Week 5 HW: Protein Design Part II
Week 5: Protein Design Part II

Part A: SOD1 Binder Peptide Design (From Pranam):
| What I know about SOD1 and its mutation: | |
|---|---|

| (Berdyński et al., 2022) |
- Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS)
- ALS is a heterogeneous, severe neurodegenerative disorder, the hallmark of which is an adult-onset loss of upper and lower motor neurons.
- It leads to a progressive paresis and atrophy of skeletal muscles, resulting in quadriplegia and fatal respiratory failure.
- The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Challenge of this week: Design short peptides that bind mutant SOD1 & then decide which ones are worth advancing toward therapy.
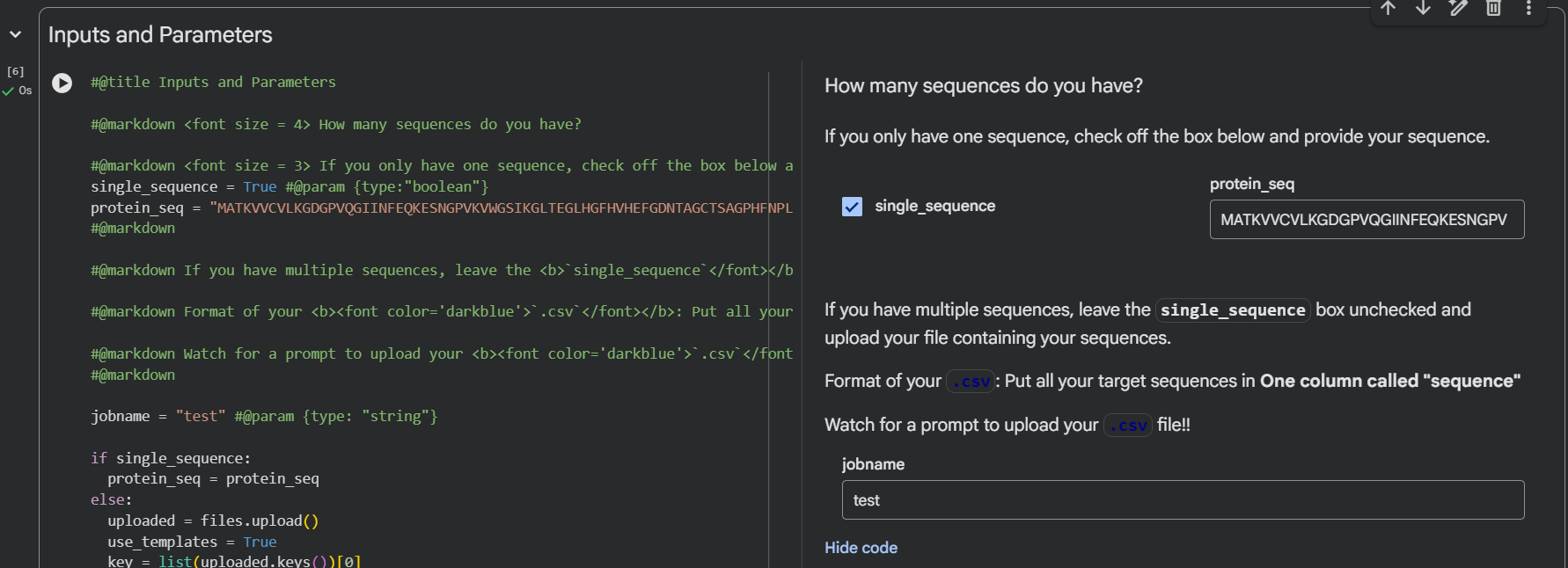
Part 1A: Generate Binders with PepMLM
To generate binders using the suggested program, it’s necessary to have the original sequence and check for the A4V mutation at that position.
Original sequence Uniprot (P00441)
A4V Mutation sequence:
After having the sequence modified, we use the Colab notebook:
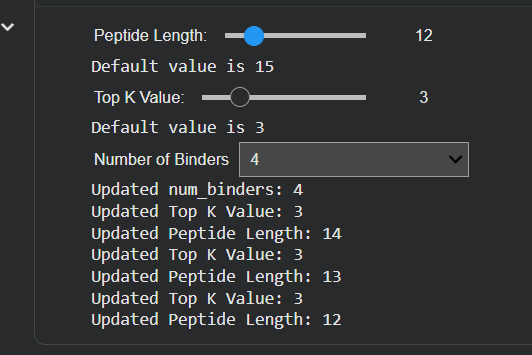
Important to know that it’s to make sure you select the number 4 of binders in the input, select the length of peptides, and then run it.
| Input | Binders and Peptide Length |
|---|---|
| 
|
Table 1. Peptides predicted
| Index | Lenght | Binder | Pseudo Perplexity (score)* |
|---|---|---|---|
| 0 | 12 | WRYPAVGARWKX | 10.660527 |
| 1 | 12 | WRYPVAAVELKX | 10.027294 |
| 2 | 12 | WLYYPAGAAHWX | 11.046032 |
| 3 | 12 | KRSYVVGVEWGX | 17.759518 |
| control** | 12 | FLYRWLPSRRGG | ——— |
Description: (*) Pseudo perplexity is an adaptation of the perplexity metric used in masked language models. The model masks each amino acid in the peptide one at a time and estimates the probability of correctly recovering it given the surrounding residues and the target protein sequence. Lower value → model assigns a higher probability to the peptide sequence and high confidence. High value → Less confidence model of sequence for the peptide. (**) Control is a known SOD1-binding peptide
Based on the results in Table 1, the candidates are in the top 2 positions. And less confidence with the last position (index 3).
Part 2A: Evaluate Binders with AlphaFold3
To evaluate the generated binders, AlphaFold3 was used to model protein–peptide complexes.

| AlphaFold Server |
For this section, AlphaFold3 does not accept the placeholder residue “X” that appeared at the terminal position of the peptide sequences generated by PepMLM. To resolve this issue, the terminal X was replaced with glycine (G) before structural modeling. Glycine was selected because it is a small and flexible residue that minimally perturbs peptide structure.
The adjusted peptide sequences used for AlphaFold3 predictions are shown in Table 2.
Table 2. Adjusted peptide sequences used for AlphaFold3 modeling
| Index | Length | Binder | Pseudo Perplexity (score) |
|---|---|---|---|
| Pep0 | 12 | WRPYAVGARWKG | 10.660527 |
| Pep1 | 12 | WRPYVAAVELKG | 10.027294 |
| Pep2 | 12 | WLYYPAGAAHWG | 11.046032 |
| Pep3 | 12 | KRSYVVGVEWGG | 17.759518 |
| Control | 12 | FLYRWLPSRRGG | — |
Description: (**) The pseudo-perplexity values reported correspond to the original PepMLM outputs before sequence adjustment. The substitution of the terminal placeholder residue (X → G) was performed only to enable compatibility with AlphaFold3 and does not affect the reported generation confidence scores.
Small tutorial AlphaFold3:
| 
| 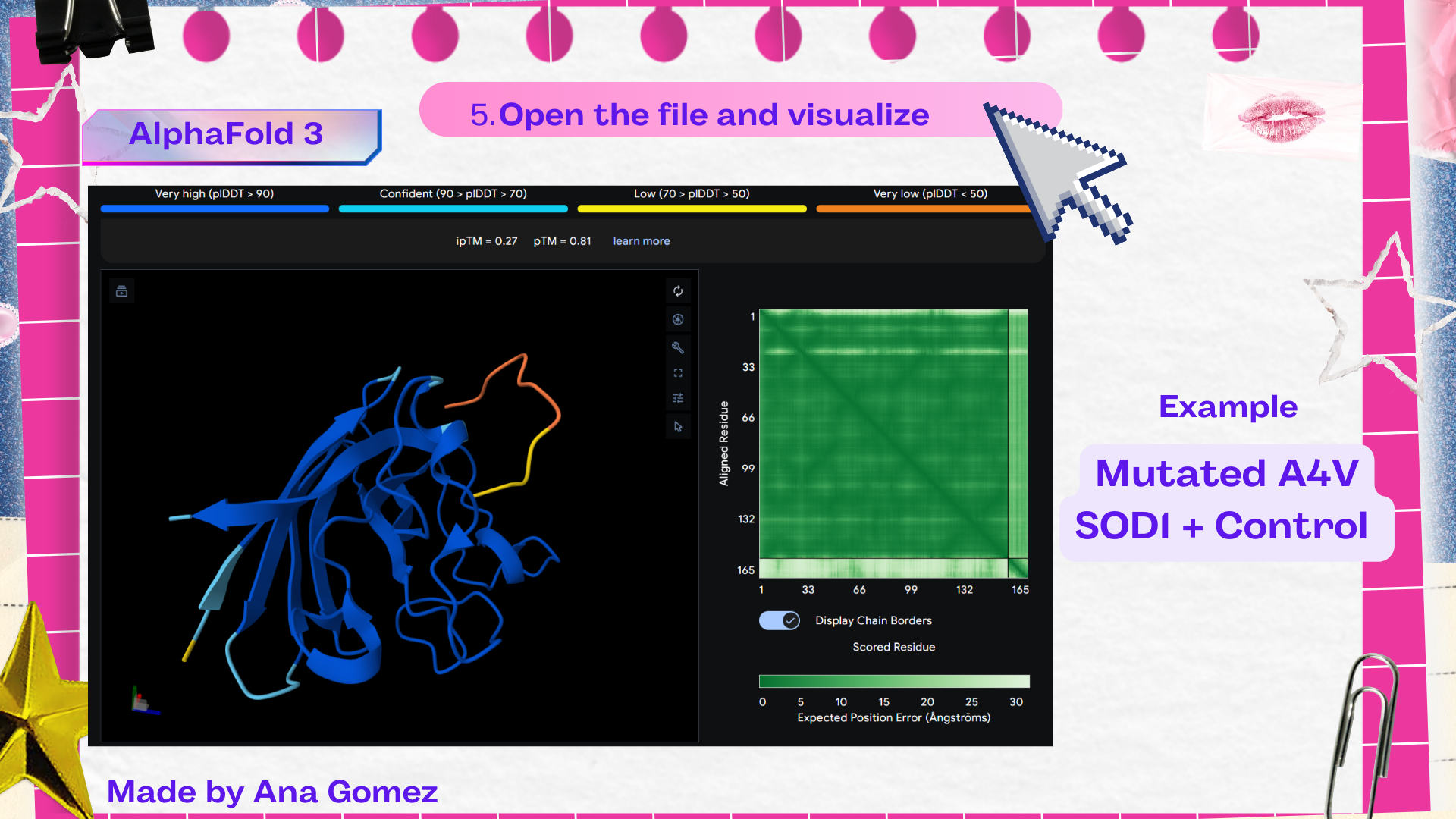
|
Table 3. Results of AlphaFold 3 SOD1 mutated A4V
| File | ipTM* | pTM** |
|---|---|---|
| Control | 0.27 | 0.81 |
| Pep0 | 0.31 | 0.85 |
| Pep1 | 0.35 | 0.8 |
| Pep2 | 0.47 | 0.82 |
| Pep3 | 0.41 | 0.89 |
Description: [ipTM]* ipTM (interface predicted TM-score) estimates the confidence of the predicted interaction between different chains in a complex. Higher ipTM values suggest a more reliable protein–peptide interface prediction. [pTM]** pTM (predicted TM-score) evaluates the overall confidence in the predicted structure of the entire protein complex. Higher pTM values indicate a more reliable structural model.
Structural interpretation of peptide binding:
Based on the AlphaFold3 models, the peptides appear to interact primarily with exposed surface regions of the SOD1 structure rather than deeply inserting into the protein core. Most peptides localize along the external surface of the β-barrel region, which forms the structural core of SOD1. In several cases the peptides appear surface-bound and loosely associated with the protein, rather than deeply buried within the structure. Some peptides also approach regions near the N-terminal segment, where the A4V mutation occurs, suggesting potential interactions with structurally sensitive areas of the mutant protein.
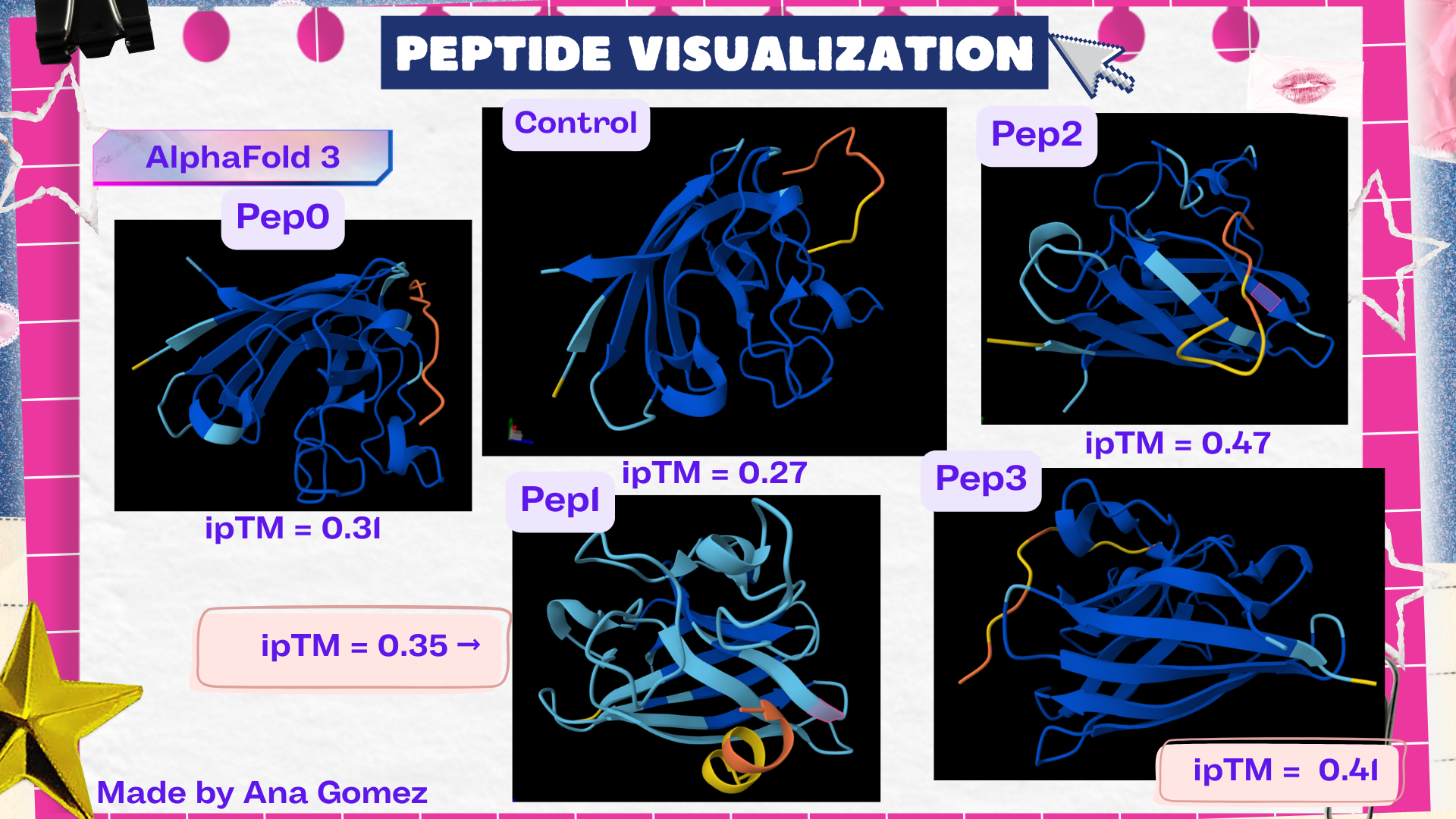
The ipTM values observed ranged from 0.27 to 0.47, indicating moderate confidence in the predicted protein–peptide interfaces. The control peptide showed the lowest interface score (ipTM = 0.27), while all PepMLM-generated peptides displayed higher ipTM values. Among them, Pep2 produced the highest interface confidence (ipTM = 0.47), followed by Pep3 (0.41). These results suggest that some peptides generated by PepMLM may interact more favorably with mutant SOD1 compared to the known binder, highlighting Pep2 as the most promising candidate for further evaluation.
Key Discoveries
All the generated peptides are superior compared with the control.
Pep2 is the best prediction.
Recommendation I recommend visualizing the extra material at the bottom of this webpage!
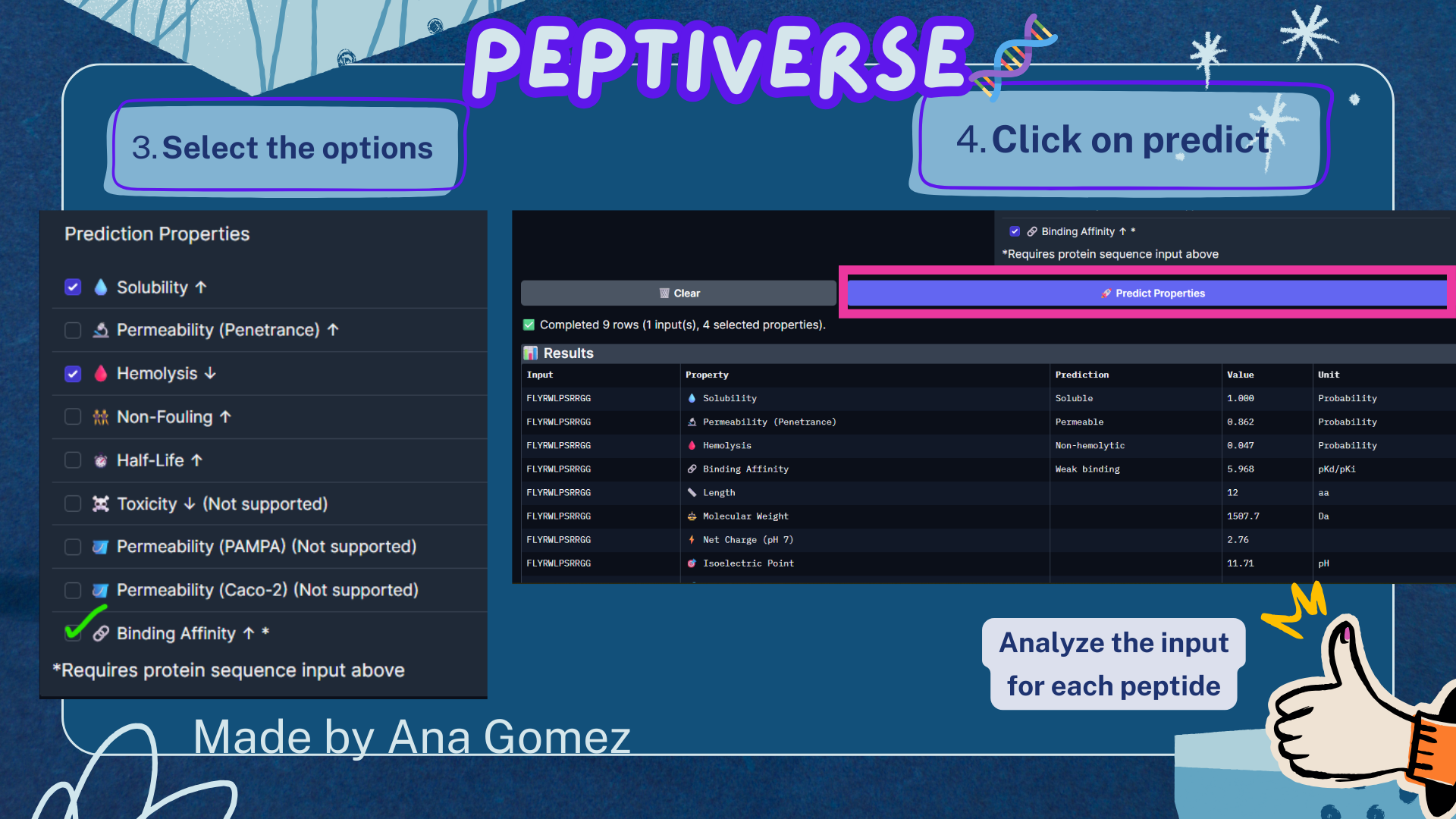
Part 3A: Evaluate Properties of Generated Peptides in the PeptiVerse
For this section, the Peptiverse website: https://huggingface.co/spaces/ChatterjeeLab/PeptiVerse
For the target, the option of binding is used:
A4V Mutation sequence:
The PeptiVerse analysis shows that all generated peptides have similar predicted binding affinities, which fall within the weak-binding range (pKd/pKi ≈ 5.7–6.4). Despite the modest affinity predictions, the peptides demonstrate favorable therapeutic properties overall. All candidates show excellent solubility (probability = 1.000) and relatively low hemolysis probabilities, suggesting acceptable safety profiles.
Table 4. Peptiverse Results
| Peptide | Predicted binding affinity (pKd/pKi) | Solubility (probability) | Hemolysis (probability) | Net charge (pH 7) | Molecular weight (Da) |
|---|---|---|---|---|---|
| Control | 5.965 [Weak binding] | 1.000 | 0.047 | 2.76 | 1507.7 |
| Pep0 | 6.152 [Weak binding] | 1.000 | 0.020 | 2.76 | 1446.7 |
| Pep1 | 5.779 [Weak binding] | 1.000 | 0.035 | 0.76 | 1388.6 |
| Pep2 | 5.825 [Weak binding] | 1.000 | 0.046 | -0.15 | 1391.5 |
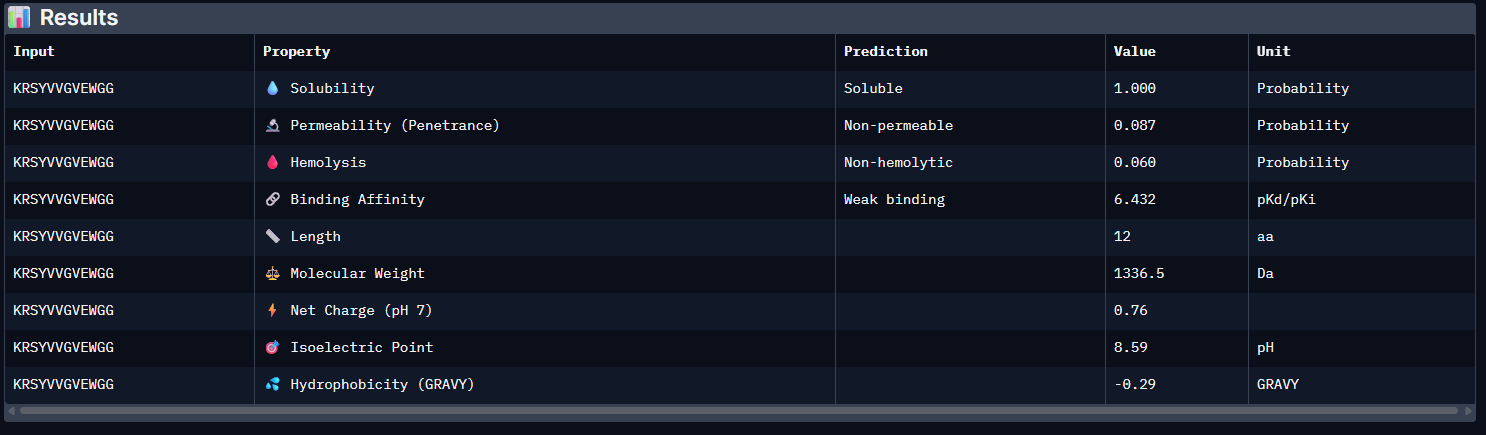
| Pep3 | 6.432 [Weak binding] | 1.000 | 0.060 | 0.76 | 1336.5 |
Comparison of structural and therapeutic predictions:
When comparing these predictions with the AlphaFold3 structural results, partial agreement can be observed. The peptide with the highest structural interface confidence, Pep2 (ipTM = 0.47), does not show the strongest predicted affinity in PeptiVerse. Instead, Pep3 displays the highest predicted binding affinity (pKd/pKi = 6.432), although it also presents the highest hemolysis probability among the candidates. Pep0 shows a relatively balanced profile, with moderate predicted affinity, the lowest hemolysis probability (0.020), and strong solubility.
Overall, these results indicate that structural confidence and predicted binding affinity do not perfectly correlate, highlighting the importance of evaluating both structural and therapeutic properties during peptide design.
Peptide selected for further evaluation:
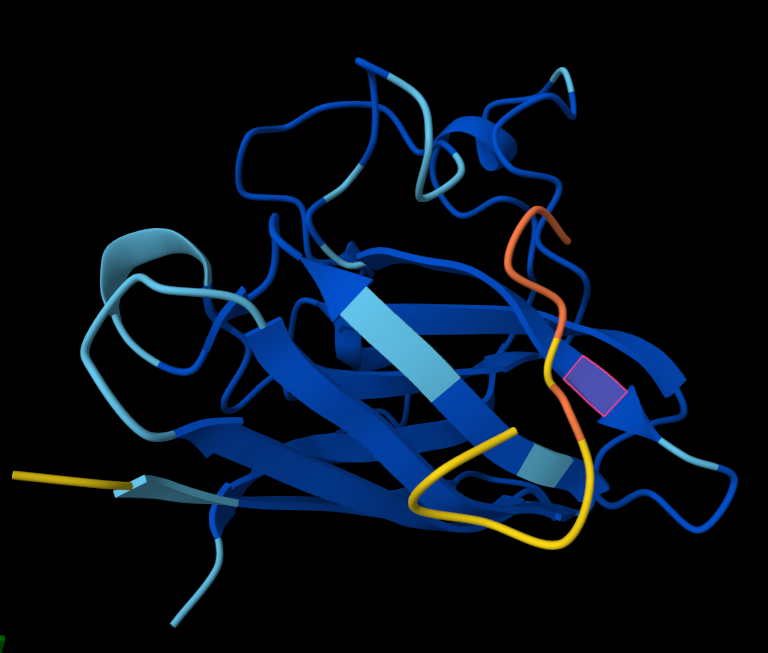
| Among the candidates, Pep2 was selected as the peptide to advance for further development. Although its predicted binding affinity is moderate, Pep2 showed the highest ipTM score in AlphaFold3, indicating the strongest predicted interaction with the mutant SOD1 structure. In addition, it maintains excellent solubility and a near-neutral net charge, while its hemolysis probability remains within an acceptable range. This balance between structural interaction and therapeutic properties makes Pep2 the most promising candidate for further optimization and experimental validation. |
Small tutorial Peptiverse:
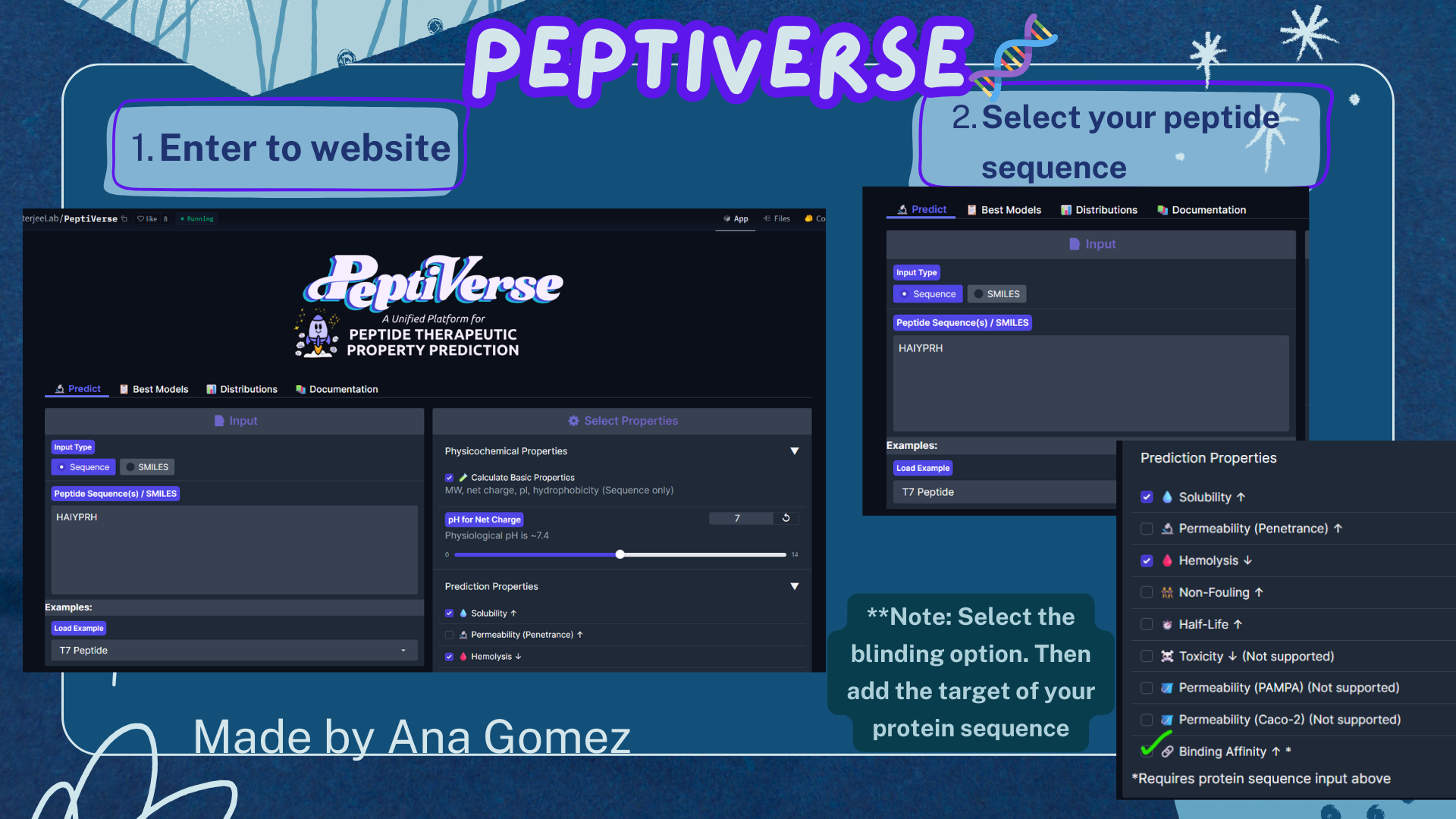
| 
|
For extra material, I recommend reading the full tables of Peptiverse at: Sources / Extra Material at the bottom of the webpage!
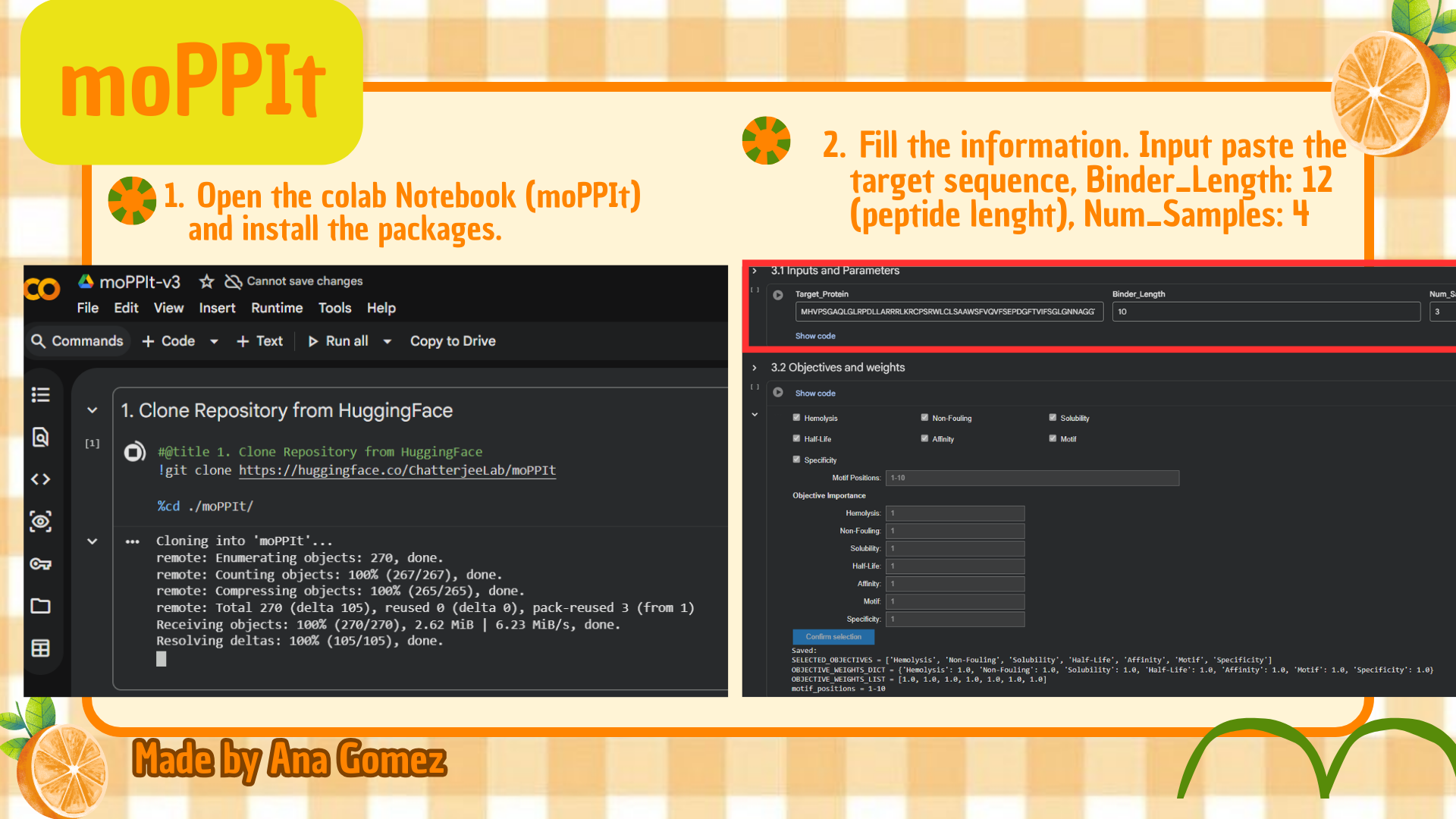
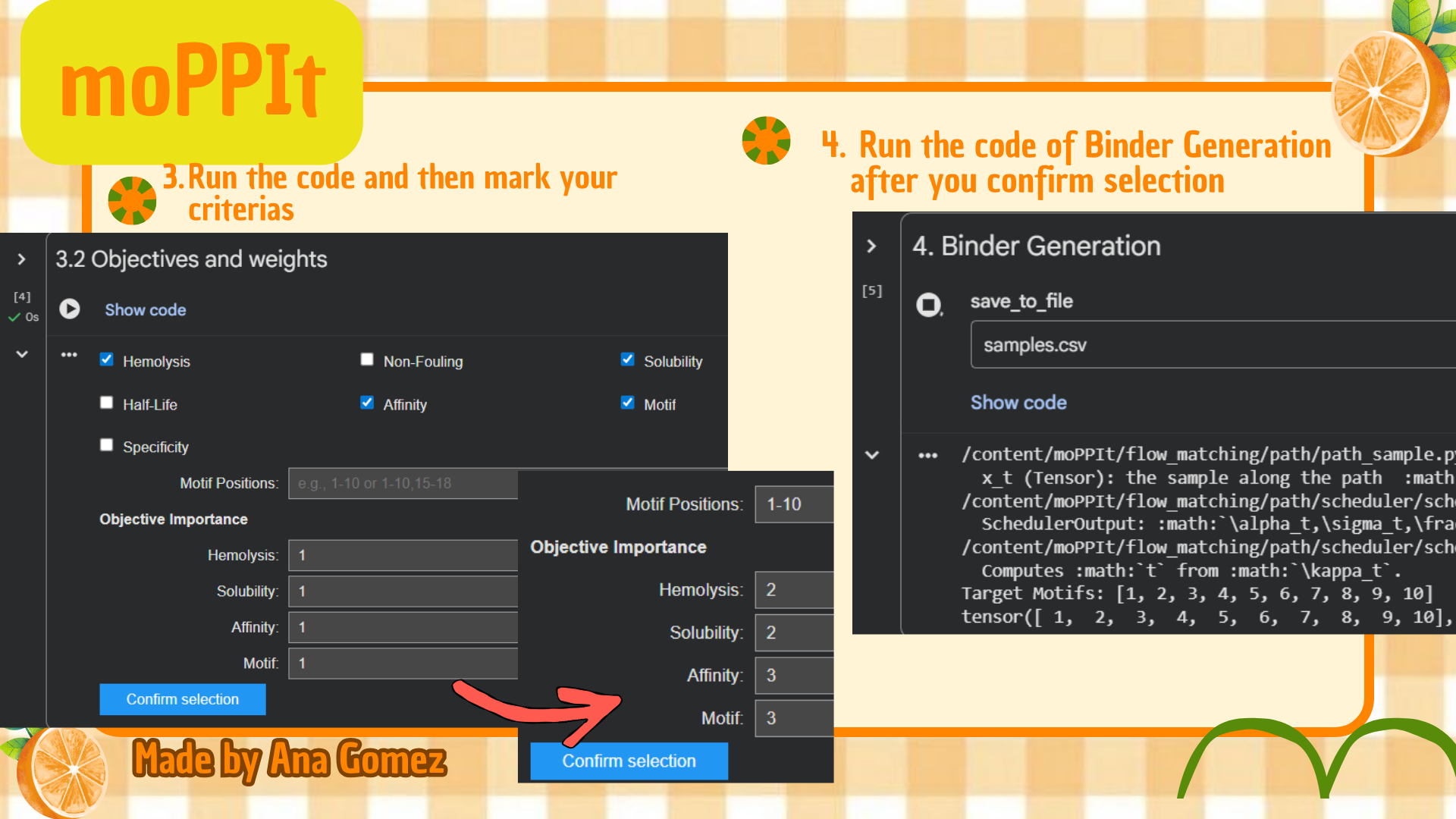
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
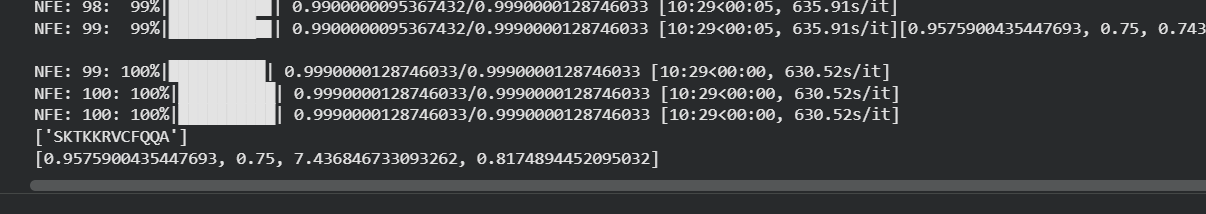
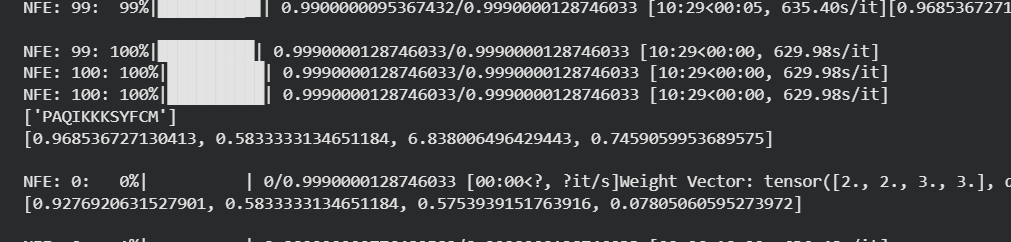
Table 5. moPPIt peptide results:
| Peptide | Output | Solubility | Hemolysis | Affinity | Motif |
|---|---|---|---|---|---|
| mPep1 | SKTKKRVFCFQA | 0.9575900435447693 | 0.75 | 7.43684673309262 | 0.81748944520095032 |
| mPep2 | PAQIKKKSYFCM | 0.968536727130413 | 0.5833333134651184 | 6.8380064964229443 | 0.7559059953689575 |
| mPep3 | GVTGSDEVKKIQ | 0.9665484763681889 | 0.75 | 5.413463115692139 | 0.44647738337516785 |
| mPep4 | YKKFKQTEKII | 0.978692626500596 | 0.83333331346551184 | 6.007977485656738 | 0.7189149856567383 |
(*) The coordinates aren’t organized. To check the outputs, please check in the Extra Material.
Compared with the PepMLM-generated peptides, the moPPIt peptides appeared more controlled and more directly optimized for the selected target region. In contrast to PepMLM outputs, the moPPIt peptides did not contain undefined terminal residues such as “X”, making them more readily usable for downstream analysis.
In addition, the moPPIt candidates appeared to show more consistent sequence patterns, with several peptides enriched in charged residues, suggesting stronger optimization toward target interaction and physicochemical constraints.
Clinical Application:
Before advancing these peptides toward clinical studies, they should first be evaluated through additional computational and experimental validation.
Structurally, the peptides should be tested in AlphaFold3 or similar protein–peptide modeling tools to verify whether they bind near the intended A4V-associated region of SOD1.
Their therapeutic properties should then be screened using predictors such as PeptiVerse, including affinity, solubility, hemolysis risk, and net charge.
Promising candidates should next be assessed with molecular dynamics simulations to evaluate complex stability, followed by experimental validation through in vitro binding assays, aggregation inhibition studies, cytotoxicity testing, and peptide stability analysis. These steps would be necessary before any preclinical or translational consideration.
(Wang et al., 2022; Barman et al., 2023)
Small tutorial moPPIt:
| (*) | |
|---|---|
| 
|
(*) Additionally: For moPPIt, the A4V mutant SOD1 sequence was used as the target protein. A binder length of 12 amino acids was selected, and peptide generation was guided toward residues 1–10 to focus on the N-terminal region containing the A4V-associated site. Affinity, motif, solubility, and hemolysis objectives were enabled to bias the design toward both target binding and therapeutic suitability.

Part C: Final Project: L-Protein Mutants
1. Selected design strategy
I selected Option 1: Mutagenesis, which combines computational mutation scoring with experimental mutational analysis of the MS2 lysis protein. This option was chosen because it provides a practical and interpretable framework for proposing candidate mutations while accounting for the limitations of structure prediction in membrane-associated proteins.
To explore potential beneficial mutations, I used the ESM-based mutation scoring notebook to estimate the tolerance of amino acid substitutions across the MS2 lysis protein sequence.
2. Sequence used
L-protein sequence used in the analysis:
This sequence contains two main structural regions:
Table 2c. Domain locations of L-protein
| Regions | Positions |
|---|---|
| soluble | 1–40 |
| transmembrane | 41–75 |
The soluble N-terminal domain interacts with host factors such as the DnaJ chaperone, while the transmembrane domain mediates membrane insertion and pore formation during lysis. Understanding the location of these domains is important when evaluating mutations, since substitutions may affect different functional aspects of the protein.
3. Mutation scoring notebook
The notebook HTGAA 2026: Using Protein Language Models (ESM).ipynb was used to generate mutation scores using a protein language model (ESM).
For each residue position, the model calculates a log-likelihood ratio (LLR) score that estimates how favorable a substitution is relative to the wild-type amino acid
Higher LLR scores indicate that the substitution is more compatible with the sequence constraints learned by the model
4. Heatmap interpretation
The mutation heatmap illustrates the predicted effects of all possible amino acid substitutions across the sequence.
- Warmer colors represent substitutions predicted to be more tolerated (yellow & green).
- Cooler colors represent substitutions predicted to be unfavorable (blue & purple).
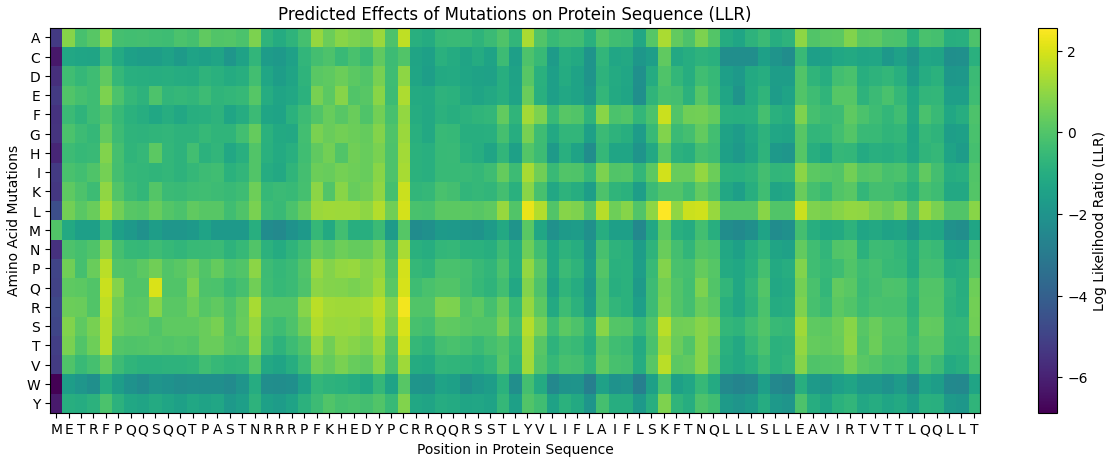
From this visualization, several mutations with relatively high predicted tolerance were observed in both the soluble and transmembrane regions of the protein.
5. Comparison with experimental mutational data
The computational mutation scores showed partial agreement with experimental mutational data obtained from previously reported MS2 lysis protein mutants.
Some substitutions predicted to be favorable by the language model correspond to mutations that experimentally maintain or improve lysis activity. This suggests that sequence-based protein language models are capable of capturing some functional and evolutionary constraints presented in the MS2 lysis protein.
6. Initial mutation ranking
The first step in the selection process was identifying the top mutations based on their LLR scores.
Table 6c. Raw mutation ranking (Top 10)
| Position | WT | Mutation | Score |
|---|---|---|---|
| 50 | K | L | 2.561468 |
| 29 | C | R | 2.395427 |
| 39 | Y | L | 2.241780 |
| 29 | C | S | 2.043150 |
| 9 | S | Q | 2.014325 |
| 29 | C | Q | 1.997049 |
| 29 | C | P | 1.971029 |
| 29 | C | L | 1.960646 |
| 50 | K | I | 1.928801 |
| 53 | N | L | 1.864932 |
Higher LLR scores indicate substitutions predicted to be more compatible with the sequence context and therefore more likely to be tolerated by the protein.
According to Zhang et al. (2025), the ESM2 score reflects the mutational tolerance of a given residue, where lower scores indicate stronger evolutionary constraints and higher scores suggest that substitutions are more likely to be tolerated.
7. Domain classification of candidate mutations
To better interpret these mutations, their positions were mapped to the structural domains of the protein, as mentioned on part 2.
Table 2c. Domain locations of L-protein
| Regions | Positions |
|---|---|
| soluble | 1–40 |
| transmembrane | 41–75 |
Using this classification, the top mutations were separated based on their structural location, as shown in the following Tables 7c1 and Table 7c2.
Table 7c1. Soluble domain candidate mutations
| Mutation | Score |
|---|---|
| C29R | 2.395427 |
| Y39L | 2.241780 |
| C29S | 2.043150 |
| S9Q | 2.014325 |
| C29Q | 1.997049 |
| C29P | 1.971029 |
| C29L | 1.960646 |
Description: Seven of the top mutations occur within the soluble domain, which may influence protein folding or interactions with host factors such as DnaJ.
Table 7c2. Transmembrane domain candidate mutations
| Mutation | Score |
|---|---|
| K50L | 2.561468 |
| K50I | 1.928801 |
| N53L | 1.864932 |
Description: Three mutations occur within the transmembrane region, which may affect membrane insertion or pore formation during lysis.
8. Final selection of Mutants
From the mutation ranking and domain analysis, five candidate mutants were selected.
Table 8c. Selected L-protein mutants
| Name | Mutation | Reason |
|---|---|---|
| LAmut1 | C29R | Selected due to a high LLR score indicating mutational tolerance. The substitution introduces a positively charged residue that may stabilize interactions in the soluble domain while preserving structural compatibility. |
| LAmut2 | Y39L | High scoring substitution predicted by the ESM2 model. Replacement of tyrosine with leucine maintains hydrophobic character while potentially improving stability in the local structural environment. |
| LAmut3 | K50L | Located in the predicted transmembrane region. Substitution from lysine to leucine increases hydrophobicity, which may improve membrane compatibility and insertion efficiency. |
| LAmut4 | K50I | High LLR score mutation within the membrane segment. Isoleucine is a hydrophobic residue commonly found in membrane helices, suggesting improved structural compatibility. |
| LAmut5 | N53L | Predicted favorable mutation according to the language model. The substitution introduces a hydrophobic residue potentially stabilizing the transmembrane segment. |
Description: The naming scheme LAmut refers to a personalized naming convention used for the designed mutants, followed by a number corresponding to the mutation order.
9. Mutation selection criteria
Amino acid substitutions are not random but are strongly influenced by physicochemical properties such as hydrophobicity, charge, and structural compatibility (Weber & Whelan, 2019; James & Lascoux, 2025 ).
Therefore, mutations were selected using three main criteria:
- High LLR scores predicted by the ESM2 model
- Compatibility with structural domains of the protein
- Physicochemical compatibility of amino acid substitutions
Hydrophobic residues are commonly enriched in transmembrane helices, suggesting that substitutions that increase hydrophobicity may enhance membrane insertion and stability. Mutations in the soluble domain may influence folding or interactions with host factors such as DnaJ, while mutations in the transmembrane region may affect membrane insertion and pore formation during bacterial lysis.
10. Conclusion
This project demonstrated how protein language models can be used to guide rational mutation design in viral proteins. Although the computational workflow was initially challenging, understanding the concepts and carefully interpreting the model outputs allowed the identification of promising mutation candidates.
By combining LLR mutation scores, structural domain information, and physicochemical reasoning, it was possible to propose several mutations that may improve the stability or functional robustness of the MS2 lysis protein. This approach highlights how computational tools can support protein engineering and help explore sequence space more efficiently.
Overall, this exercise illustrates how integrating computational predictions with biological reasoning can provide a practical strategy for designing and evaluating potential protein variants.

Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
Extra credit (Coming Soon!)

Weekly Reflection:
Testing these different peptide prediction tools was really interesting because each one approaches the problem differently. I noticed that the usefulness of each tool depends a lot on how confident the model is and how interpretable the results are. PeptiVerse was probably the most user-friendly tool, since the interface made it easy to quickly evaluate different peptide properties. In contrast, AlphaFold3 required a bit more effort, but it has a big advantage because it allows us to visualize the interaction between the peptide and the protein, which helps a lot when trying to interpret the structural results
moPPIt was the tool I struggled with the most**.** The inputs were actually straightforward, but the runtime was quite long, and the outputs were harder to interpret compared to the other tools. Waiting for the computation also made the workflow slower compared to PeptiVerse or AlphaFold
Finally, I really appreciate that this class is connected to real research questions, especially in areas like phage therapy and antibiotic resistance. Knowing that our work could potentially contribute to ongoing research efforts or even collaborative publications with MIT researchers makes the assignments feel much more meaningful and motivating
Thanks for reading! This info is also posted in my personal Notion. For more info, enter here! Notion W5
References & Sources:
PART A:
Barman, P., Joshi, S., Sharma, S., Preet, S., Sharma, S., & Saini, A. (2023). Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics. International journal of peptide research and therapeutics, 29(4), 61. https://doi.org/10.1007/s10989-023-10524-3
Berdyński, M., Miszta, P., Safranow, K. et al. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci Rep 12, 103 (2022). https://doi.org/10.1038/s41598-021-03891-8
Wang, L., Wang, N., Zhang, W. et al. Therapeutic peptides: current applications and future directions. Sig Transduct Target Ther 7, 48 (2022). https://doi.org/10.1038/s41392-022-00904-4
PART C:
Claudia C Weber, Simon Whelan, Physicochemical Amino Acid Properties Better Describe Substitution Rates in Large Populations, Molecular Biology and Evolution, Volume 36, Issue 4, April 2019, Pages 679–690, https://doi.org/10.1093/molbev/msz003
James, J. E., & Lascoux, M. (2025). Amino Acid Properties, Substitution Rates, and the Nearly Neutral Theory. Genome biology and evolution, 17(3), evaf025. https://doi.org/10.1093/gbe/evaf025
Zhang Yumeng, Zheng Jared, Zhang Bin (2025) Protein Language Model Identifies Disordered, Conserved Motifs Implicated in Phase Separation eLife 14:RP105309 https://doi.org/10.7554/eLife.105309.2
Sources / Extra Material:
Part 2A
- Gallery View of AlphaFold peptides
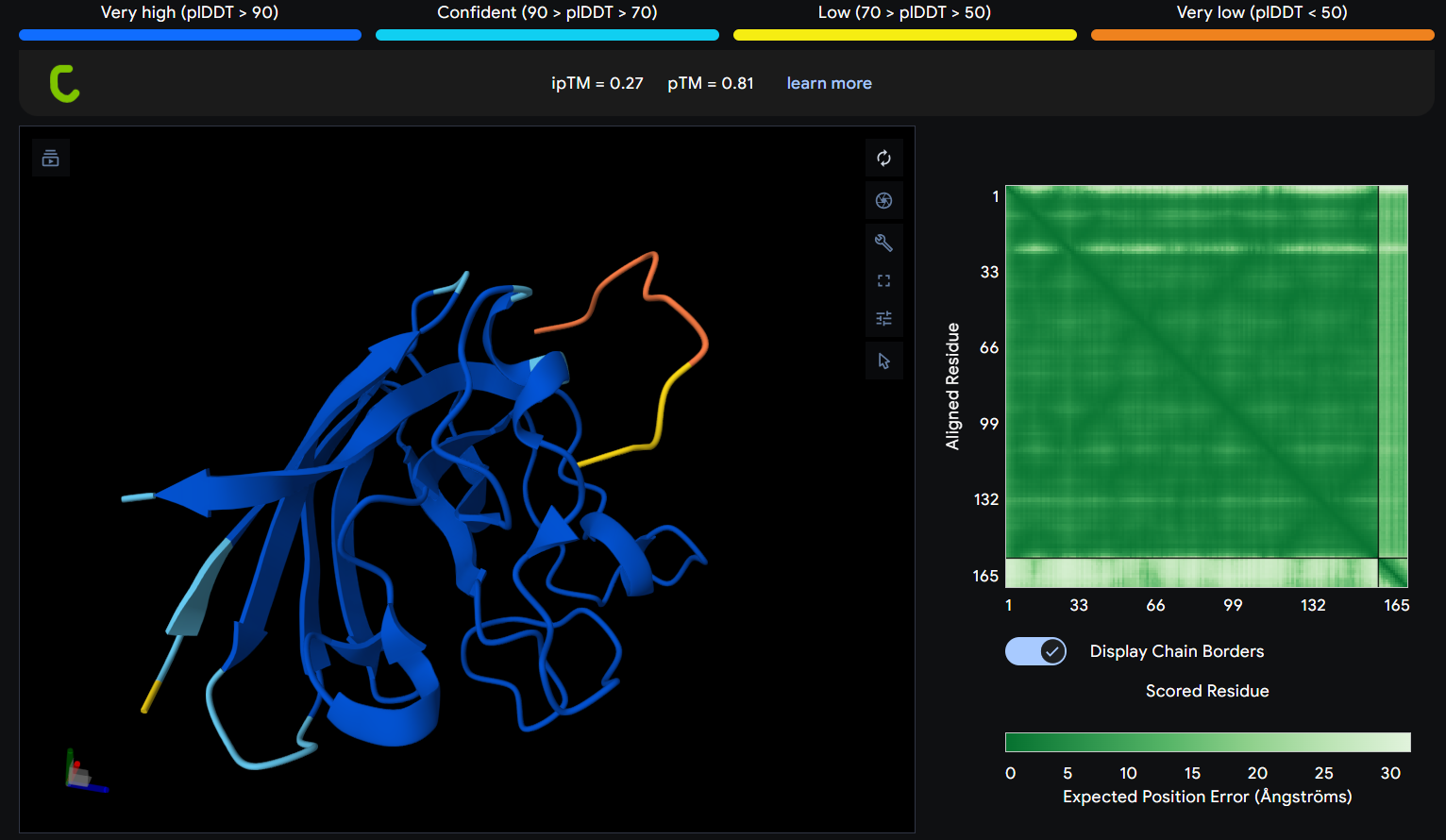
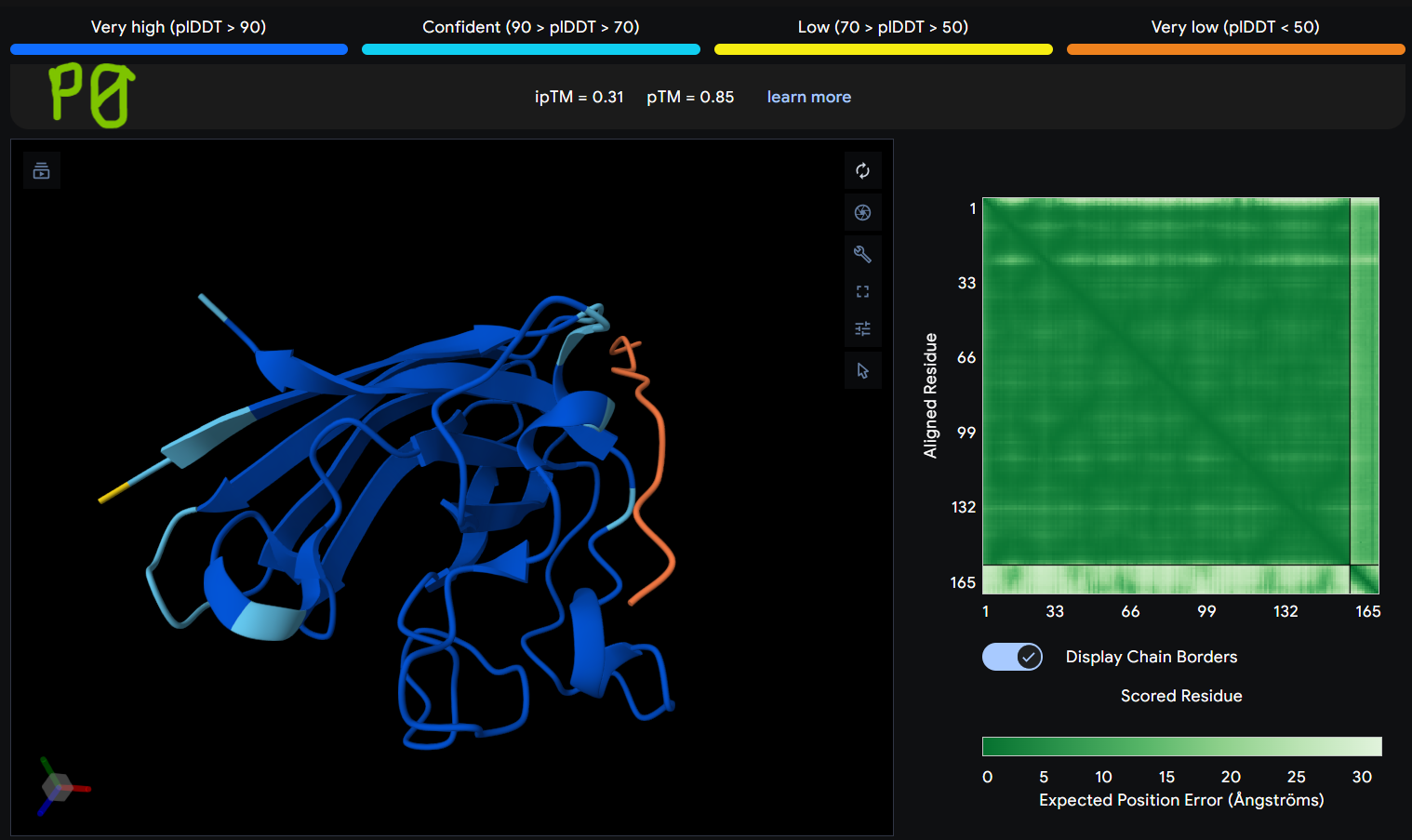
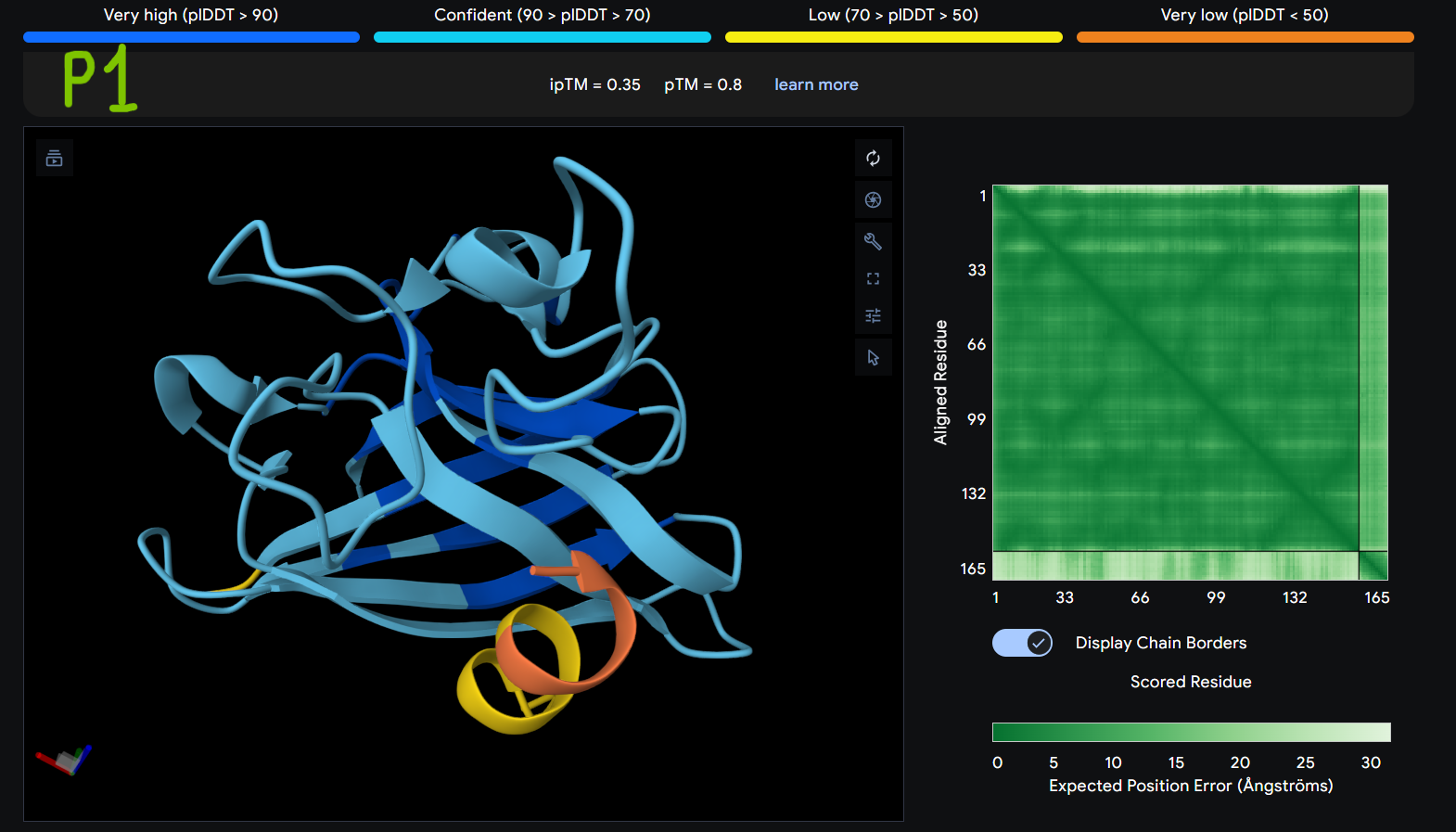
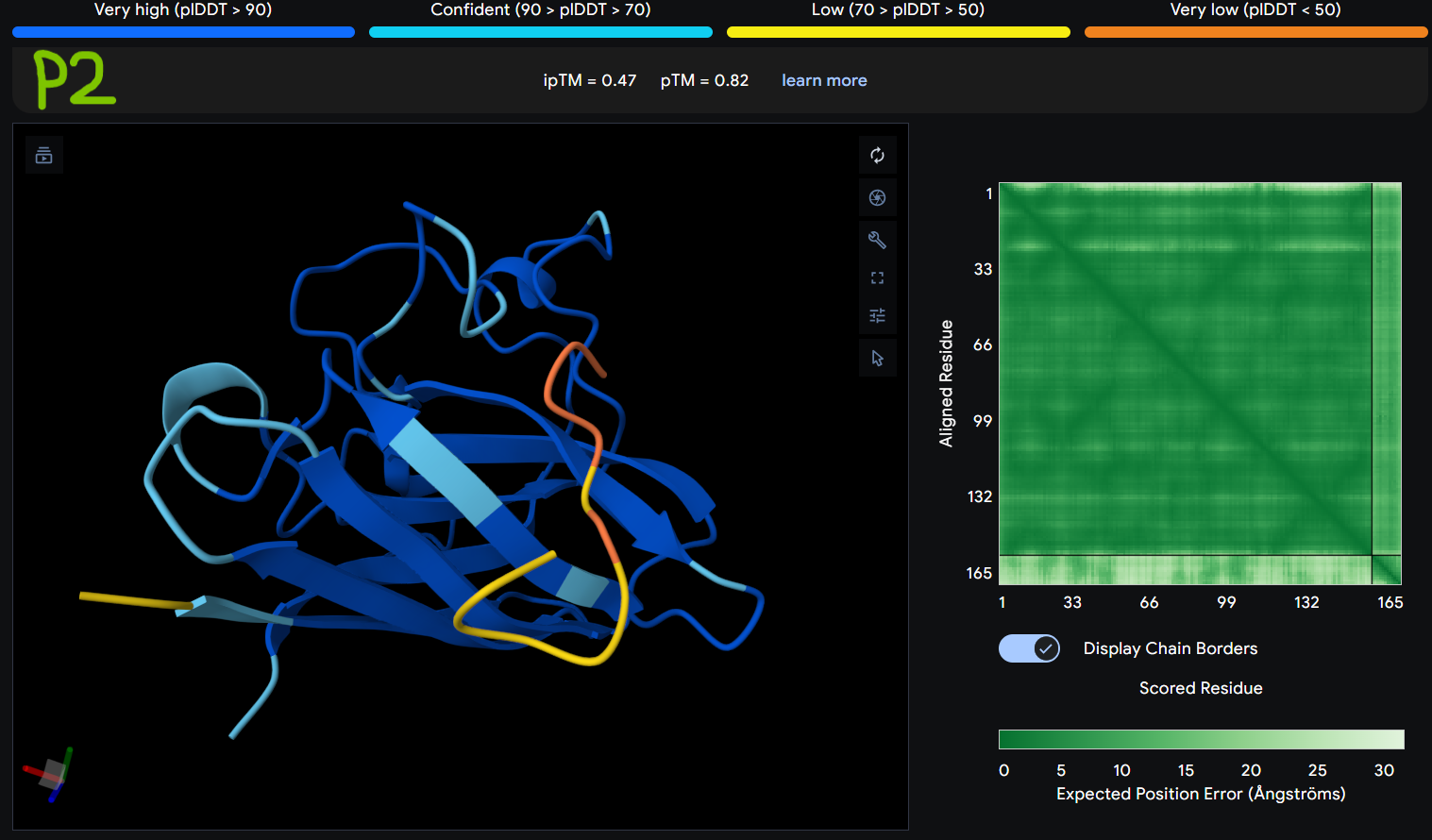
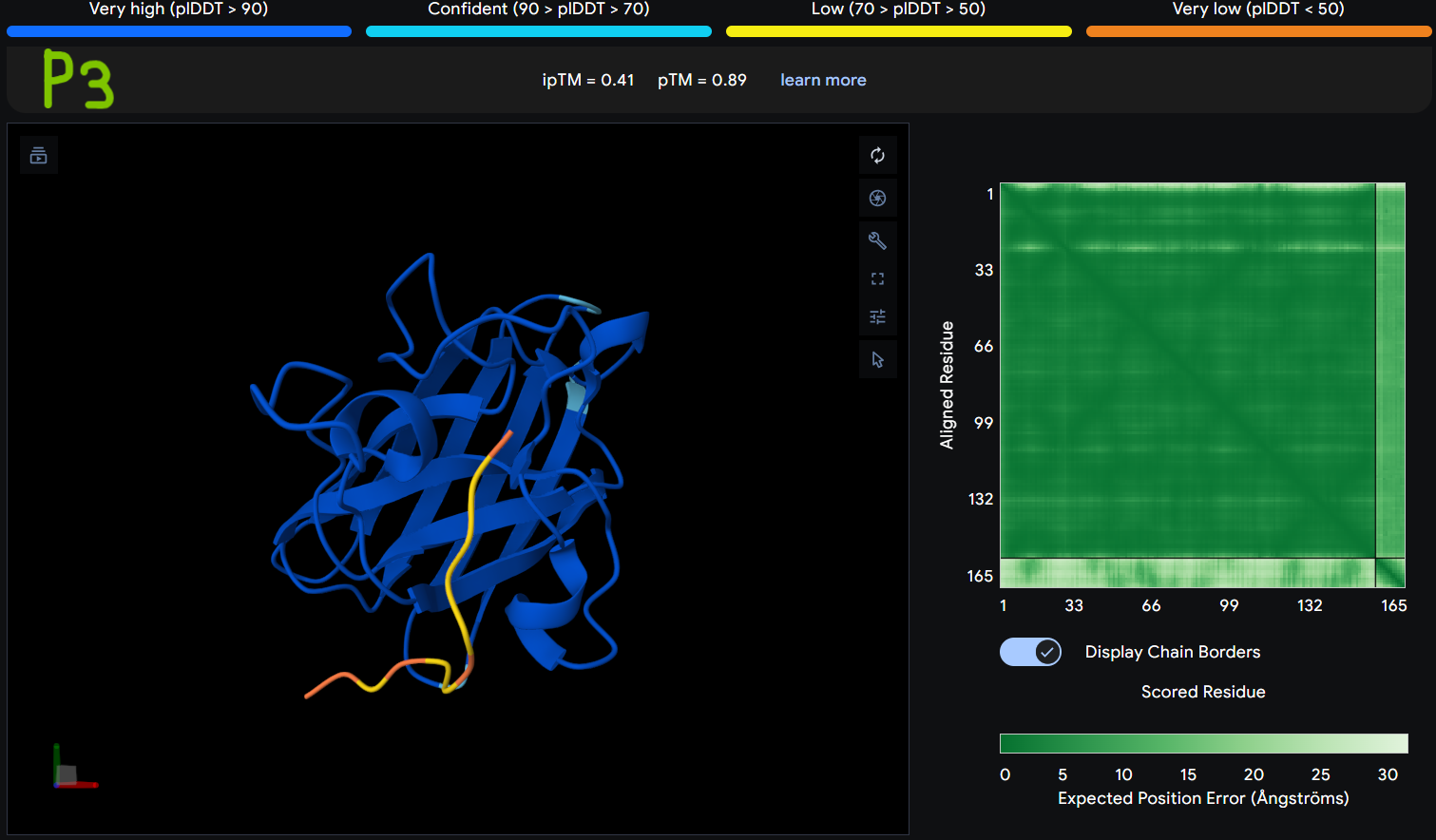
Description: The order of the images is
- “c”= SDO1 (Peptide control)
- “P0” = SOD1 (Pep0)
- “P1” = SOD1 (Pep1)
- “P2” = SOD1 (Pep2)
- “P3” = SOD1 (Pep3)
The Predicted Aligned Error (PAE) heatmap (right side) shows the expected positional error between residue pairs in the predicted structure. Darker green regions indicate lower predicted error and therefore higher confidence in the relative positioning of residues. In this model, the protein core displays low error values, suggesting a reliable fold for SOD1, while the peptide region shows slightly higher uncertainty, which is expected for flexible short peptides interacting with protein surfaces.
Gallery of Tables from Peptiverse for Part 3A
Control:
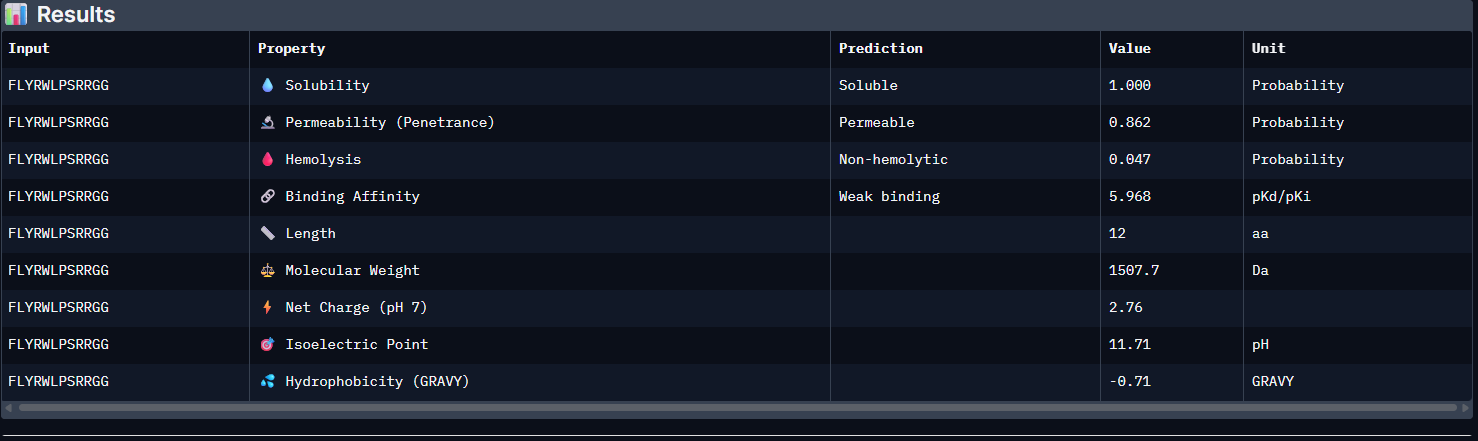
Pep0:
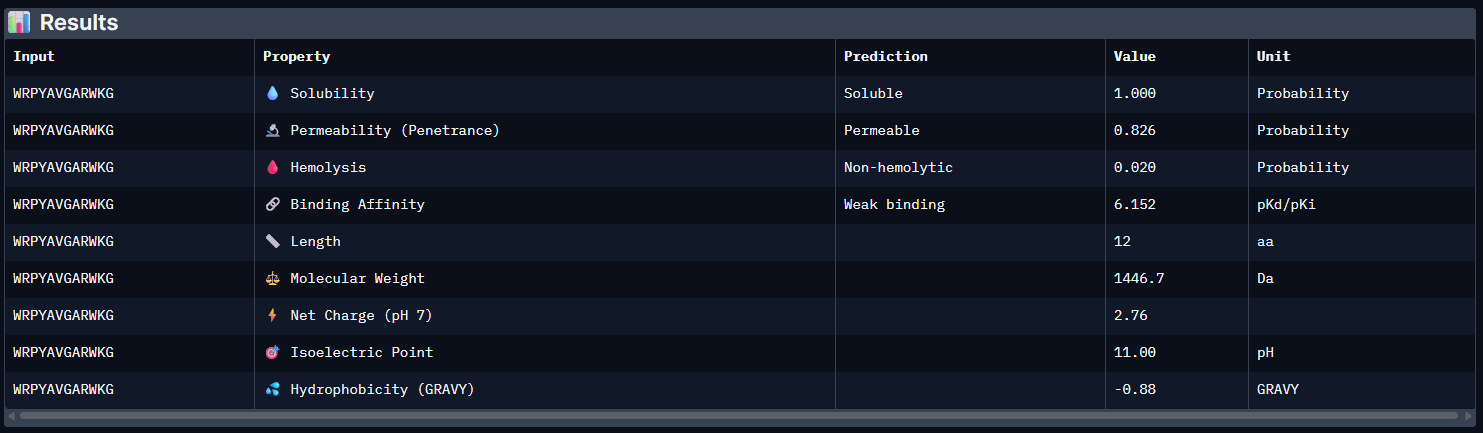
Pep1:
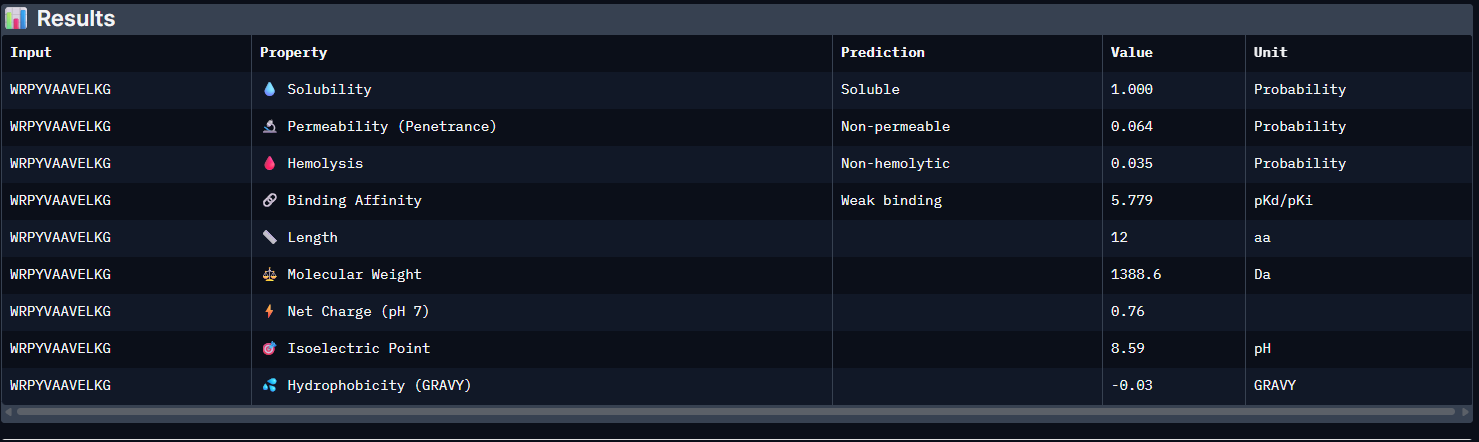
Pep2:
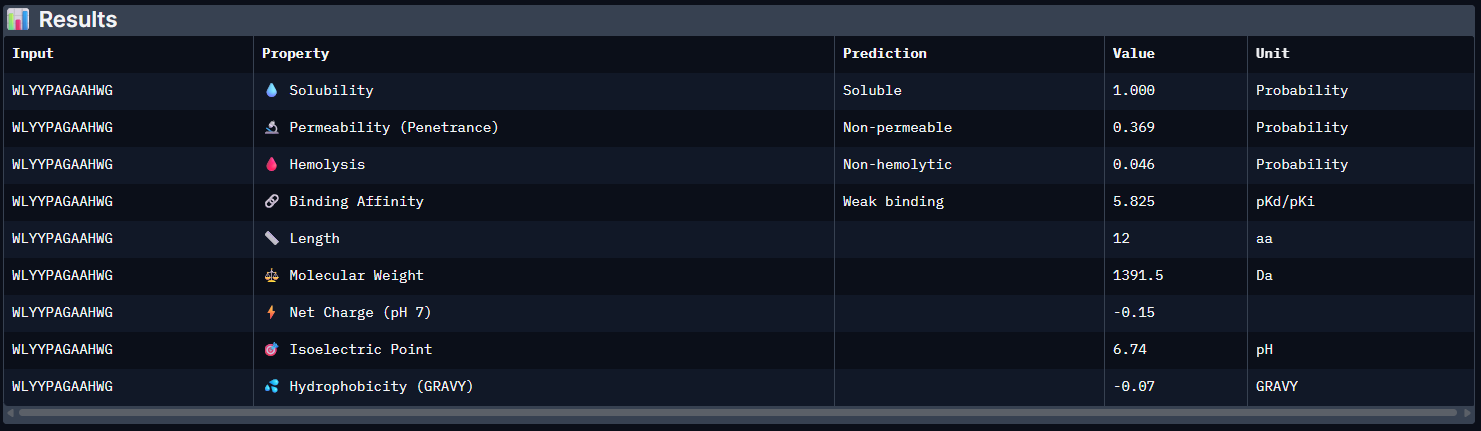
Pep3:
Part 4A: moPPDIt
Outputs: