W2: Assignment
Week 2: Dna-read-write-and-edit Assignment
Part 0: Basics of Gel Electrophoresis:
Documentation:
Make sure to document every step of the in-silico and lab experiments. Make sketches, screenshots, notes, drawings… anything that helps you - and others - understand the experiment. Your documentation should help you - and others - to understand the topic. Don’t be afraid to add things that don’t work. Show your failures - and how you overcame them. Your Documentation should be a description of the amazing journey you are on!
- Gel electrophoresis is a laboratory technique used to separate biomolecules such as DNA, RNA, or proteins based on their size and electrical charge as they migrate through a porous gel matrix under an electric field.
- Smaller molecules move faster through the gel pores, while larger fragments migrate more slowly and tend to remain closer to the wells.
- Some applications of the electrophoresis are:
flowchart TD
C{Electrophoresis Applications}
C --> D[Clinical diagnostics: Parenting tests]
C --> E[Forensic investigations]
C --> F[Transformation and insertions of plasmids]
C --> G[Genetic Maps: Detecting species]- From my own laboratory experience, early electrophoresis runs are rarely perfect. During a previous project involving Lactobacillus strains from commercial probiotics, I had to amplify bacterial DNA using PCR and then verify the products by gel electrophoresis before sequencing. Initially, achieving clear and well-defined bands was challenging.
Some of the mistakes I made in previous assays were:
- Applying too much pressure on the gel.
- Loading low PCR product on the well.
- Leaving the gel running for too long.
- Or preparing an agarose gel with distilled water instead of using a buffer 💀
Each of these errors affected band clarity or migration, but they also became valuable learning moments. By the time, I learned to be more careful with gel handling, optimize PCR concentrations, monitor run times, and always prepare gels with the appropriate buffer.
This process reminded me that electrophoresis is not only a technical protocol but also a skill developed through practice, troubleshooting, and patience. Making mistakes and understanding why they happen. This is part of building confidence at the bench and developing experimental intuition.
Here are some pictures comparing my own process of learning how to charge a gel before (top) and after (bottom):
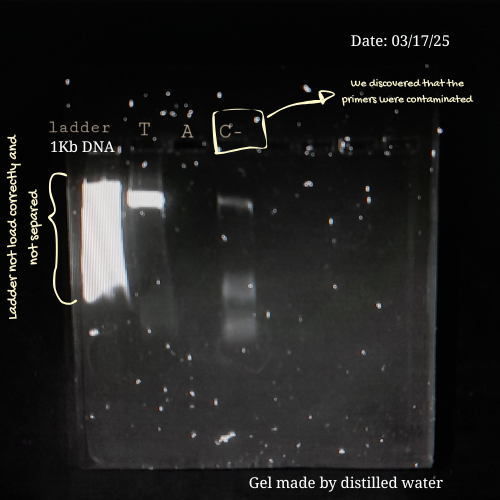
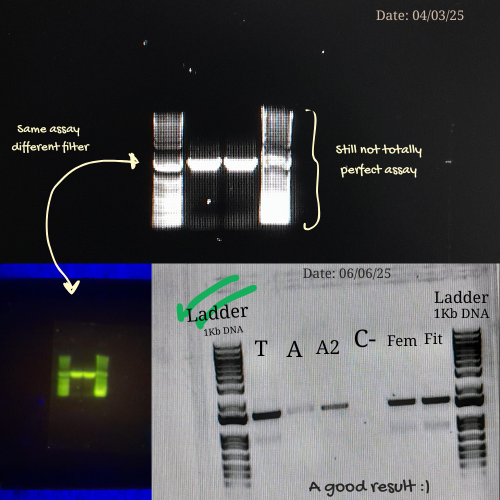
These are my volunteer pictures from my Molecular Biology experiments at the Biomedical Research Center (CENBIO-UTE).
Part 1: Benchling & In-silico Gel Art
Creating Gel Art- in silico using Benchling
- First, I searched for the Lambda phage genome using the NCBI Nucleotide database by entering Enterobacteria phage lambda or directly the accession number NC_001416.1. From the available results, I selected the complete genome sequence and downloaded it in FASTA format. (Figure 1)
- Next, the FASTA file was imported into Benchling using the Create DNA/RNA → Upload files option. Once uploaded, the Lambda DNA sequence was opened and visualized in linear map mode. (Figure 2)
Following the assignment instructions, I used the following restriction enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. After selecting the enzymes, a virtual restriction digest was performed using Benchling’s Run Digest tool. This generated simulated fragment patterns that were visualized as in-silico gel electrophoresis bands.
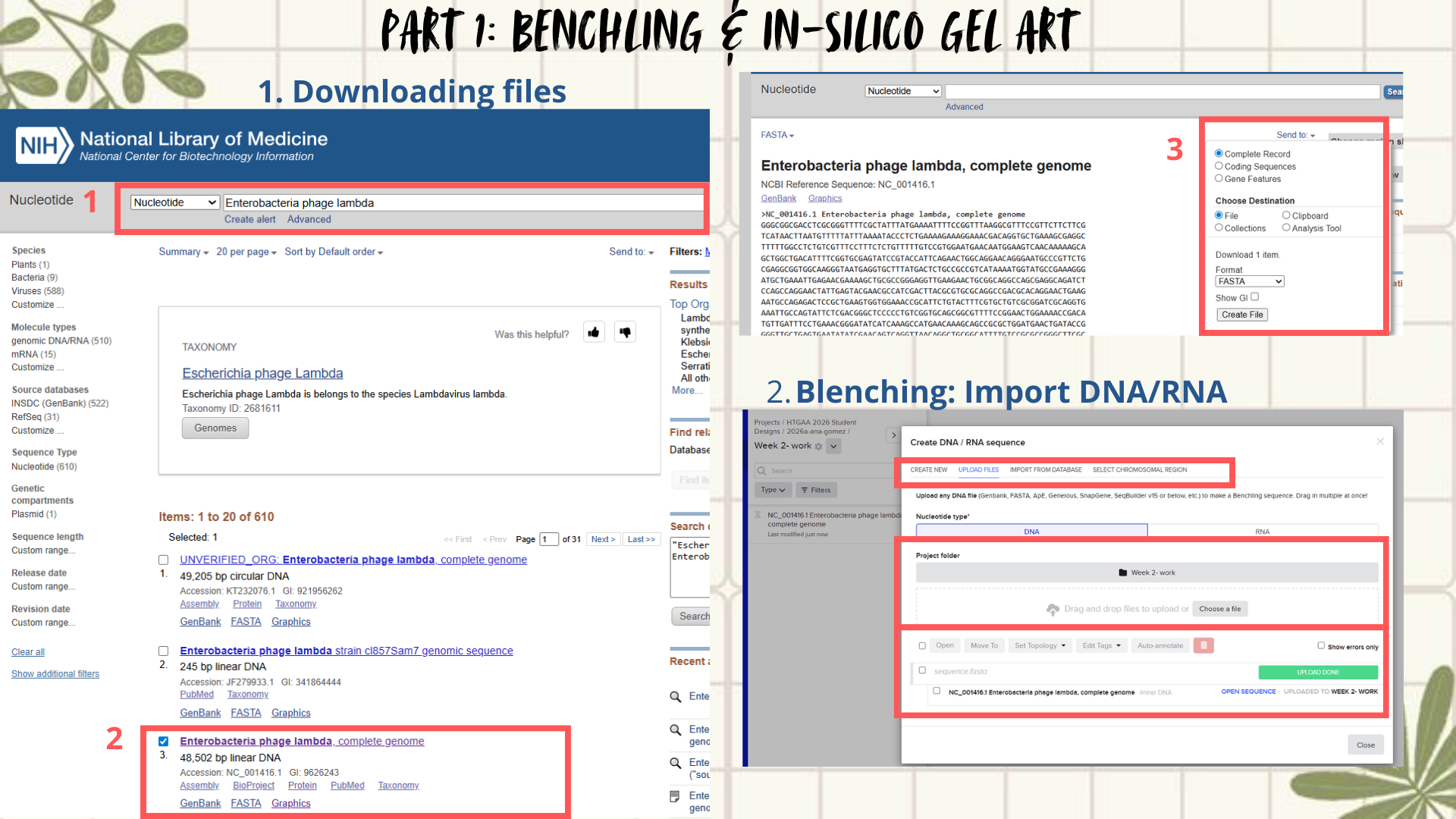
Figure 1: Workflow part 1- image 1
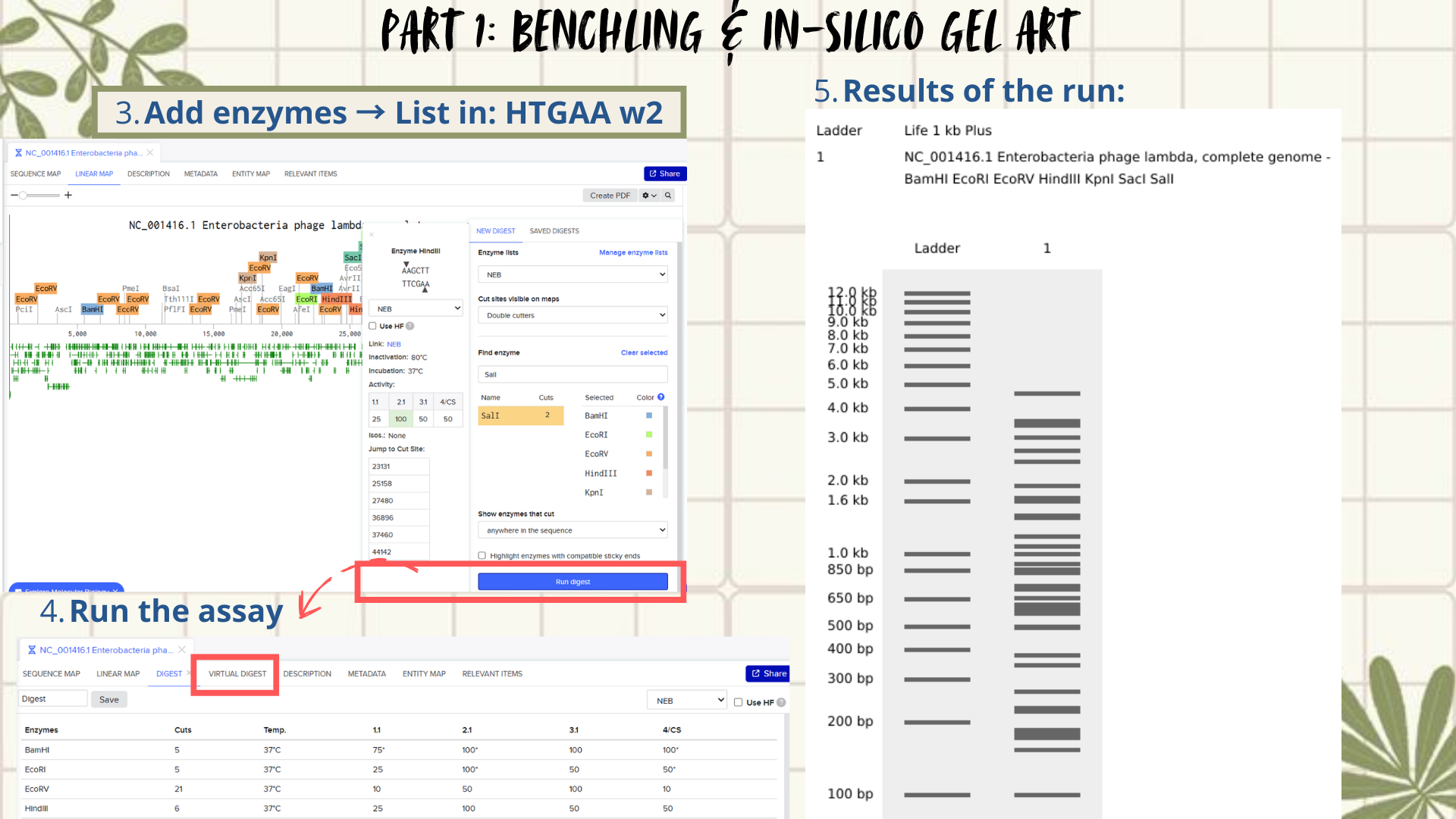
Figure 2: Workflow part 1- image 2
Creative exploration:
Initial attempts focused on creating typographic shapes, like the letter “A” (for Ana or Anita). But honestly, I got frustrated because the bands didn’t line up the way I expected. Benchling doesn’t “order” the runs like a design tool, so I assumed that it reflects the natural distribution of fragments, so the patterns kept turning into round shapes. Plus, I decided to create an enzyme catalog to visualize it. (Figure 3)
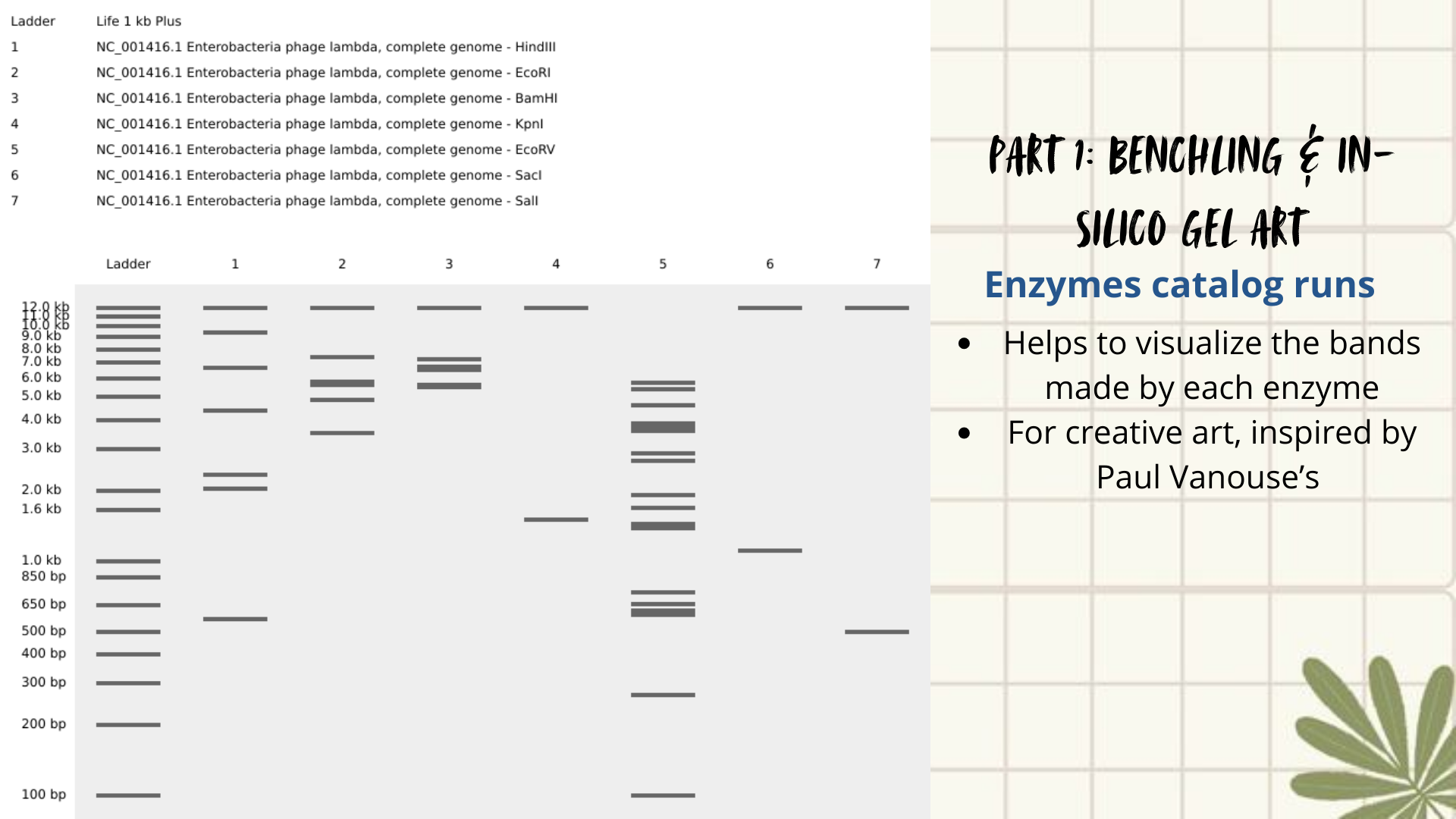
Figure 3: enzyme catalog
Then I remembered Paul Vanouse’s webpage, where gel images are shown inverted. So, I tried flipping my gel image too, and that small change completely shifted how I saw it. Suddenly, the band pattern looked like a landscape: a skyline that reminded me of Quito, with the Andean forest covering the mountains. (Figures 4 and 5)
The next slides show the Benchling work step-by-step and how I got to this final sketch:
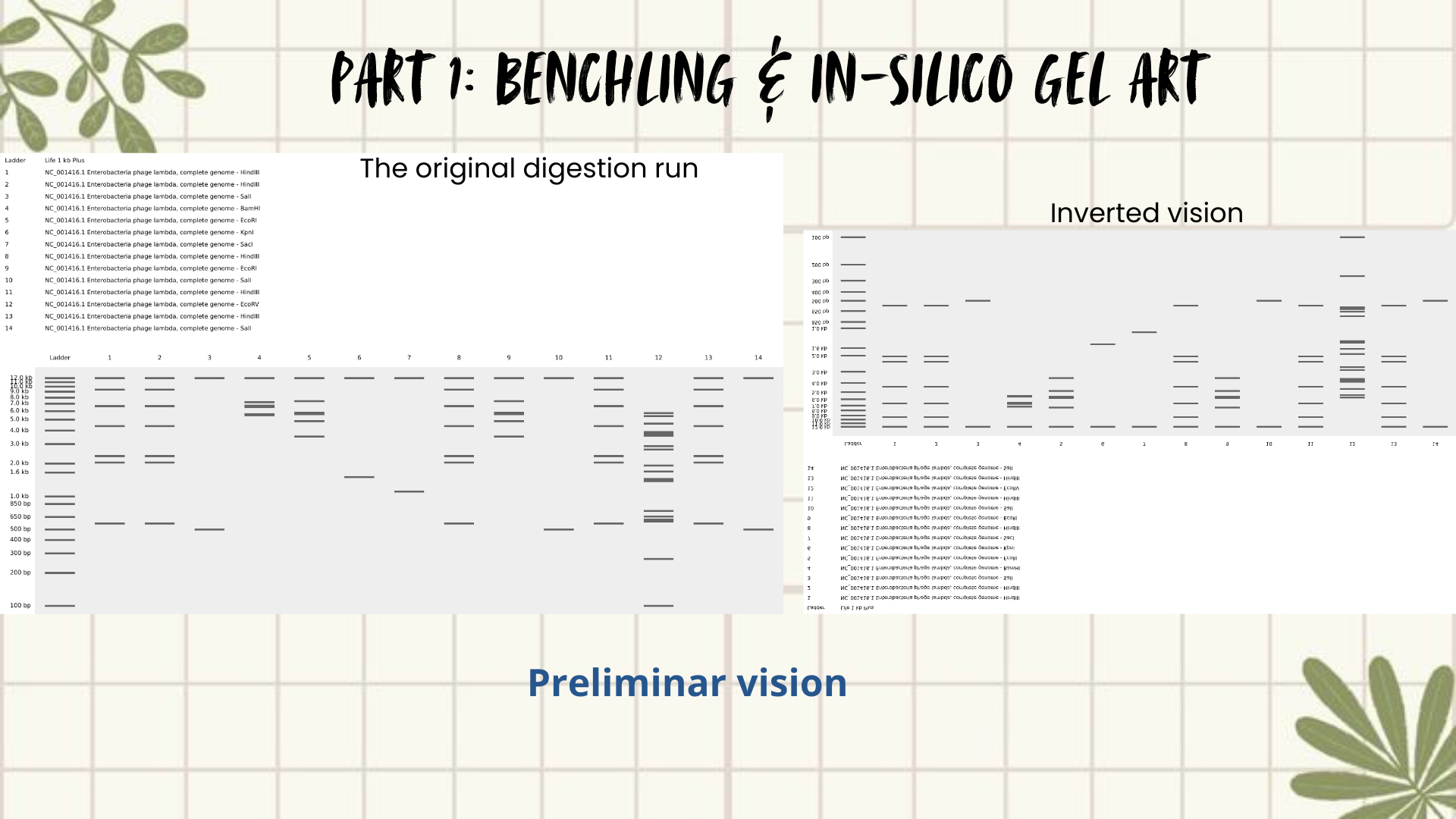
Figure 4: Preliminar design

Figure 5: Final result
Part 2:Gel Art - Restriction Digests and Gel Electrophoresis
Not available since I’m not in a node yet
Part 3: DNA Design Challenge
3.1. Choose your protein: In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
I chose L-lactate dehydrogenase (LDH) from Lactobacillus plantarum because it is a key enzyme in lactic acid fermentation, one of the most characteristic metabolic pathways of Lactobacillus. Since I’m interested in probiotics, LDH seems like an important protein to work with for this DNA design challenge.
Sequence from: https://www.uniprot.org/uniprotkb/F9USS9/entry
Lactiplantibacillus plantarum (strain ATCC BAA-793 / NCIMB 8826 / WCFS1) (Lactobacillus plantarum): MVAIDLPYDKRTITAQIDDENYAGKLVSQAATYHNKLSEQETVEKSLDNPIGSDKLEELARGKHNIVIISSDHTRPVPSHIITPILLRRLRSVAPDARIRILVATGFHRPSTHEELVNKYGEDIVNNEEIVMHVSTDDSSMVKIGQLPSGGDCIINKVAAEADLLISEGFIESHFFAGFSGGRKSVLPGIASYKTIMANHSGEFINSPKARTGNLMHNSIHKDMVYAARTAKLAFIINVVLDEDKKIIGSFAGDMEAAHKVGCDFVKELSSVPAIDCDIAISTNGGYPLDQNIYQAVKGMTAAEATNKEGGTIIMVAGARDGHGGEGFYHNLADVDDPKEFLDQAINTPRLKTIPDQWTAQIFARILVHHHVIFVSDLVDPDLITNMHMELAKTLDEAMEKAYAREGQAAKVTVIPDGLGVIVK
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence: Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
I used an online reverse translation tool Bioinformatic.org to convert the protein sequence into a coding DNA sequence. Because the genetic code is degenerate (multiple codons can encode the same amino acid), the generated sequence represents one possible nucleotide sequence compatible with the selected protein, rather than its original genomic DNA.
Tool: Bioinformatic.org
Result:
reverse translation of sample to a 1272 base sequence of most likely codons.
atggtggcgattgatctgccgtatgataaacgcaccattaccgcgcagattgatgatgaaaactatgcgggcaaactggtgagccaggcggcgacctatcataacaaactgagcgaacag gaaaccgtggaaaaaagcctggataacccgattggcagcgataaactggaagaactggcgcgcggcaaacataacattgtgattattagcagcgatcatacccgcccggtgccgagccatattattaccccgattctgctgcgccgcctgcgcagcgtggcgccggatgcgcgcattcgcattctggtggcgaccggctttcatcgcccgagcacccatgaagaactggtgaacaaatatggcgaagatattgtgaacaacgaagaaattgtgatgcatgtgagcaccgatgatagcagcatggtgaaaattggccagctgccgagcggcggcgattgcattattaacaaagtggcggcggaagcggatctgctgattagcgaaggctttattgaaagccatttttttgcgggctttagcggcggccgcaaaagcgtgctgccgggcattgcgagctataaaaccattatggcgaaccatagcggcgaatttattaacagcccgaaagcgcgcaccggcaacctgatgcataacagcattcataaagatatggtgtatgcggcgcgcaccgcgaaactggcgtttattattaacgtggtgctggatgaagataaaaaaattattggcagctttgcgggcgatatggaagcggcgcataaagtgggctgcgattttgtgaaagaactgagcagcgtgccggcgattgattgcgatattgcgattagcaccaacggcggctatccgctggatcagaacatttatcaggcggtgaaaggcatgaccgcggcggaagcgaccaacaaagaaggcggcaccattattatggtggcgggcgcgcgcgatggccatggcggcgaaggcttttatcataacctggcggatgtggatgatccgaaagaatttctggatcaggcgattaacaccccgcgcctgaaaaccattccggatcagtggaccgcgcagatttttgcgcgcattctggtgcatcatcatgtgatttttgtgagcgatctggtggatccggatctgattaccaacatgcatatggaactggcgaaaaccctggatgaagcgatggaaaaagcgtatgcgcgcgaaggccaggcggcgaaagtgaccgtgattccggatggcctgggcgtgattgtgaaa
To validate the result, I also used a second online tool Cusabio | Protein to DNA Sequence Converter http://cusabio.com/Protein-to-DNA-Sequence-Generator.html?srsltid=AfmBOopcO_Hr9FvnMWNA5QgjWDa1m5YP1YSDWoOOFiNarKxvim5XEA5t, which produced a highly similar nucleotide sequence, confirming consistency between platforms.
Results:
ATGATGGTTGCTATTGATTTACCTTATGATAAACGTACTATTACTGCTCAAATTGATGATGAAAATTATGCTGGTAAATTAGTTTCTCAAGCTGCTACTTATCATAATAAATTATCTGAACAAGAAACTGTTGAAAAATCTTTAGATAATCCTATTGGTTCTGATAAATTAGAAGAATTAGCTCGTGGTAAACATAATATTGTTATTATTTCTTCTGATCATACTCGTCCTGTTCCTTCTCATATTATTACTCCTATTTTATTACGTCGTTTACGTTCTGTTGCTCCTGATGCTCGTATTCGTATTTTAGTTGCTACTGGTTTTCATCGTCCTTCTACTCATGAAGAATTAGTTAATAAATATGGTGAAGATATTGTTAATAATGAAGAAATTGTTATGCATGTTTCTACTGATGATTCTTCTATGGTTAAAATTGGTCAATTACCTTCTGGTGGTGATTGTATTATTAATAAAGTTGCTGCTGAAGCTGATTTATTAATTTCTGAAGGTTTTATTGAATCTCATTTTTTTGCTGGTTTTTCTGGTGGTCGTAAATCTGTTTTACCTGGTATTGCTTCTTATAAAACTATTATGGCTAATCATTCTGGTGAATTTATTAATTCTCCTAAAGCTCGTACTGGTAATTTAATGCATAATTCTATTCATAAAGATATGGTTTATGCTGCTCGTACTGCTAAATTAGCTTTTATTATTAATGTTGTTTTAGATGAAGATAAAAAAATTATTGGTTCTTTTGCTGGTGATATGGAAGCTGCTCATAAAGTTGGTTGTGATTTTGTTAAAGAATTATCTTCTGTTCCTGCTATTGATTGTGATATTGCTATTTCTACTAATGGTGGTTATCCTTTAGATCAAAATATTTATCAAGCTGTTAAAGGTATGACTGCTGCTGAAGCTACTAATAAAGAAGGTGGTACTATTATTATGGTTGCTGGTGCTCGTGATGGTCATGGTGGTGAAGGTTTTTATCATAATTTAGCTGATGTTGATGATCCTAAAGAATTTTTAGATCAAGCTATTAATACTCCTCGTTTAAAAACTATTCCTGATCAATGGACTGCTCAAATTTTTGCTCGTATTTTAGTTCATCATCATGTTATTTTTGTTTCTGATTTAGTTGATCCTGATTTAATTACTAATATGCATATGGAATTAGCTAAAACTTTAGATGAAGCTATGGAAAAAGCTTATGCTCGTGAAGGTCAAGCTGCTAAAGTTACTGTTATTCCTGATGGTTTAGGTGTTATTGTTAAATAA
3.3. Codon optimization: Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
For this section, I use the same Blenching to help with the codon optimization. It is important to make this step because different organisms prefer different synonymous codons, even though they encode the same amino acids. Without optimization, heterologous genes may be poorly expressed due to rare codons, inefficient tRNA availability, or unstable mRNA structures. In this case, I select Escherichia coli K-12, since it’s a versatile bacteria, also is recognized as a research model, and specific for Escherichia coli K-12 is useful for detailed information on: enzymes, metabolites, transporters, and metabolic pathways. (Booster, 2024)
3.4. You have a sequence! Now what?: What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
The next step would be to produce the protein through recombinant expression, the optimized gene could be chemically synthesized and cloned into an expression plasmid containing essential regulatory elements such as a promoter, ribosome binding site (RBS), and terminator (for example, using a T7 or lac promoter system).
Once assembled, the plasmid would be introduced into Escherichia coli through transformation. Inside the bacterial cell, the DNA is transcribed into mRNA by RNA polymerase, and the mRNA is translated by ribosomes into a protein. Because the sequence was codon-optimized for E. coli, protein expression efficiency would be improved. Expression can be induced using an inducible promoter, and the resulting protein can later be purified, for example, using affinity chromatography if a His-tag was included in the design (Rosano & Ceccarelli, 2014).
Alternatively, the protein could also be produced using a cell-free expression system, where the DNA (or mRNA) is added directly to a reaction mixture containing ribosomes, enzymes, nucleotides, and amino acids, allowing protein synthesis without living cells. This process can be produced faster and nowadays is used for the construction of genetic circuits (Perez et al., 2016).
3.5. (Optional) How does it work in nature/biological systems?: Describe how a single gene codes for multiple proteins at the transcriptional level, and try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!!.
For this alignment, I used the codon-optimized DNA sequence designed for expression in Escherichia coli. Although the original protein comes from Lactiplantibacillus plantarum, the sequence was reverse-translated and optimized to match E. coli codon usage, simulating a synthetic biology workflow.
A short fragment of the optimized DNA was aligned with its transcribed RNA and translated protein to illustrate the central dogma.
DNA: ATGGTGGCAATCGACCTGCCATATGATAAGCGTACTATCACCGCCCAGATCGACGATGAA RNA: AUGGUGGCAAUCGACCUGCCAUAUGAUAAGCGUACUAUCACCGCCCAGAUCGACGAUGAA PROTEIN: (show below in the figure)

Part 4: Prepare a Twist DNA Synthesis Order
Creating a Plasmid using Blenching and Twist
Following the previous steps, my goal was to design an expression plasmid for Escherichia coli carrying a codon-optimized Lactobacillus lactate dehydrogenase (LDH) gene.
To build the DNA insert (expression cassette), I assembled the following genetic elements in Benchling using a linear DNA topology:
Table 1. Linear map table
| Type | Name | Sequence |
|---|---|---|
| Promoter | BBa_J23106 | TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC |
| RBS | BBa_B0034 | CATTAAAGAGGAGAAAGGTACC |
| Start codon | – | ATG |
| Coding sequence | LDH (codon optimized) | (full sequence as shown above) |
| 7× His tag | C-terminus | CATCACCATCACCATCATCAC |
| Stop codon | – | TAA |
| Terminator | BBa_B0015 | CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA |
Each component was annotated in Benchling (promoter, RBS, CDS, His-tag, terminator) to clearly define the structure of the expression cassette.
After assembling the sequence, I visualized the construct using the Linear Map tool:
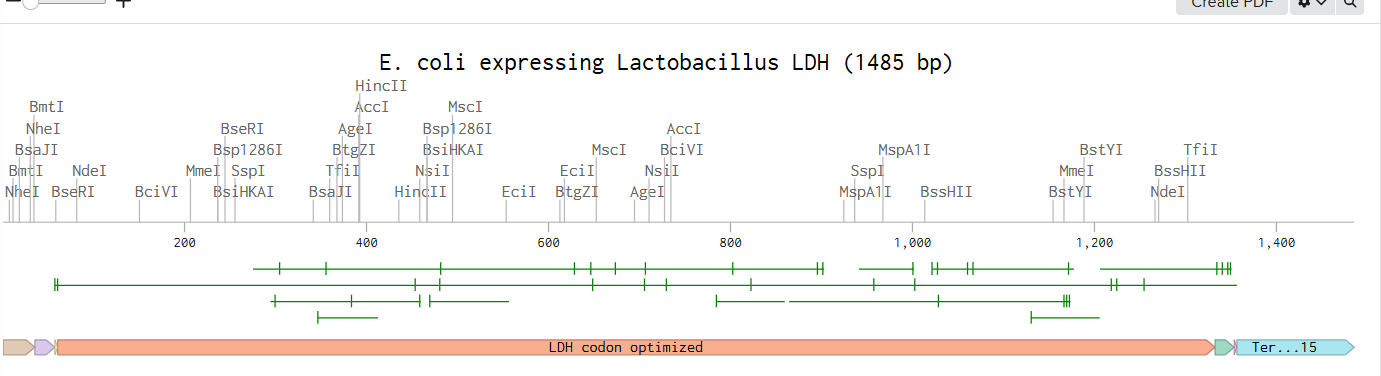
As an extra, here is a link to my Blenching project: Linemap Blenching
Plasmid construction:
The complete expression cassette was exported as a FASTA file and uploaded to Twist Bioscience using the Clonal Genes option. For the backbone vector, I selected pTwist Amp High Copy, which provides ampicillin resistance and a high-copy origin of replication suitable for protein expression in E. coli.
The resulting plasmid contains the LDH expression cassette inserted into the pTwist vector:
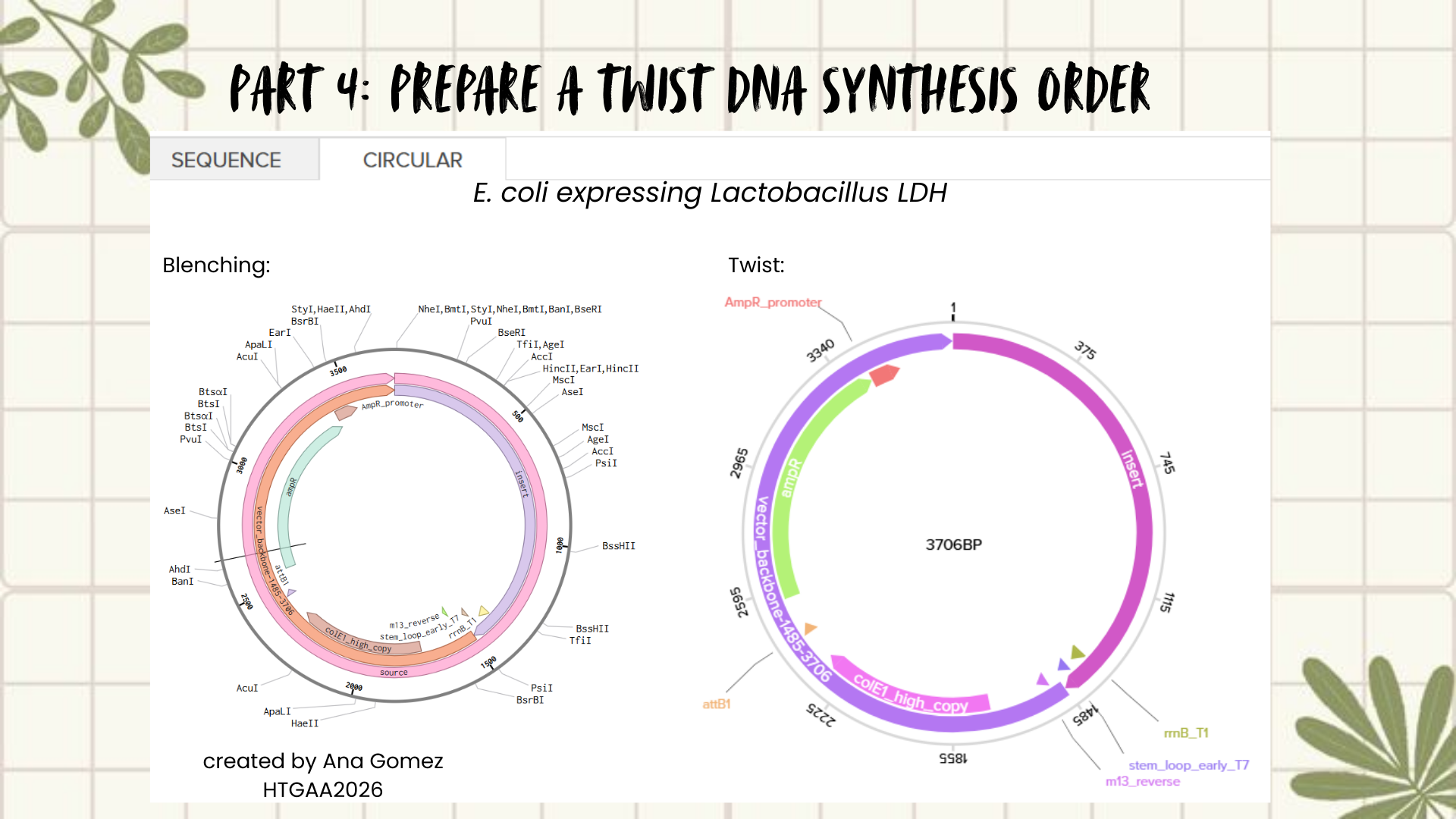 This is the result of transforming E. coli for recombinant LDH production.
This is the result of transforming E. coli for recombinant LDH production.
Part 5: DNA Read/Write/Edit

5.1 DNA Read:
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would be interested in sequencing Lactobacillus strains involved in probiotic activity, particularly those capable of producing antimicrobial compounds or enzymes such as lactate dehydrogenase (LDH). These bacteria are compatible with human physiology and play important roles in gut health. Additionally, understanding their genetic background could help identify mechanisms related to adhesion and biofilm formation.
Biofilms represent a major challenge in clinical settings, especially on medical devices, where they contribute to persistent infections. Similarly, in the food industry, biofilm formation is associated with contamination and spoilage, posing risks to public health. Sequencing these strains could therefore support both biomedical and industrial applications by enabling the identification of genes involved in antimicrobial activity and biofilm regulation. (Cangui-Panchi et al., 2022; Pang et al., 2023)
In this project, constructing and sequencing a plasmid expressing Lactobacillus LDH in E. coli would allow verification of correct gene insertion, absence of mutations after synthesis or cloning, and confirmation of reading frame integrity. Sequencing would also validate promoter–RBS–CDS junctions and His-tag fusion, ensuring proper protein expression. Such validation is essential for recombinant protein production workflows and quality control in synthetic biology.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
I would use Illumina short-read sequencing (second-generation sequencing).
Here are some reasons that are summarized in the following table
Table 2: Characteristics of Illumina sequencing
| Category | Description |
|---|---|
| Advantages | • High base accuracy (>99.9%) • Cost-effective for plasmids and bacterial constructs • Well-suited for constructs <10 kb |
| Generation | Second-generation (massively parallel sequencing with amplified fragments). |
| Input and preparation | 1. Plasmid extraction from E. coli 2. DNA fragmentation 3. Adapter ligation 4. Cluster generation on flow cell |
| Essential sequencing steps | • Sequencing-by-synthesis using fluorescently labeled nucleotides • Base calling is performed by detecting emitted fluorescence during nucleotide incorporation |
| Output | • FASTQ files containing millions of short reads • Reads assembled against reference plasmid to verify sequence integrity |
(Based on Emiyu & Lelisa, 2022; Sanderson et al., 2023)
5.2 DNA Write:
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! 😊
I am interested in synthesizing DNA for two main applications: a genetic biosensor circuit for lactate detection and recombinant enzyme production. First, inspired by my Week 1 project, I would like to design a lactate-responsive genetic circuit that could eventually be integrated into a wearable biosensor (like a temporary tattoo) for competitive swimmers. This biosensor would detect lactate levels, providing an alternative to repetitive blood sampling, reducing pain and laboratory dependency while allowing real-time metabolic monitoring.
Also, for this work, I focused on expressing Lactobacillus LDH in E. coli as a proof-of-concept for recombinant protein production. Building on this, it might be a way to design lactate-responsive genetic circuits for wearable biosensors, such as a temporary tattoo for competitive swimmers.
Additionally, I am also interested in DNA origami as a creative and structural application of DNA synthesis, exploring how programmed DNA folding could be used for nanoscale architectures and bio-art.
(ii) What technology or technologies would you use to perform this DNA synthesis and why? Also answer the following questions:
- What are the essential steps of your chosen sequencing methods?
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
To synthesize the designed genetic circuits, I would use commercial DNA synthesis platforms such as Twist Bioscience, which allow accurate construction of gene fragments or clonal plasmids from digitally designed sequences.
Process:
- In silico design of the genetic circuit (promoter, RBS, coding sequence, reporter).
- Codon optimization for E. coli expression.
- Chemical or enzymatic DNA synthesis of fragments.
- Assembly of fragments using Gibson Assembly or Golden Gate cloning.
- Transformation into E. coli for amplification and expression.
- Sequence verification using Illumina sequencing.
This approach allows rapid prototyping of biosensor constructs with high sequence fidelity.
Limitations include synthesis length constraints, potential sequence errors in long constructs, and cost when scaling multiple variants. Additionally, DNA origami applications require precise strand design and may be limited by folding efficiency and structural stability. (based on Hoose et al., 2023)
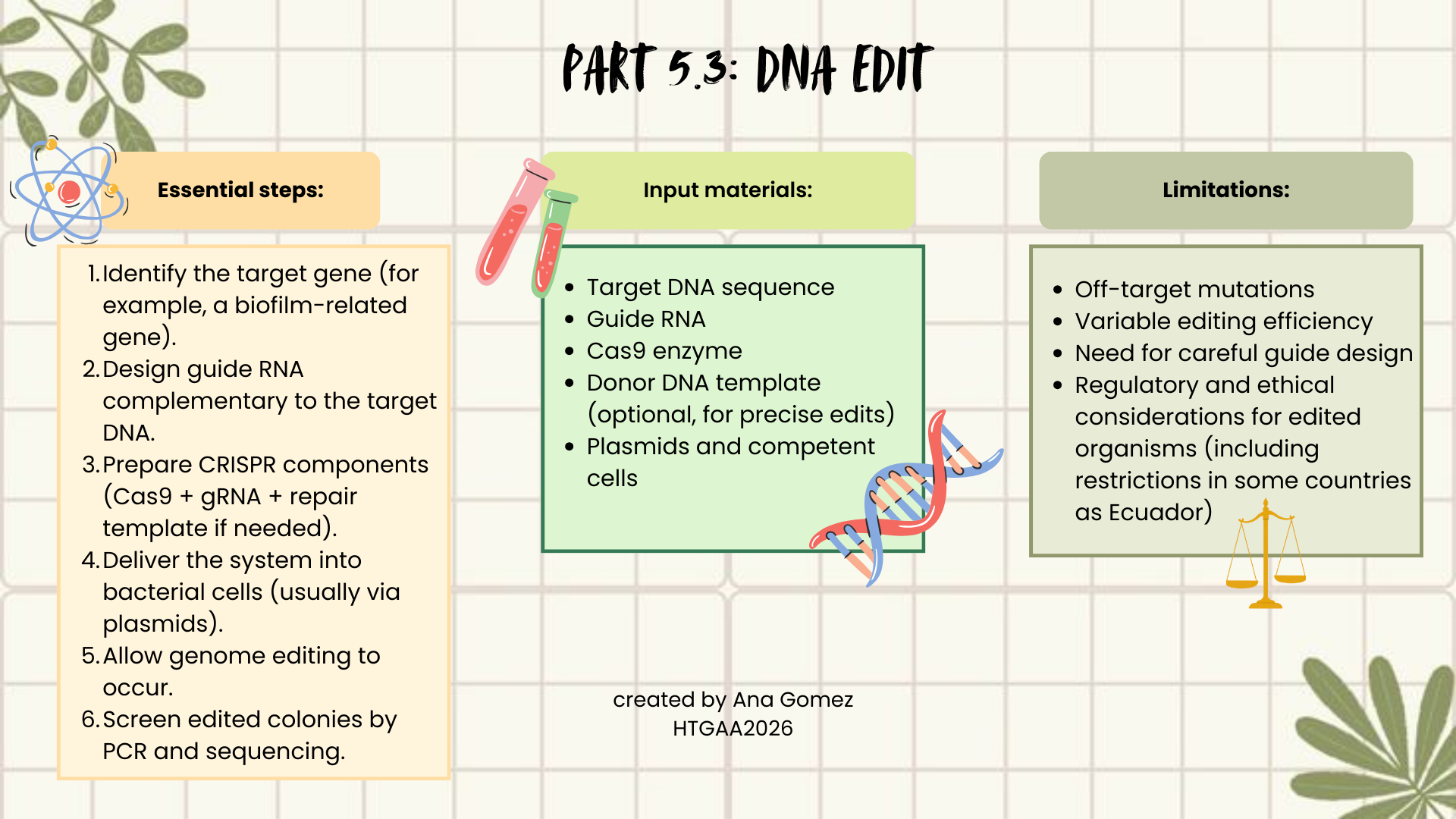
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
As I mentioned before, I would be interested in editing genes related to biofilm formation or antimicrobial production in Lactobacillus strains. Biofilms are a major problem in hospital environments and medical devices, and they also affect food safety. By modifying regulatory genes or metabolic pathways, it could be possible to reduce biofilm formation or enhance antimicrobial compound production. This could contribute to public health, infection prevention, and safer food systems.
Additionally, editing probiotic strains could help improve adhesion to intestinal surfaces or increase beneficial metabolite production, strengthening their therapeutic potential.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I would use CRISPR–Cas9, because it is precise, relatively easy to design, and widely used in bacteria. CRISPR works by using a guide RNA (gRNA) that matches a target DNA sequence. The Cas9 enzyme follows this guide and creates a double-strand break at the selected genomic location. The cell then repairs this break either by non-homologous end joining (NHEJ), which may introduce mutations, or homology-directed repair (HDR), if a repair template is provided, allowing precise edits.
 (Diagram)
(Diagram)

Weekly reflection:
- I enjoyed this homework because it allowed me to combine creativity with molecular biology tools. I liked being able to design gel art and work with DNA sequences. It was surprising to discover that platforms I had previously used for volunteering or simple visualization, such as Benchling, also contain useful functions for enzyme digestion, codon optimization, and plasmid design.
- This project also reminded me that not every experiment works perfectly the first time, just like real gel electrophoresis runs. Mistakes, unexpected results, and trial-and-error are part of the learning process. Repeating steps, understanding errors, and refining designs are essential to improve outcomes.
- Working with tools like Benchling and Twist helped me realize how accessible synthetic biology has become, and how digital platforms can support creative biological design. This experience helped clarify where future projects could begin: starting from a biological question, translating it into DNA design, and then imagining real applications such as biosensors, antimicrobial systems, or therapeutic constructs.
Thanks for reading!
This webpage is also upload in my personal Notion, if you want to visit it, please click in the next link! :) Notion week 2
References and Resources:
Part 0:
- Smith M (2026) Electrophoresis. National Human Genome Research Institute. https://www.genome.gov/genetics-glossary/Electrophoresis
- Tramini J (2023) Electrophoresis in Molecular Biology: Principles and Applications. J Biol Res Ther. 12:335. https://www.walshmedicalmedia.com/open-access/electrophoresis-in-molecular-biology-principles-and-applications.pdf
Part 1
- William (2024). Ciudad y bosque (Quito, Ecuador). Shutterstock. [image]. https://www.shutterstock.com/es/image-photo/city-forest-quito-ecuador-2440373267?dd_referrer=https%3A%2F%2Fwww.google.com%2F
Part 3:
- Booster. (2024). E. coli as a Model Organism. Booster antibody and ELISA experts. https://www.bosterbio.com/blog/post/e-coli-as-a-model-organism?srsltid=AfmBOooc_L8Ggou7g3cUyxTlNc1DTtfu-aYeu4mwmyqqThmy4lZTC0Gx
- Fang, S., Song, X., Cui, L., Bai, J., Lu, H., & Wang, S. (2023). The lactate dehydrogenase gene is involved in the growth and metabolism of Lacticaseibacillus paracasei and the production of fermented milk flavor substances. Frontiers In Microbiology, 14, 1195360. https://doi.org/10.3389/fmicb.2023.1195360
- Li, X., Wang, G., Wang, J., Song, X., Xiong, Z., Xia, Y., & Ai, L. (2024). The ldh Gene Plays a Crucial Role in Mediating the Pathogen Control of Lactiplantibacillus plantarum AR113. Foodborne pathogens and disease, 21(9), 578–585. https://doi.org/10.1089/fpd.2024.0028
- Perez, J. G., Stark, J. C., & Jewett, M. C. (2016). Cell-Free Synthetic Biology: Engineering Beyond the Cell. Cold Spring Harbor perspectives in biology, 8(12), a023853. https://doi.org/10.1101/cshperspect.a023853
- Rosano GL and Ceccarelli EA (2014) Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 5:172. doi: https://doi.org/10.3389/fmicb.2014.00172
Part 5:
- Aljabali, A. A. A., El-Tanani, M., & Tambuwala, M. M. (2024). Principles of CRISPR-Cas9 technology: Advancements in genome editing and emerging trends in drug delivery. Journal of Drug Delivery Science and Technology, 92(105338), 105338. https://doi.org/10.1016/j.jddst.2024.105338
- Cangui-Panchi, S. P., Ñacato-Toapanta, A. L., Enríquez-Martínez, L. J., Reyes, J., Garzon-Chavez, D., & Machado, A. (2022). Biofilm-forming microorganisms causing hospital-acquired infections from intravenous catheter: A systematic review. Current research in microbial sciences, 3, 100175. https://doi.org/10.1016/j.crmicr.2022.100175
- Emiyu, K., & Lelisa, K. (2022). Review on illumina sequencing technology. Austin Journal of Veterinary Science & Animal Husbandry, 9(1), 1088-1091. d1wqtxts1xzle7.cloudfront.net
- Hoose, A., Vellacott, R., Storch, M., Freemont, P. S., & Ryadnov, M. G. (2023). DNA synthesis technologies to close the gene writing gap. Nature reviews. Chemistry, 7(3), 144–161. https://doi.org/10.1038/s41570-022-00456-9
- Pang, X., Hu, X., Du, X., Lv, C., & Yuk, H. G. (2023). Biofilm formation in food processing plants and novel control strategies to combat resistant biofilms: the case of Salmonella spp. Food science and biotechnology, 32(12), 1703–1718. https://doi.org/10.1007/s10068-023-01349-3
- Sanderson, H., McCarthy, M. C., Nnajide, C. R., Sparrow, J., Rubin, J. E., Dillon, J. A. R., & White, A. P. (2023). Identification of plasmids in avian-associated Escherichia coli using nanopore and illumina sequencing. BMC genomics, 24(1), 698. https://doi.org/10.1186/s12864-023-09784-6
Resources A webpage that helped me to visualized flowcharts for markdown was: Online Flowchart