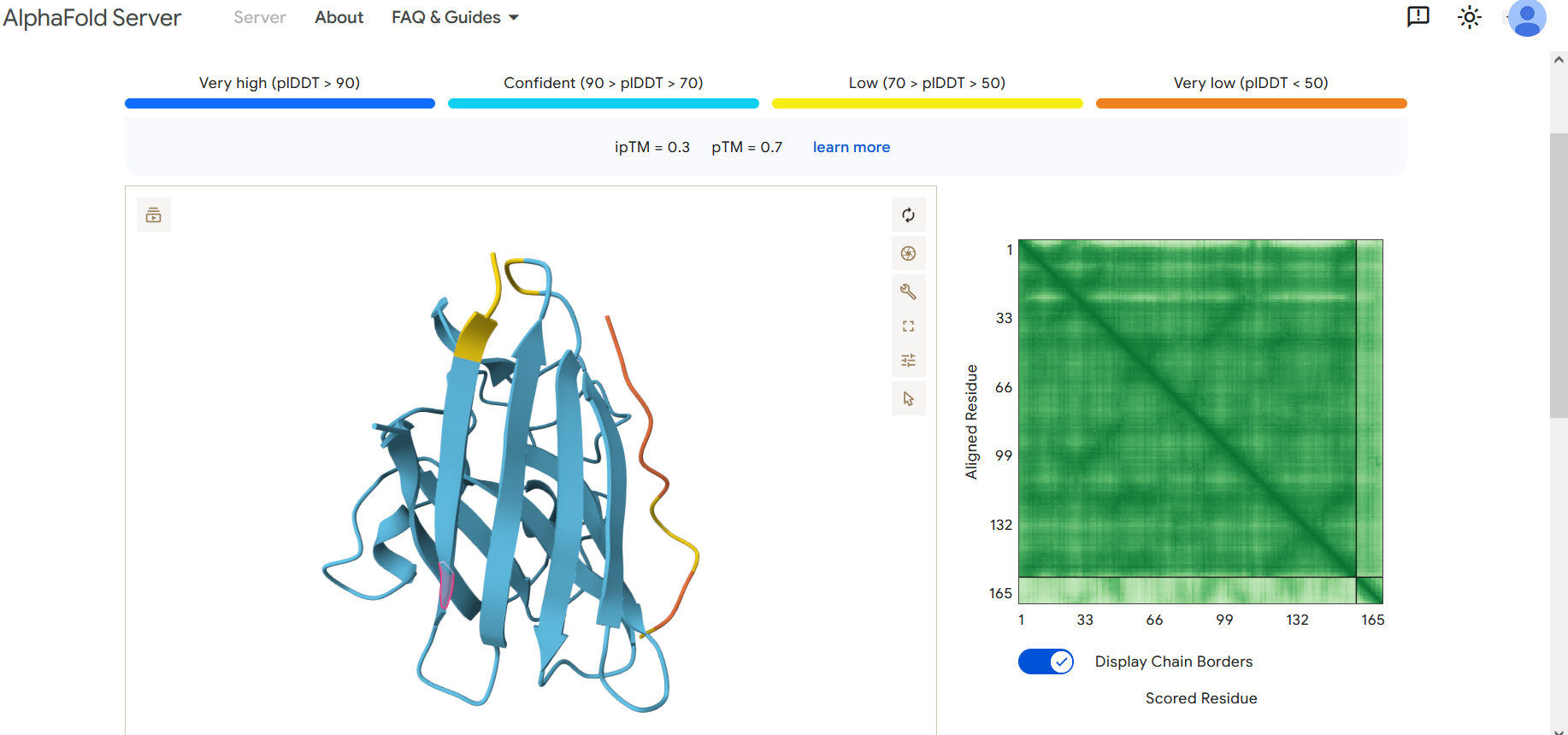
Part 2: Evaluate Binders with AlphaFold3
To evaluate the predicted binders, the mutant SOD1 sequence containing the A4V mutation was submitted to the AlphaFold server together with each peptide as separate chains to model the protein–peptide complex. The predicted interaction confidence was assessed using the ipTM score. The peptides generated showed ipTM values between about 0.30 and 0.36. When visualizing the structures, the peptides appear to bind on the surface of the SOD1 protein, mainly around the β-barrel region, and some approach the N-terminal area where the A4V mutation is located. The interactions appear surface-bound rather than buried inside the protein. The known SOD1-binding peptide produced an ipTM of about 0.31, while one PepMLM-generated peptide showed a slightly higher value (~0.36), suggesting a potentially stronger predicted interaction.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Annealing temperature depends mainly on primer Tm (melting temperature), GC content, and primer length. Tm is the temperature at which half of the DNA strands separate. Higher GC content increases Tm because GC bonds are stronger. If the temperature is too low, primers bind nonspecifically, and if it is too high, they fail to bind.
https://academic.oup.com/nar/article/18/21/6409/2388653?login=false
PCR and restriction digestion both generate linear DNA fragments, but they differ in approach. PCR amplifies a specific DNA region using primers and allows modification of sequences, such as adding overlaps or mutations. Restriction digestion uses enzymes to cut DNA at specific recognition sites, producing defined fragments without amplification.
PCR is more flexible and useful when designing constructs or preparing fragments for Gibson assembly. Restriction digestion is more straightforward but depends on the availability of suitable restriction sites in the DNA sequence.
To make sure the DNA fragments work for Gibson assembly, the main thing is that they have overlapping sequences, usually around 20–40 base pairs. These overlaps are not random, they need to match exactly between adjacent fragments so they can base-pair during the reaction. In practice, these overlaps are usually added through primer design during PCR.
Another important point is using a high-fidelity polymerase, because even small mutations in the overlap region can prevent proper assembly. It’s also a good idea to check fragment size on a gel and confirm sequence accuracy before moving forward. If the overlaps are not correct or the DNA quality is poor, the assembly simply won’t work, since Gibson relies entirely on sequence homology rather than restriction sites.
https://www.nature.com/articles/nmeth.1318
From what I understood in the protocol, the plasmid doesn’t enter the cells through any active mechanism. The cells are first made competent using calcium chloride, which helps reduce the repulsion between the DNA and the membrane. Then during heat shock, the sudden temperature change kind of destabilizes the membrane and creates temporary openings, so the DNA can slip inside.
So it’s really more of a physical process rather than something controlled by the cell. After that, the cells recover and start expressing the plasmid. This also explains why transformation efficiency depends a lot on how well the cells were prepared.
https://www.sciencedirect.com/science/article/abs/pii/S0022283683802848?via%3Dihub
Golden Gate Assembly is different from Gibson because it depends on restriction enzymes instead of sequence homology. It uses Type IIS enzymes like BsaI, which cut outside of their recognition site. This is useful because it lets you design specific overhangs, so different fragments can attach to each other in a defined order.
What I found interesting is that digestion and ligation happen in the same reaction, so the system basically cycles between cutting and joining until the correct construct forms. Also, the recognition sites are removed during the process, so the final DNA doesn’t contain extra unwanted sequences.
This makes it especially useful when assembling multiple fragments at once, since you can control how everything connects.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0003647
Week 07 – Genetic Circuits Part II
Part 1: Intracellular Artificial Neural Networks
Q.1. Traditional genetic circuits mostly behave like Boolean logic, meaning everything is either ON or OFF. That works for simple designs like AND or OR gates, but it doesn’t really match what actually happens inside cells.
In real biology, nothing is strictly binary. Gene expression can be low, medium, or high, and signals are noisy and constantly changing. So forcing everything into ON/OFF makes the system too limited.
IANNs are different because they allow the cell to deal with inputs more flexibly. Instead of just switching ON or OFF, the system can combine multiple inputs with different strengths. One input might have a stronger effect, another might weaken the output, and the final result depends on the overall combination.
Also, these systems can be layered, meaning one step produces something that regulates the next step. This makes the behavior more complex and closer to how real biological systems actually work.
So overall, IANNs are more useful because they allow continuous, tunable, and more realistic decision-making inside cells, instead of forcing everything into simple logic gates.
Q.2. If I think about where an IANN is actually useful, it’s when one signal is not enough and you need some kind of decision inside the cell, not just detection.
A simple example would be something like stress sensing. Cortisol alone is not reliable, and the same goes for any single marker. So instead of building one sensor per molecule, the idea is to combine multiple inputs and only produce an output when the combination actually makes sense biologically.
From the kind of systems shown in work like CRISPR gene regulation (Nissim et al., 2017) and RNA-based control systems (Green et al.), we already know cells can integrate multiple inputs at the gene expression level.
Part 2: Fungal Materials
Q.1. Fungal materials, particularly mycelium-based composites, are already being used in several applications. Studies such as those on mycelium materials show that these materials can be used for packaging, leather alternatives, insulation panels, and lightweight structural components. In these systems, fungal mycelium grows through agricultural waste and binds it together into a solid material, meaning the material is grown rather than manufactured.
The main advantages of fungal materials compared to traditional materials are related to sustainability. They are biodegradable, require low energy to produce, and can use waste as a substrate. They also have useful properties such as low density and good insulation. Because of this, they are considered strong alternatives to petroleum-based materials like plastics and foams.
However, they also have limitations. Their mechanical strength is generally lower than traditional construction materials, making them unsuitable for load-bearing applications. They are also sensitive to moisture, which can affect their stability and durability. In addition, their properties can vary depending on growth conditions, which makes standardization difficult. Production is also slower compared to conventional manufacturing processes.
Q.2. Genetically engineering fungi offers an opportunity to move beyond passive materials and create systems with controlled or responsive behavior. Instead of only improving growth, fungi could be engineered to have enhanced material properties, such as increased strength or reduced water absorption. More importantly, they could be engineered to respond to environmental signals, for example by changing color, producing a detectable signal, or altering their structure under certain conditions.
This opens the possibility of combining material formation with sensing and decision-making, especially if integrated with systems similar to IANNs. In this case, fungal materials would not only exist as structures but could also act as responsive systems that process inputs and produce outputs.
Compared to bacteria, fungi offer several advantages for this type of application. While bacteria are easier to engineer and grow faster, fungi naturally form complex three-dimensional structures and are better suited for material-based applications. They can grow on low-cost substrates and are capable of producing and secreting complex molecules. This makes them more suitable for applications where physical structure and environmental interaction are important.
However, bacteria remain preferable for simpler and faster genetic systems due to their ease of manipulation and well-established tools. Therefore, the choice between fungi and bacteria depends on the application: bacteria are more suitable for controlled molecular systems, while fungi are more suitable for structural and material-based systems.
Refrences:
Nissim, L., Perli, S. D., Fridkin, A., Perez-Pinera, P., & Lu, T. K. (2014).
Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells.
Molecular Cell, 54(4), 698–710.
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., & Lim, W. A. (2013).
Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression.
Nature Biotechnology, 31(3), 233–239.
Green, A. A., Silver, P. A., Collins, J. J., & Yin, P. (2014).
Toehold switches: De novo-designed regulators of gene expression.
Cell, 159(4), 925–939.
Jones, M., Bhat, T., Huynh, T., Kandare, E., Yuen, R., Wang, C. H., & John, S. (2020).
Engineered mycelium composite construction materials: A review.
Fungal Biology Reviews, 34(4), 162–173.
Appels, F. V. W., Camere, S., Montalti, M., Karana, E., Jansen, K. M. B., Dijksterhuis, J., Krijgsheld, P., & Wösten, H. A. B. (2019).
Fabrication factors influencing mechanical, moisture- and water-related properties of mycelium-based composites.
Scientific Reports, 9, 1–11.
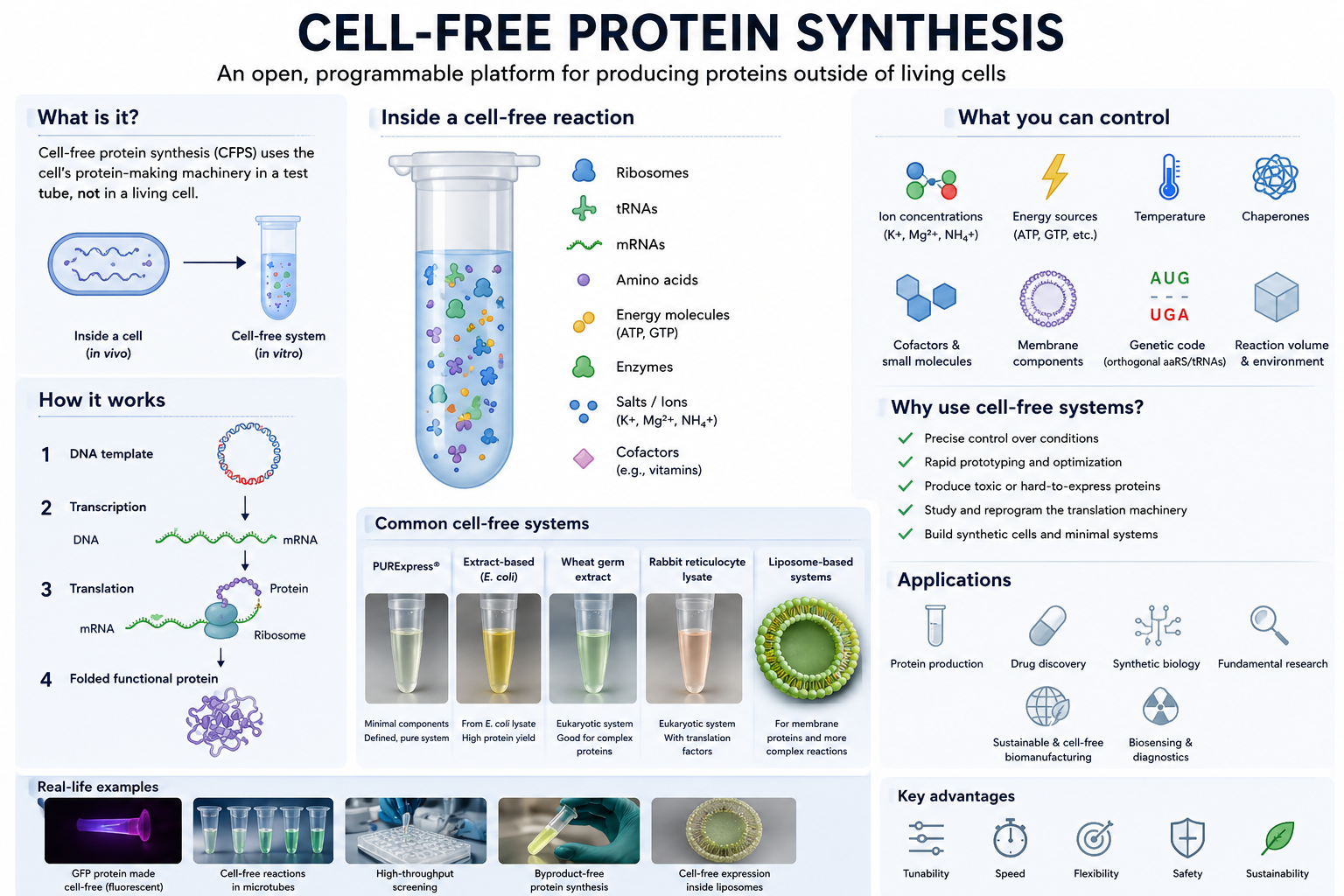
Week 09 – Cell-Free Systems
Part A: General and Lecturer-Specific Questions
Q.1. One of the main advantages of cell-free protein synthesis over traditional in vivo methods is the flexibility and level of control over the system. In normal in vivo systems, cells are still trying to survive, grow, divide, regulate metabolism, and maintain their own functions. Because of that, researchers are working with the cell’s own priorities and limitations.
In a cell-free setup, that limitation becomes much smaller because the system is open. Researchers can directly adjust variables like ion concentrations, energy sources, cofactors, temperature, chaperones, membrane components, and even parts of the genetic code itself without worrying about keeping a cell alive. I think this is where the engineering side of synthetic biology becomes very clear because instead of only observing biology, we can start designing and tuning the system for a specific purpose.
Cell-free expression is also useful in situations where living cells usually struggle. One example is toxic proteins because these proteins can harm or kill host cells during production. Another example is membrane proteins, which are often difficult because they can misfold or aggregate inside living cells. In cell-free systems, membrane-like environments such as liposomes, detergents, or nanodiscs can be added directly during synthesis to help improve folding and function.
Q.2. The most important component is the cell extract or lysate because it contains the core machinery needed for protein synthesis, including ribosomes, tRNAs, translation factors, enzymes, and other cellular components.
The system also needs a DNA or mRNA template containing the gene of interest. This acts as the instruction set that tells the system which protein should be produced.
Amino acids are another essential component because they serve as the building blocks used to assemble the protein.
An energy source is also required because protein synthesis consumes a large amount of ATP and GTP. Energy regeneration systems help maintain the reaction so protein production can continue for a longer period of time.
Salts and buffers are important because they maintain the correct chemical environment. Ribosomes and enzymes are sensitive to factors such as pH and ion concentrations, particularly magnesium levels, so these conditions need to remain stable.
Additional components can also be added depending on the goal of the experiment. For example, liposomes or detergents can help support membrane protein production, chaperones can assist with protein folding, cofactors can improve protein activity, and non-natural amino acids can introduce new properties into the final protein.
Q.3. Energy regeneration is critical because protein synthesis requires a continuous energy supply. Processes such as transcription, translation, and amino acid charging continuously consume ATP and GTP during the reaction. If ATP becomes depleted, protein production will stop even if all the other components are still present.
Unlike living cells, cell-free systems do not naturally sustain long-term metabolic activity unless the reaction is intentionally designed to support it. That is why energy regeneration becomes an important part of the system rather than just an additional component.
One commonly used strategy is phosphoenolpyruvate (PEP), which acts as a phosphate donor and regenerates ATP from ADP to keep the reaction running. Other systems may also use energy substrates such as glucose or maltodextrin because enzymes already present in the extract can process them gradually, allowing a more sustained energy supply during longer reactions.
Q.4. Prokaryotic systems, especially E. coli-based systems, are usually faster, cheaper, and easier to optimize. They can produce high protein yields relatively quickly, which makes them useful for rapid prototyping and for proteins that do not require complex processing.
I would use an E. coli system to produce GFP because it generally folds well in bacterial systems and does not require complicated post-translational modifications. It is also easy to monitor since fluorescence can quickly show whether the protein is being expressed successfully.
Eukaryotic systems are more complex, but they are often better for proteins that need proper folding, disulfide bond formation, chaperone support, or post-translational modifications such as glycosylation.
For a eukaryotic system, I would produce erythropoietin (EPO) because it is a human glycoprotein that depends on proper glycosylation for normal biological function. Since bacterial systems cannot perform the same type of processing as mammalian systems, a eukaryotic environment would be more suitable.
Overall, I would choose the system based on what the protein actually needs rather than automatically choosing the more complex option.
Q.5. Membrane proteins are difficult mainly because of their hydrophobic regions. Once they are outside a membrane environment, they can misfold or aggregate pretty easily.
If I were designing a cell-free experiment for a membrane protein, I would make sure the membrane-like environment is already present during protein synthesis instead of trying to add it later.
I would probably test different conditions using:
• liposomes
• detergents
• nanodiscs
• synthetic membrane systems
Nanodiscs seem especially interesting because they provide a stable lipid bilayer environment while still remaining relatively controlled experimentally.
One challenge would definitely be aggregation, so I would optimize factors such as:
Another issue would be proper insertion and folding of the protein into the membrane environment. Chaperones or slower translation conditions could help give the protein more time to fold correctly.
Q.6. It could be poor DNA quality or an inefficient template design. I would first check the DNA integrity and then test different construct designs or template concentrations.
Or it could be issues with the lysate itself. Since the lysate contains the machinery needed for protein production, including ribosomes, enzymes, and translation factors, poor extract quality or non-optimal reaction conditions could affect protein yield. I would probably test a fresh lysate preparation and optimize factors such as magnesium or salt concentrations.
A third possibility is protein aggregation or incorrect folding, especially for membrane proteins or larger proteins. In that case, I would try lower temperatures, add chaperones, or include membrane mimetics such as nanodiscs or liposomes to improve protein stability and folding.
Part B: Homework question from Kate Adamala
A synthetic minimal cell would function as a stress sensor for biological soil crust environments. Since biocrust organisms experience environmental stress from drought and increasing salinity, the system would detect osmotic stress conditions before the soil ecosystem becomes severely damaged.
Input: High salt concentration / osmotic stress signal
Output of SMC: GFP fluorescence signal
The idea is that when osmotic stress increases, the synthetic cell activates a stress-responsive system and produces a measurable fluorescent signal.
B. Partially yes. Cell-free Tx/Tl could generate the sensing reaction, but encapsulation would help organize the system into a defined compartment and better mimic environmental sensing conditions.
C. Yes. A bacterial cell could be engineered to detect osmotic stress. However, natural cells introduce their own metabolism and stress responses, which may affect the signal. Using a synthetic minimal cell would provide more direct control over the system.
I would expose the synthetic minimal cells to different salt concentrations and measure GFP fluorescence.
Conditions:
• low salt concentration (control)
• moderate salt concentration
• high salt concentration
If the system works correctly, GFP expression should increase when osmotic conditions activate the sensing system. I would compare the fluorescence intensity across the different conditions to determine whether the synthetic cell responds to environmental changes.
Adamala et al. (2017) – Engineering genetic circuit interactions within and between synthetic minimal cells
Adamala, K. P., Martin-Alarcon, D. A., Guthrie-Honea, K. R., & Boyden, E. S. Nature Chemistry, 9(5), 431–439.
Supports: synthetic minimal cells, liposome encapsulation, genetic circuits.
Mellies et al. (1994) – The Escherichia coli proU promoter element and its contribution to osmotically signaled transcription activation
Mellies, J., Bremer, E., & Villarejo, M. Journal of Bacteriology, 176(12), 3638–3645.
Supports: osmotic-response system (proU promoter) used in your sensing design.
Hilburger et al. (2019) – Controlling Secretion in Artificial Cells with a Membrane AND Gate
Hilburger, C. E., Jacobs, M. L., Lewis, K. R., Peruzzi, J. A., & Kamat, N. P. ACS Synthetic Biology, 8(6), 1224–1230.
Supports: α-hemolysin pores and membrane communication in artificial cells.
Shin & Noireaux (2012) – An E. coli cell-free expression toolbox: Application to synthetic gene circuits and artificial cells
Shin, J., & Noireaux, V. ACS Synthetic Biology, 1(1), 29–41.
Supports: bacterial cell-free transcription/translation systems.
Guo et al. (2017) – Insights on osmotic tolerance mechanisms in Escherichia coli gained from omics studies
Guo, Y., Winkler, J., & Kao, K. C. Biotechnology for Biofuels, 10, 38.
Supports: osmotic stress biology and bacterial responses to salt changes.
Homework question from Peter Nguyen
I trust that a wearable bracelet or skin patch with freeze-dried cell-free biosensors built into the material that activate with moisture and detect environmental heavy metal exposure through a visible color change.
The bracelet or patch would contain small freeze-dried cell-free reaction spots embedded inside a hydrogel or flexible wearable material. Sweat or environmental moisture would naturally reactivate the biological reactions. Heavy metals from polluted air particles, contaminated water, or environmental exposure could then interact with the sensing system. Once activated, the biosensors could detect heavy metals such as lead, mercury, or cadmium and produce a visible color response.
Heavy metal exposure can happen gradually and people may not realize they are being exposed until symptoms start appearing later on. Long-term exposure can also contribute to oxidative damage and broader health effects. A wearable biosensing material could help provide easier real-time monitoring for people who work in polluted or higher-risk environments.
One limitation is that freeze-dried systems need hydration to become active. In this design, sweat or environmental moisture would naturally activate the system only when the bracelet or patch is being worn. Another limitation is that many cell-free reactions are still single-use. To help with this, the material could contain replaceable sensing layers or small compartments that activate gradually instead of all at once. Protective hydrogels or polymer coatings could also help improve stability and reduce damage from heat, oxidation, or UV exposure.
References
Pardee, K., et al. (2014). Paper-based synthetic gene networks. Cell, 159(4), 940–954. DOI:10.1016/j.cell.2014.10.004
Carlson, E. D., et al. (2012). Cell-Free Protein Synthesis: Applications Come of Age. Biotechnology Advances, 30(5), 1185–1194. DOI:10.1016/j.biotechadv.2011.09.016
Shin, J., & Noireaux, V. (2012). An E. coli cell-free expression toolbox: Application to synthetic gene circuits and artificial cells. ACS Synthetic Biology, 1(1), 29–41. DOI:10.1021/sb200016s
Heikenfeld, J., et al. (2018). Wearable sensors: modalities, challenges, and prospects. Lab on a Chip, 18(2), 217–248. DOI:10.1039/C7LC00914C
Homework question from Ally Huang
Background information
Long-duration space missions will likely depend on portable biotechnology systems such as freeze-dried cell-free reactions for diagnostics and on-demand protein production. One major challenge in space is exposure to solar and cosmic radiation, which can damage DNA and interfere with biological function. NASA has linked deep-space radiation exposure to increased risks of cancer, nervous system damage, degenerative diseases, and acute radiation sickness. Since BioBits systems rely on DNA templates to produce proteins, I became curious about whether DNA used in these systems would still function properly after radiation exposure. Since cosmic radiation is difficult to reproduce in a classroom setting, UV-B exposure will be used as an accessible model of DNA damage.
Molecular or genetic target
UV-B-exposed DNA templates encoding GFP and RFP fluorescent proteins used in the BioBits cell-free protein expression system.
Relationship of target to the space biology challenge
BioBits reactions use DNA templates as the instructions for transcription and translation. The freeze-dried pellets were described as containing the molecular machinery needed for protein production once DNA and water are added. If radiation damages the DNA template, the system may produce lower amounts of fluorescent protein. Measuring GFP and RFP fluorescence provides a simple way to test whether radiation-exposed DNA remains functional and whether different DNA templates show different sensitivity to radiation exposure.
Hypothesis
I trust that ncreasing UV-B exposure of DNA templates will reduce fluorescent protein production in BioBits reactions because damaged DNA may no longer provide accurate instructions for transcription and protein production. I also want to compare GFP and RFP templates to see whether different DNA templates show different sensitivity to UV-B exposure.
This question is important because future astronauts will rely on portable cell-free systems for diagnostics, research, or on-demand protein production during long-duration missions. If radiation affects the DNA used by these systems, it could influence how reliable they remain in space. Understanding how radiation changes DNA function could help improve future biological systems used during space missions.
Experimental plan
Identical GFP and RFP DNA templates would be exposed to different UV-B exposure levels before being added to separate BioBits reactions. Non-UV-exposed GFP and RFP templates would serve as positive controls, while no-DNA reactions would serve as negative controls. After hydration and incubation, fluorescence intensity would be measured using the P51 Molecular Fluorescence Viewer. If available, miniPCR could also be used to amplify DNA templates after exposure to compare whether UV-B treatment influences amplification performance. The main data collected would be fluorescence intensity across different UV-B conditions.
I would mainly want to see whether the redesigned GR-LBD protein is successfully produced and whether introducing the Q642K mutation improves cortisol selectivity compared with cortisone. Since the DNA construct was designed and ordered through Twist Bioscience, I would first verify the received construct sequence before moving into expression studies.
I would then express the construct in E. coli BL21(DE3) and purify the protein using affinity chromatography. SDS-PAGE gel electrophoresis would be used to check whether the expected protein band is present and whether expression was successful.
I would also compare cortisol versus cortisone binding using fluorescence-based ligand binding assays to determine whether the redesigned protein shows improved selectivity. Since the long-term goal is a saliva-based biosensor, I would also test the protein under saliva-like conditions to see whether proteins, enzymes, or salts interfere with signal performance.
The main technologies I would use include DNA sequencing verification, protein expression in E. coli BL21(DE3), affinity chromatography, SDS-PAGE gel electrophoresis, fluorescence-based ligand binding assays, and saliva-like testing.
Homework: Waters Part I — Molecular Weight
Theoretical Molecular Weight of eGFP
The theoretical molecular weight of eGFP was first calculated using the ExPASy Compute pI/Mw tool based on the provided amino acid sequence, including the LE linker and the 6× His purification tag.
The calculated molecular weight was: 28,006.60 Da
Since mature eGFP undergoes chromophore cyclization during folding, a small mass loss of approximately 20 Da was considered.
[28006.60 - 20 = 27986.60]
Therefore, the corrected theoretical molecular weight of mature eGFP is approximately:
27,986.60 Da
Two adjacent peaks were selected from Figure 1:
• 903.7148
• 933.8044
Using the adjacent charge state equation, the charge state was calculated to be:
z ≈ 30
Therefore:
• 933.8044 = 30+
• 903.7148 = 31+
Using the relationship between m/z, MW, and z, the experimental molecular weight of eGFP was calculated to be:
27,983.91 Da
The accuracy compared to the theoretical molecular weight (27,986.60 Da) was:
~0.01% error
Yes, the charge state can be roughly estimated from the zoomed-in peak because the isotope peaks are very closely spaced. Based on Lindsay’s explanation during the recitation that smaller isotope spacing corresponds to higher charge states, this peak appears to have a relatively high charge, approximately between 15+ and 20+
Using the ExPASy PeptideMass tool with trypsin and 0 missed cleavages, 19 peptides were generated under the selected settings (>500 Da displayed peptides).
Based on Figure 5a, I observed approximately 18 chromatographic peaks above 10% relative abundance between 0.5 and 6 minutes in the eGFP peptide map.
The number of major chromatographic peaks is close to the number of predicted peptides. However, if smaller low-abundance peaks are also counted, the chromatogram appears to contain more peaks than the 19 predicted peptides. This is likely due to background signals, partially overlapping peptides, noise, or additional low-intensity peptide fragments detected in the LC-MS data.
[(525.76712 \times 2) - 1.0073 = 1050.52]
Therefore, the singly charged peptide mass was calculated to be approximately 1050.52 Da.
The experimental mass from Figure 5b was approximately 1050.52 Da.
When I compared it with the expected peptide masses from PeptideMass, the closest match was:
FEGDTLVNR = 1050.5214 Da
So the peptide is: FEGDTLVNR
Mass error: (∣1050.5269-1050.5214∣)/1050.5214×10^6=5.2" ppm
Mass accuracy/error ≈ 5.2 ppm
Based on Figure 6, approximately 88% of the eGFP sequence was confirmed by peptide mapping.
Homework: Waters Part IV — Oligomers
I used the given KLH subunit masses and multiplied them by the number of subunits in each oligomer. Since a decamer contains 10 subunits, the didecamer, 3-decamer, and 4-decamer correspond to 20, 30, and 40 subunits.
7FU Decamer
10 × 340 kDa = 3400 kDa = 3.4 MDa
Matches the peak around 3.4 MDa.
8FU Didecamer
20 × 400 kDa = 8000 kDa = 8.0 MDa
Matches the peak around 8.33 MDa.
8FU 3-Decamer
30 × 400 kDa = 12000 kDa = 12.0 MDa
Matches the peak around 12.67 MDa.
8FU 4-Decamer
40 × 400 kDa = 16000 kDa = 16.0 MDa
Expected near 16 MDa on the spectrum.
Homework: Waters Part V — Did I make GFP?
Protein
Theoretical Molecular Weight (kDa)
Observed/Measured on Intact LC-MS (kDa)
PPM Mass Error
eGFP
27.9866
27.9839
~96
The observed molecular weight measured from the intact LC-MS analysis was very close to the theoretical molecular weight predicted from the eGFP sequence, suggesting accurate detection of the intact mature eGFP protein.
Week 11 – Building Genomes
For the collaborative bioart project, I contributed to several wells including K10, K11, K12, K13, K15, K16, L14, and L15. Most of my contributions appeared in the right-side region of the artwork.
What I liked most about this project was seeing how many people from different backgrounds worked together to create one large scientific artwork. It honestly felt chaotic at first, but super cool once the final pattern started forming. I also liked how science and art were combined together in a very interactive way.
One thing that could make the project even more fun next year is adding a little more guidance or visualization for first-time participants to better follow how the final image develops collectively over time. I really enjoyed the project overall, but at the beginning it took me some time to fully understand how everything was coming together. I wish I have participated more and on time!
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Provides the cellular machinery needed for protein synthesis, including ribosomes, enzymes, and T7 RNA polymerase for transcription. It essentially acts as the “engine” of the cell-free system.
Salts/Buffer
Potassium Glutamate
Helps maintain ionic conditions similar to the inside of a cell and supports efficient protein synthesis.
HEPES-KOH pH 7.5
Acts as a buffer to maintain a stable pH during the reaction since cell-free systems are highly pH sensitive.
Magnesium Glutamate
Provides magnesium ions required for ribosome activity, enzyme function, and transcription/translation.
Potassium phosphate monobasic
Helps maintain phosphate balance and contributes to buffering capacity in the reaction.
Potassium phosphate dibasic
Works together with monobasic phosphate to stabilize pH and maintain proper reaction conditions
Energy / Nucleotide System
Ribose
Serves as a precursor for nucleotide synthesis in the NMP-ribose system.
Glucose
Provides an energy source that helps sustain protein production for longer periods.
AMP
Acts as a nucleotide precursor that can eventually be converted into ATP inside the lysate.
CMP
Serves as a precursor for CTP synthesis during transcription.
GMP
Acts as a precursor for GTP production used in RNA synthesis.
UMP
Serves as a precursor for UTP production during transcription.
Guanine
Can be enzymatically converted into GMP and later into GTP within the lysate system.
Translation Mix (Amino Acids)
17 Amino Acid Mix
Provides most of the amino acids required for protein translation.
Tyrosine
Added separately because it can become limiting during protein synthesis reactions.
Cysteine
Added separately since it is relatively unstable and easily oxidized in solution.
Additives
Nicotinamide
Supports metabolic enzyme activity and helps improve reaction stability and protein production.
Backfill
Nuclease Free Water
Used to adjust the final reaction volume without introducing nucleases that could damage nucleic acids.
Based on the HTGAA recitation discussion, the 1-hour PEP-NTP system directly supplies nucleotide triphosphates (NTPs) and phosphoenolpyruvate (PEP), allowing rapid protein production but at a higher cost and for a shorter duration. In contrast, the 20-hour NMP-ribose-glucose system uses simpler precursor molecules such as NMPs, ribose, and glucose, which support longer and more sustainable protein production through enzymatic regeneration inside the lysate.
Bonus question: Transcription can still occur because guanine can be enzymatically converted into GMP inside the lysate. The GMP can then be further phosphorylated into GTP, which is required for RNA synthesis during transcription.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
sfGFP is known for very efficient folding, which makes it reliable in cell-free protein expression systems. Its fluorescence still depends on proper chromophore maturation, which requires oxygen and enough incubation time.
mRFP1 is an older red fluorescent protein and is generally less bright than newer red variants. Slower maturation and folding efficiency can affect the strength of its final fluorescence signal.
mKO2 produces a bright orange fluorescence, but its signal can be sensitive to pH changes. Because of this, maintaining stable reaction conditions may help preserve fluorescence during longer incubations.
mTurquoise2 is a cyan fluorescent protein with strong brightness and relatively good stability. Its lower acid sensitivity makes it useful for longer cell-free reactions where conditions may gradually change over time.
mScarlet-I is a newer red fluorescent protein with improved brightness and maturation compared with older red fluorescent proteins. Proper folding and chromophore maturation are still important for achieving strong fluorescence output.
Electra2 is a blue fluorescent protein, and blue fluorescent signals are sometimes harder to detect clearly compared with green or red proteins. Stable reaction conditions and efficient folding may help improve its fluorescence readout.
These descriptions were based on the HTGAA cell-free recitation discussions, the fluorescent protein information provided in the Benchling folder, and general fluorescent protein properties related to folding, maturation, pH sensitivity, and oxygen-dependent chromophore formation.
I would focus on sfGFP because its fluorescence depends on efficient folding and proper chromophore maturation over time. Based on the discussion about longer incubations and sustaining cell-free reactions, my hypothesis is that supplementing the reaction with additional amino acids and improving the glucose/ribose energy-support system could help maintain protein production during the 36-hour incubation, leading to stronger and more stable green fluorescence.
For the second phase, I would use the Cell-Free Optimization Interface to test small changes based on my sfGFP hypothesis. I would mainly focus on amino acid supplementation and improving the glucose/ribose energy-support system to see if it helps maintain stronger green fluorescence during the longer incubation.
For the final part, I am still waiting for the email and assigned instructions so it is TBD.
Design of a Glucocorticoid Receptor (GR-LBD)-Based Biosensor for Cortisol Detection Section1:Abstract Cortisol is one of the main hormones we look at when studying stress, circadian rhythm, and endocrine function, and it can be measured easily from saliva. It plays an important role in regulating multiple systems in the body, including metabolism, immune response, and cardiovascular function, which makes accurate measurement of cortisol important for understanding physiological stress. The problem is that most current detection systems rely on antibodies, which are not always stable, can be expensive, and do not perform well in complex environments like saliva. On top of that, cortisol is very similar in structure to cortisone, which makes it difficult to distinguish between the two and leads to noise and cross-reactivity in many biosensors.
From Brainstorming to Project Direction During our first group brainstorming session, We kept talking about the L protein stability, how the MS2 L protein interacts with the bacterial chaperone DnaJ, whether specific amino acid changes affect lysis behavior, and how modifying certain regions could potentially improve protein function. We also discussed improving lysis efficiency and helping MS2 overcome possible host resistance.
Subsections of Projects
Individual Final Project
Design of a Glucocorticoid Receptor (GR-LBD)-Based Biosensor for Cortisol Detection
Section1:Abstract
Cortisol is one of the main hormones we look at when studying stress, circadian rhythm, and endocrine function, and it can be measured easily from saliva. It plays an important role in regulating multiple systems in the body, including metabolism, immune response, and cardiovascular function, which makes accurate measurement of cortisol important for understanding physiological stress. The problem is that most current detection systems rely on antibodies, which are not always stable, can be expensive, and do not perform well in complex environments like saliva. On top of that, cortisol is very similar in structure to cortisone, which makes it difficult to distinguish between the two and leads to noise and cross-reactivity in many biosensors.
The goal of this project is to design a protein-based system using the ligand-binding domain of the human glucocorticoid receptor (GR-LBD, NR3C1), which naturally binds the ligand cortisol. Instead of designing a completely new protein, the idea is to start from a real biological system and improve it. The hypothesis is that a targeted mutation in the GR-LBD, combined with a dual-layer system design, will improve selectivity for cortisol over cortisone.
To achieve this, the project involves designing a codon-optimized DNA sequence encoding the GR-LBD for expression in E. coli, introducing a single point mutation (Q→K) based on literature showing that mutations in this domain can change receptor behavior without completely disrupting ligand binding, and evaluating the design using computational tools such as AlphaFold and ESMFold to assess structural stability and compare the wild-type and mutated versions.
In addition, the project includes a proposed dual approach, where a simple “scavenger” step could help remove interfering molecules before detection.
Overall, this project is trying to see whether a slightly engineered natural receptor can be a more stable and selective alternative to antibody-based cortisol sensors in saliva.
Section2
Aim 1: Experimental Aim
The first aim of my project is to design a modified glucocorticoid receptor ligand-binding domain by introducing a glutamine to lysine mutation and evaluating its structural impact. A codon-optimized DNA sequence was generated for expression in Escherichia coli and submitted for synthesis. Structural prediction tools such as AlphaFold and Evolutionary Scale Modeling Fold will be used to analyze changes in the binding region. The goal is to assess whether this mutation could influence selectivity toward cortisol over cortisone based on structural behavior.
Aim 2: Development Aim
The second aim of this project is to move from computational design to experimental validation by expressing and testing the engineered glucocorticoid receptor ligand-binding domain protein in a laboratory setting at the Designer Cells node. The protein will be expressed in Escherichia coli BL21(DE3), purified using an appropriate chromatography method, and tested using a fluorescence-based assay to measure its interaction with cortisol. Cortisone will also be tested to evaluate selectivity.
Additional testing will be performed under saliva-like conditions to assess how the protein behaves in a more realistic environment. The goal of this aim is to determine whether the mutation has a measurable effect on ligand selectivity and to identify any limitations related to protein expression, folding, or stability.
Aim 3: Visionary Aim
The third aim of this project is to explore how the engineered glucocorticoid receptor ligand-binding domain protein could be used in a real-time, non-invasive cortisol sensing system. If the protein shows sufficient selectivity and stability, it could be linked to a fluorescence-based signal to convert binding into a measurable output.
The long-term goal is to enable continuous monitoring of cortisol in biological samples such as saliva. This would address current limitations in cortisol detection, which are not well suited for real-time use in complex environments. By modifying a natural receptor instead of designing a fully synthetic system, this approach could provide a more stable and adaptable platform for biosensing. In the future, this could support the development of portable or wearable systems for stress monitoring.
Section3: Background
Recent work from the Baker Lab shows that protein biosensors can be designed computationally to respond to specific targets and generate measurable outputs such as fluorescence or luminescence. These systems are powerful and flexible, especially for detecting larger targets like proteins. However, their application to small molecules like cortisol is still limited, mainly because it is difficult to achieve high selectivity when molecules are structurally very similar. In addition, these systems often lose stability when moved from controlled laboratory conditions to complex environments such as saliva, where non-specific interactions can interfere with detection.
On the other hand, studies on the glucocorticoid receptor ligand-binding domain (GR-LBD) show that it naturally binds cortisol with high affinity, making it a strong starting point for biosensing. However, the receptor is structurally flexible and can also bind similar molecules like cortisone, which explains the issue of cross-reactivity. Research has shown that small mutations in this domain can modify receptor behavior without completely disrupting binding, suggesting that selectivity can be improved through targeted changes.
Despite this, the GR-LBD alone is not optimized for use in complex biological environments, highlighting a gap between strong natural binding and the level of specificity needed for reliable biosensing.
This project looks at measuring cortisol to better understand stress, but that also raises some concerns about how this kind of biological data is used. If the measurements are not fully accurate, especially in saliva, people might misunderstand their stress levels and make decisions based on incomplete information. Stress is also more complex than a single hormone, so relying only on cortisol could be misleading. Because of this, it is important to follow the principle of non-maleficence, meaning we should avoid causing harm by overinterpreting the results. At the same time, responsibility matters in how the data is explained, and justice is important if this type of technology becomes more widely used, so it is not limited to only certain groups.
To handle these issues, the project will focus on clearly defining the limits of the system and carefully validating its performance in real samples like saliva. The results should be presented in a way that makes it clear that this is not a full measure of stress, but only one indicator. One possible issue is that people may assume the measurement fully reflects their stress level, which is not always true. There is also uncertainty in the design itself, since modifying the protein may not completely fix problems like cross-reactivity or stability. As an alternative, cortisol measurements could be used together with other indicators instead of on their own. This is relevant to public health because improving how stress is monitored could support earlier awareness and help reduce long-term health effects.
Section4: EXPERIMENTAL DESIGN
The DNA construct encoding the mutated glucocorticoid receptor ligand-binding domain, where glutamine is replaced with lysine, will be submitted to Twist Bioscience for synthesis and cloning into the pTwist Chlor high-copy vector. Since I am a committed listener, the construct will be delivered to my node, where the experimental work will be carried out. This is expected to take around two weeks, although delays may occur depending on node logistics.
Once plasmid is received, the sequence will be verified to confirm that it matches the design, which usually takes one to two days. The plasmid will then be introduced into Escherichia coli BL21(DE3), and within two days successful colonies should show that the construct is stable. The bacteria will then be grown in liquid culture, and over the next three days protein expression will be induced using isopropyl beta-D-1-thiogalactopyranoside, with the expectation that the target protein will be produced. The cells should be harvested and lysed within two days, resulting in a crude protein extract.
The protein will be purified using a chromatography-based method within few days, depending on the construct behavior, to reduce contamination from other proteins. The purified sample will then be analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis, which takes about one to two days, where a band at the expected size would confirm protein identity and purity.
A fluorescence-based binding assay will be used during several days to test interaction with cortisol, where successful binding should produce a measurable signal. The same assay will be performed with cortisone, where a lower signal would suggest improved selectivity. Since a wild-type construct is not included, the results will be interpreted based on existing literature.
Binding experiments will also be repeated under saliva-like conditions over a few additional days to evaluate performance in a more complex environment. Some signal reduction may occur due to interference, but detectable binding would indicate stability. Repeating the measurements will help assess consistency.
In addition, structural prediction tools such as AlphaFold and Evolutionary Scale Modeling Fold will be used over two days to examine changes in the binding region. These results will help explain experimental observations.
Overall, the aim is to determine whether the mutation improves selectivity toward cortisol while maintaining stability under realistic conditions. Any limitations, such as weak signal or instability, will be noted and documented well for future improvement.
DNA Gel Art
☐ DNA Sequencing
☐ DNA Editing
☑ DNA Construct Design
☐ Restriction Enzyme Digestion
☑ Gel Electrophoresis
☐ DNA Purification From Gel
☑ Databases (e.g., GenBank, NCBI, Ensembl, UCSC Genome Browser)
Lab Automation
☐ Creating Code for Laboratory Automation
☐ Using Liquid Handling Robots (e.g., Opentrons)
☑ Designing a Twist Order
☐ Creating a plan to use the Autonomous lab at Ginkgo Bioworks
Protein Design
☑ Protein Design
☐ Use of Boltz or PepMLM
☐ Use of Asimov Kernel
☑ Use of Benchling
☐ Models and Notebooks
☑ Databases
Cell-Free Systems
☐ Cell Free Reactions
☐ Freeze-Dried Cell Free Systems
☐ miniPCR Tools
☑ Protein Purification
Gibson Assembly
☐ Primer Design or Selection
☐ PCR Reactions
☐ Gibson Assembly
☐ Other Cloning Methods
CRISPR
☐ CRISPR/Cas9
☐ Designing Prime Editing gRNA
DNA Construct Design: One of the main techniques used in this project is DNA construct design. I designed a codon-optimized sequence of the glucocorticoid receptor ligand-binding domain and introduced a specific mutation, where glutamine was replaced with lysine, to explore its effect on ligand selectivity. This step was important because it directly determines how the protein will be expressed and whether it can potentially distinguish between cortisol and cortisone. The final construct was prepared for synthesis and cloning into an expression vector through Twist Bioscience, allowing it to be used in downstream experimental steps.
Protein Purification: Another important technique in this project is protein purification. After expressing the mutated protein in Escherichia coli, the protein needs to be isolated from other cellular components to ensure accurate testing. A chromatography-based purification method would be used to obtain a cleaner protein sample, which is necessary before performing binding assays. This step is critical because any contamination could affect the fluorescence signal and lead to incorrect conclusions about binding and selectivity.
Section5: Results & Quantitative Expectations
In this project, I designed and verified the GR-LBD DNA construct containing the Q642K mutation. The goal was to ensure that the sequence was correctly modified and ready for future expression studies. Preliminary structural analysis showed that Q642 is positioned close to the cortisol binding region, with ~55 local contacts and a measured distance of ~3.86 Å. No direct hydrogen bonds were observed. Following technical guidance from Mikael Hwang, these findings suggested that Q642 may influence ligand recognition through indirect steric or electrostatic effects rather than direct hydrogen-bond interactions. This provided additional rationale for further exploring the Q642K mutation.
Protocol
I started with the GR-LBD protein sequence and converted it into a DNA sequence.
The sequence was codon-optimized for E. coli.
I imported the sequence into Benchling and checked the reading frame.
A point mutation (Q → K) was introduced around base pairs 365–369.
I confirmed that the mutation does not disrupt the sequence or translation.
The final construct (777 bp) was reviewed and prepared as a Twist order.
The main technique used here is DNA construct design, where I created a codon-optimized sequence with a specific mutation. The starting point was a known GR-LBD protein sequence, which was converted into DNA before modification. Benchling was used to visualize and edit the sequence and confirm correct translation. Designing the Twist order was also part of the process to make sure the construct can be synthesized. These steps are important because they affect whether the protein can be expressed correctly later.
Data + analysis: The data comes from the DNA sequence and its visualization in Benchling. The sequence confirms that the mutation is correctly introduced, the reading frame is intact, and the construct is suitable for expression.
Challenges / limitations: One limitation is that this validation is based only on sequence design and has not been tested experimentally yet. The protein may not express well or may behave differently in E. coli. Also, a single mutation might not be enough to create a strong effect. If needed, additional mutations or redesign of the sequence could be considered.
Schaaf MJM, Lewis-Tuffin LJ, Cidlowski JA. Ligand-selective targeting of the glucocorticoid receptor to nuclear subdomains is associated with decreased receptor mobility. Molecular Endocrinology. 2005. https://doi.org/10.1210/me.2005-0050
Note: ChatGPT (GPT-5.3 instant) was used to help generate experimental design figure and to support refining the design and summarizing some of the references. The main idea, scientific reasoning, and all design decisions were developed independently by me.
Supplies & Estimated Cost
• DNA synthesis (Twist Bioscience construct): ~$150–$300
• E. coli BL21(DE3) competent cells: ~$100
• Growth media (LB broth, agar plates): ~$50
• Antibiotics (chloramphenicol): ~$30
• Protein expression inducer (IPTG): ~$50
• Cell lysis reagents (buffers, enzymes): ~$80
• Protein purification materials (affinity column + buffers): ~$200
• SDS-PAGE materials (gels, buffers, staining): ~$100
• Fluorescence-based assay reagents: ~$150
• General lab consumables (pipette tips, tubes): ~$100
Estimated total: ~$1,160
Group Final Project
From Brainstorming to Project Direction
During our first group brainstorming session, We kept talking about the L protein stability, how the MS2 L protein interacts with the bacterial chaperone DnaJ, whether specific amino acid changes affect lysis behavior, and how modifying certain regions could potentially improve protein function. We also discussed improving lysis efficiency and helping MS2 overcome possible host resistance.
stability alone was too broad. The bigger question became: Can we help the L protein maintain function while reducing its dependence on host processing? This became important because proteins that cannot maintain proper folding or structural integrity may lose function before successfully carrying out membrane-associated activity.
Previous studies identified Lodj mutants that could overcome DnaJ-related lysis defects, suggesting that modifications in specific regions of the L protein may influence how strongly the protein depends on host processing.
Goal: For this project, we want to investigate whether selected modifications can help the MS2 L protein maintain function while preserving the biological properties required for lysis.
Previous studies showed that relatively small amino acid changes in the MS2 L protein can significantly affect lysis behavior and host dependence. To build on this, we will first identify regions predicted to contribute to membrane-associated activity, DnaJ-related processing, and overall structural stability while distinguishing regions that may tolerate modification. We will use Lodj variants as a reference point because previous studies suggest that changes in these variants can influence how strongly the L protein depends on DnaJ-assisted processing.
Sequence conservation analysis: We will compare available MS2 L protein sequences and related lysis proteins to identify:
• highly conserved residues that are likely important for function
• less conserved regions that may better tolerate modification
• candidate regions for mutation selection
Because highly conserved residues are often linked to important biological functions, these regions may be less suitable targets for engineering. Less conserved regions may provide safer locations for introducing modifications while minimizing disruption of essential activity.
Structural prediction and visualization: We will use AlphaFold, ESMFold & ChimeraX to examine:
• predicted structural organization of the L protein
• membrane-associated regions
• candidate residues selected from conservation analysis
• flexible regions that may contribute to instability
The goal is not only to visualize the protein structure, but also to determine whether candidate mutations are located in regions likely to tolerate modification without disrupting membrane-associated activity.
Computational stability analysis: Computational prediction tools will be used to compare wild-type and candidate mutant designs and estimate whether modifications are predicted to stabilize or destabilize protein behavior before moving into experimental testing.
Note: This project direction was built from ideas discussed during our first group brainstorming session and our shared discussion notes. Since we had limited time for additional discussions, I continued developing and expanding the project while keeping our original ideas and overall direction as the foundation of the work.
References
Chamakura KR, Tran JS, Young R. (2017). MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ. Journal of Bacteriology, 199(12): e00058–17.
Chamakura KR et al. (2017). Mutational Analysis of the MS2 Lysis Protein L. Microbiology.
Mezhyrova J, et al. (2023). In vitro Characterization of the Phage Lysis Protein MS2-L. Microbiome Research Reports, 3:28.
Jumper J, Evans R, Pritzel A, et al. (2021). Highly Accurate Protein Structure Prediction with AlphaFold. Nature, 596:583–589.
Lin Z, Akin H, Rao R, et al. (2023). Evolutionary-scale Prediction of Atomic-Level Protein Structure with a Language Model. Science, 379(6637):1123–1130.