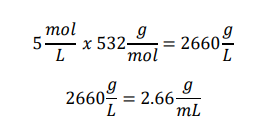Introduction to Pipetting and Dilutions 1. Concentration of the 5 M stock solution in g/mL Given:
Stock concentration = 5 M Molecular weight = 532 g/mol Calculation:
Answer The concentration of the stock solution is 2.66 g/mL.
Week 6 — Genetic Circuits Part I Protocol: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix, and what is their purpose? The Phusion High-Fidelity PCR Master Mix contains several components required for efficient and accurate DNA amplification. The key components are:
Phusion DNA Polymerase: A high-fidelity thermostable polymerase derived from Pyrococcus. It has proofreading (3’→5’ exonuclease) activity, which reduces the error rate during DNA synthesis
Bioproduction of Beta-Carotene and Lycopene 1. Enzymes of the carotene pathway CrtE, CrtB, CrtI, and CrtY are key enzymes involved in carotenoid biosynthesis. CrtE participates in geranylgeranyl pyrophosphate synthesis, CrtB catalyzes phytoene formation, CrtI mediates phytoene desaturation, and CrtY converts lycopene into beta-carotene. Although these enzymes are essential for carotenoid production, recent metabolic engineering studies suggest that carotenoid-specific enzymes do not solely limit lycopene yield. Instead, precursor availability, metabolic flux through the MEP pathway, cofactor balance, and overall cellular metabolism strongly influence production efficiency. (Huang et al., 2025; Jing et al., 2021; Sandmann & Misawa, 1992)
Subsections of Labs
Week 1 Lab: Pipetting
Introduction to Pipetting and Dilutions
1. Concentration of the 5 M stock solution in g/mL
Given:
Stock concentration = 5 M
Molecular weight = 532 g/mol
Calculation:
Answer
The concentration of the stock solution is 2.66 g/mL.
2. Serial dilution plan from 5 M to 100 µM
Because the dilution factor is extremely large, serial dilutions are more accurate than a single-step dilution.
Steps:
Step 1: Dilute 5M to 10 mM (10,000 µM)
Add 2 µL of 5M
Add the rest to complete 1000 µL (998 µL of 𝑑𝐻2𝑂)
𝐷𝑖𝑙𝑢𝑡𝑖𝑜𝑛 1:500
Step 2: Dilute 10.000 µM to 100 µM
Add 10 µL from the previous step
Add 990 µL of 𝑑𝐻2𝑂
𝐷𝑖𝑙𝑢𝑡𝑖𝑜𝑛 1:1000
Tubes & Pipettes - Use 1,5 mL microcentrifuge tubes
Why prepare 100 µM first instead of directly preparing 40 µM?
Preparing 100 µM first improves dilution accuracy and reproducibility. Very large dilution factors increase pipetting error, especially when extremely small volumes are required. Using an intermediate concentration allows more reliable serial dilutions and reduces experimental variability.
What are some components in the Phusion High-Fidelity PCR Master Mix, and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains several components required for efficient and accurate DNA amplification. The key components are:
Phusion DNA Polymerase: A high-fidelity thermostable polymerase derived from Pyrococcus. It has proofreading (3’→5’ exonuclease) activity, which reduces the error rate during DNA synthesis
dNTPs (deoxynucleotide triphosphates): These are the building blocks used by the polymerase to synthesize the new DNA strand
Reaction buffer: Provides optimal ionic strength and pH for polymerase activity. It usually contains Mg²⁺, which is required as a cofactor for DNA polymerase
MgCl₂: Magnesium ions stabilize primer-template interactions and are essential for the catalytic activity of the polymerase
Stabilizers and additives: These help maintain enzyme stability and improve amplification efficiency
The master mix in PCR is prepared to simplify the reaction setup and ensure consistency across multiple reactions. Instead of adding each component separately, the master mix contains the essential reagents required for DNA amplification, such as the polymerase, buffer, Mg²⁺ ions, and dNTPs.
In practice, the total master mix volume is calculated based on the number of reactions to be performed, and it is recommended to prepare additional volume (usually enough for one or two extra reactions) to account for a negative control and possible pipetting errors.
In general, using a master mix also helps reduce variability between reactions and ensures high specificity and low mutation rates during amplification when using high-fidelity enzymes such as Phusion polymerase.
What are some factors that determine primer annealing temperature during PCR?
The primer annealing temperature (Ta) is critical for PCR specificity and efficiency. Some factors that determine the annealing temperature include:
Primer melting temperature (Tm): The annealing temperature is typically 3–5°C below the primer Tm
Primer length: Longer primers generally have higher melting temperatures
GC content: Primers with higher GC content have stronger hydrogen bonding and therefore higher Tm
Primer sequence composition: Runs of GC or secondary structures can affect annealing behavior
Salt concentration in the buffer: Ionic conditions influence primer-template hybridization
Template complexity: Complex templates or repetitive regions may require optimized annealing temperatures
(Rychlik et al., 1990)
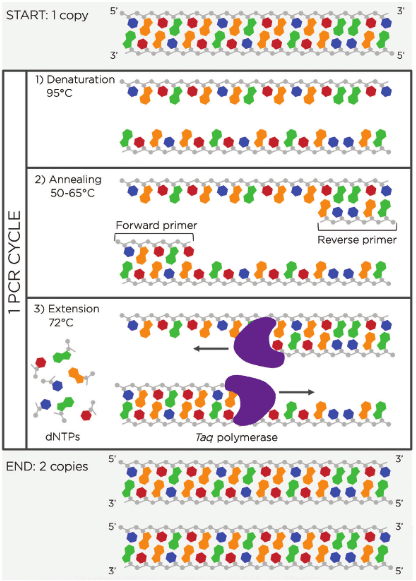
The important part is choosing the correct annealing temperature, which helps ensure that primers bind specifically to the intended target sequence. As well, remember the principal steps of PCR in Figure 1.
Figure 1. MiniPCR graphic ‘Depiction of one PCR cycle.’
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both PCR amplification and restriction enzyme digestion can generate linear DNA fragments, but they differ significantly in methodology and application. In Table 1, the differences between PCR and restriction enzyme digest are summarized.
Table 1. Comparison of PCR vs. Restriction enzyme digest
Category
PCR
Restriction Enzyme Digest
Principle
DNA amplification using primers and polymerase
Cutting DNA using sequence-specific enzymes
DNA requirement
Small amount of template DNA
Requires plasmid or DNA containing restriction sites
Specificity
Determined by primer design
Determined by enzyme recognition sequences
Output
Amplified fragment of specific length
Linearized DNA or defined fragments
Flexibility
Can add overhangs or modifications through primers
Limited to existing restriction sites
When is PCR preferable?
When amplifying a specific gene or fragment
When introducing mutations or overhangs
When restriction sites are not present
When are restriction enzymes preferable?
When cutting plasmids or large DNA constructs
When working with known restriction maps
When cloning using classical restriction-ligation methods
PCR offers greater flexibility, while restriction enzyme digestion provides precise cleavage at defined sequences. Also, PCR amplifies many copy of the DNA or genetic material, while restriction enzymes cut a specific region as mentioned before.
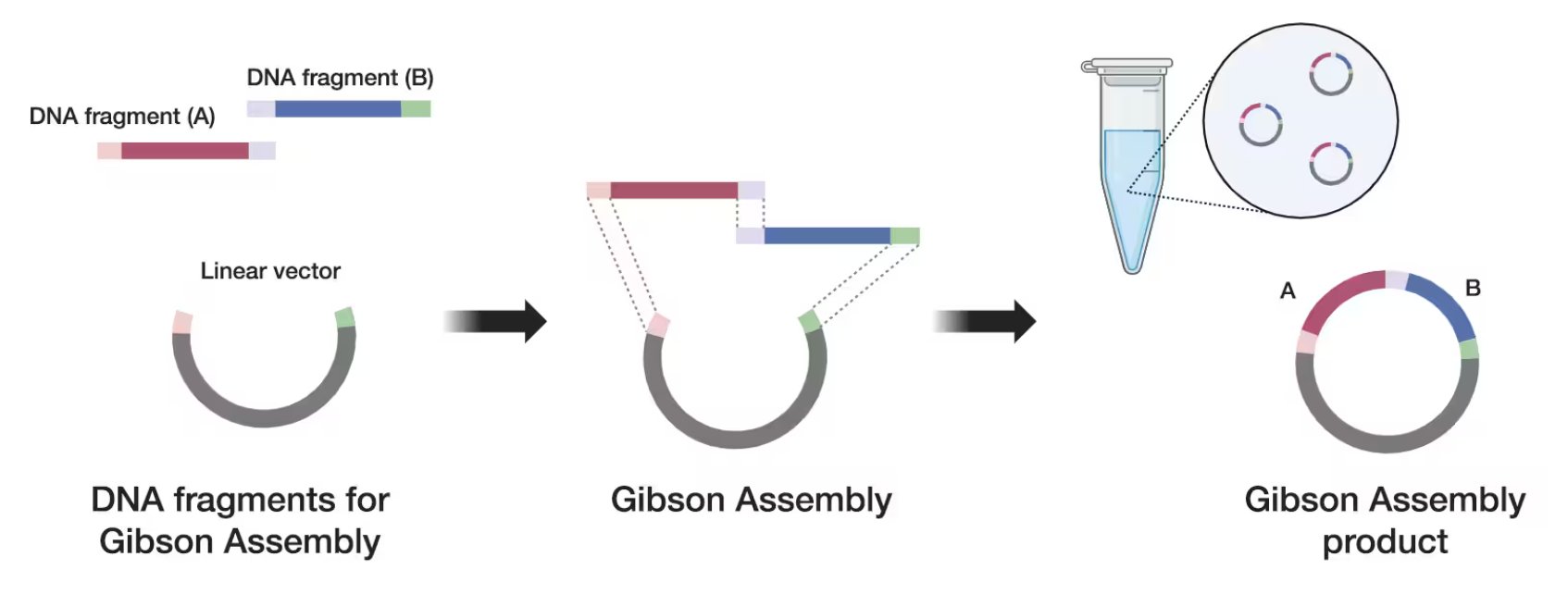
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson Assembly, DNA fragments must contain overlapping homologous regions.
Table 2. Requirements for Gibson Assembly
To ensure compatibility:
Design PCR primers with 20–40 bp overlaps matching adjacent fragments.
Verify sequences in silico using tools such as Benchling.
Ensure fragments are linear and free of secondary structures.
Confirm correct fragment size via gel electrophoresis.
Remove template plasmid contamination if necessary (e.g., DpnI digestion).
Then, those overlapping sequences allow the Gibson reaction enzymes to:
Generate single-stranded overlaps
Anneal complementary regions
Fill gaps and ligate the fragments.
Extra: Thermofisher graphic:
Figure 2. Gibson Assembly 101: Expert Cloning Tips You Need to Know (thermofisher)
How does the plasmid DNA enter the E. coli cells during transformation?
During transformation, plasmid DNA enters E. coli cells that have been made competent, the most common methods are: Chemical transformation & Electroporation.
Table 3. Chemical transformation vs. Electroporation
Chemical transformation
Electroporation
Cells are treated with CaCl₂, which neutralizes negative charges on DNA and the cell membrane
Cells are washed to remove salts
DNA is added to the competent cells
A high-voltage electric pulse creates temporary pores in the membrane
A heat shock (~42°C) briefly disrupts the membrane
After transformation, cells are plated on selective media containing antibiotics to identify successful transformants.
Describe another assembly method in detail (such as Golden Gate Assembly):
a. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a molecular cloning technique that allows the simultaneous assembly of multiple DNA fragments in a single reaction. The method uses Type IIS restriction enzymes, such as BsaI, which cut DNA outside of their recognition sequence to generate customizable overhangs. These overhangs allow DNA fragments to ligate together in a specific and predetermined order. During the reaction, the restriction enzyme first digests the DNA fragments, generating compatible sticky ends, and DNA ligase then joins the fragments together. Because the recognition sites are removed during digestion, the final construct does not contain unwanted restriction sequences, resulting in scarless cloning. This technique is widely used in synthetic biology because it enables efficient multi-fragment assembly in a single tube (Bird et al., 2018).
Diagram:
flowchart LR
A[Fragment A] --> D[BsaI digestion]
B[Fragment B] --> D
C[Fragment C] --> D
D --> E[Sticky overhangs]
E --> F[Ligation]
F --> G[Final plasmid<br>A-B-C]
b. Model this assembly method with Benchling or Asimov Kernel!
Assembly Method with Benchling
For this activity, the Golden Gate Assembly method was modeled using Benchling. The project folder used for this exercise can be accessed through the following link:
To perform the assembly, the plasmid pUC19 was selected as the backbone sequence. The full plasmid sequence was obtained from Addgene, which provides verified plasmid maps and DNA sequences commonly used in molecular biology. Sequence
This fragment includes recognition sites for the Type IIS restriction enzyme BsaI, which is required for Golden Gate Assembly. And its structure has a short alanine-rich peptide used as a synthetic example insert. Golden Gate Assembly uses Type IIS restriction enzymes that cut outside their recognition sequence, allowing the creation of custom overhangs. These overhangs guide the directional ligation of DNA fragments, enabling multiple pieces of DNA to be assembled in a single reaction.
In Benchling, the pUC19 backbone and the synthetic insert were added to the assembly workspace. The Golden Gate cloning method was selected, and BsaI was defined as the restriction enzyme. The insert fragment was generated using a primer pair to simulate PCR amplification, which allows the introduction of compatible overhangs for the assembly.
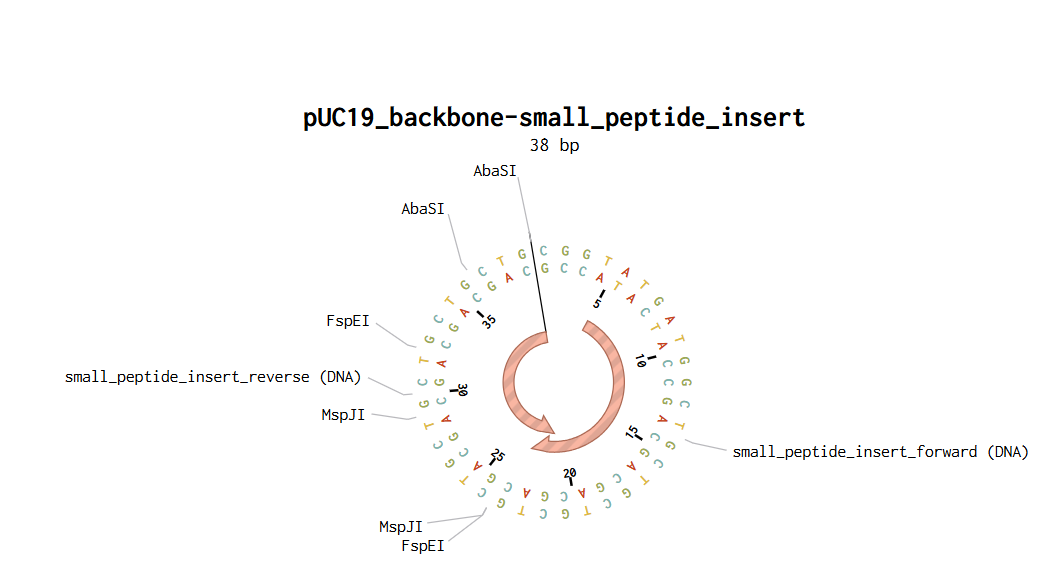
After defining the fragments, Benchling automatically generated compatible sticky ends between the backbone and the insert. Once the fragments were validated, the assembly was finalized to generate the construct pUC19_backbone–small_peptide_insert.
Final Golden Gate Assembly plasmid:
Figure 3. pUC19_backbone–small_peptide_insert by Golden Gate Assembly method
Tutorial for assembly method with Benchling
Additionally, I created a document that shows the process I used to create the plasmid.
Next part of the homework is at: Homework- Week 6 section
References & sources:
Asif, A., Mohsin, H., Tanvir, R., & Rehman, Y. (2017). Revisiting the Mechanisms Involved in Calcium Chloride Induced Bacterial Transformation. Frontiers in microbiology, 8, 2169. https://doi.org/10.3389/fmicb.2017.02169Open Access
Bird, J. E., Marles-Wright, J., & Giachino, A. (2022). A User’s Guide to Golden Gate Cloning Methods and Standards. ACS synthetic biology, 11(11), 3551–3563. https://doi.org/10.1021/acssynbio.2c00355Open Access
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., 3rd, & Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods, 6(5), 343–345. https://doi.org/10.1038/nmeth.1318Open Access
Liu, J., Chang, W., Pan, L., Liu, X., Su, L., Zhang, W., Li, Q., & Zheng, Y. (2018). An Improved Method of Preparing High Efficiency Transformation Escherichia coli with Both Plasmids and Larger DNA Fragments. Indian journal of microbiology, 58(4), 448–456. https://doi.org/10.1007/s12088-018-0743-zOpen Access
Rychlik, W., Spencer, W. J., & Rhoads, R. E. (1990). Optimization of the annealing temperature for DNA amplification in vitro. Nucleic acids research, 18(21), 6409–6412. https://doi.org/10.1093/nar/18.21.6409Open Access
Week 12 Lab: Bioproduction of Beta-Carotene and Lycopene
Bioproduction of Beta-Carotene and Lycopene
1. Enzymes of the carotene pathway
CrtE, CrtB, CrtI, and CrtY are key enzymes involved in carotenoid biosynthesis. CrtE participates in geranylgeranyl pyrophosphate synthesis, CrtB catalyzes phytoene formation, CrtI mediates phytoene desaturation, and CrtY converts lycopene into beta-carotene. Although these enzymes are essential for carotenoid production, recent metabolic engineering studies suggest that carotenoid-specific enzymes do not solely limit lycopene yield. Instead, precursor availability, metabolic flux through the MEP pathway, cofactor balance, and overall cellular metabolism strongly influence production efficiency. (Huang et al., 2025; Jing et al., 2021; Sandmann & Misawa, 1992)
2. Rate-determining step
Although phytoene desaturation mediated by CrtI has historically been considered an important control point in carotenoid biosynthesis, recent studies suggest that carotenoid production is often limited by precursor supply and metabolic flux rather than by a single carotenoid-specific enzyme. In engineered E. coli systems, optimization of the MEP pathway, carbon metabolism, and cofactor availability can significantly influence lycopene and β-carotene yields (Wang et al., 2019; Liu et al., 2026).
DNA Construct Design
1. Choice of production organism
I would choose Escherichia coli as the production organism for this construct because the goal of this lab is to design a microbial system for carotenoid bioproduction, especially lycopene or β-carotene. E. coli is a convenient chassis because it grows quickly, is inexpensive to culture, and has well-established plasmid-based expression systems. In addition, several metabolic engineering strategies for carotenoid production in E. coli focus on improving precursor supply through the MEP pathway, optimizing central carbon metabolism, and balancing expression of heterologous carotenoid genes. For this reason, E. coli would be a practical host for a first plasmid-based design.
(Liu et al., 2026; Wu et al., 2020)
2. Example enzyme for expression
For the expression construct, I would choose CrtY, the lycopene β-cyclase enzyme. This enzyme converts lycopene into β-carotene, so it is directly relevant if the desired final product is β-carotene. In this design, lycopene can be considered the precursor, and CrtY would redirect the pathway toward β-carotene accumulation. This choice also makes the construct easy to explain because the presence or absence of CrtY activity helps determine whether the engineered system accumulates lycopene or produces β-carotene.
(Wang et al., 2020; Liu et al., 2026)
A promoter is a regulatory DNA sequence that controls the initiation of transcription by recruiting RNA polymerase and transcriptional machinery. In engineered microbial systems, promoters are essential for regulating the expression level of heterologous genes involved in biosynthetic pathways.
2. Types of promoters
Promoters can be classified as constitutive, inducible, or repressible promoters. Constitutive promoters continuously drive gene expression, whereas inducible promoters activate transcription in response to specific molecules such as IPTG or arabinose. Repressible promoters decrease transcription in response to regulatory signals or metabolites. In metabolic engineering, inducible promoters are commonly used to balance cell growth and product synthesis while minimizing metabolic burden.
3. Promoter response to metabolites
If the goal is to suppress transcription in response to a metabolite, repressible promoters would be useful because they decrease gene expression after sensing a specific molecule. Conversely, inducible promoters are useful when gene expression should increase in the presence of a metabolite or inducer. Dynamic promoter regulation is important in metabolic engineering because excessive expression of biosynthetic genes can impose a metabolic burden and reduce cell growth.
4. Promoter choice
For this construct, I would choose a strong inducible promoter such as the T5 promoter. Recent metabolic engineering studies demonstrated that replacing native promoters of MEP pathway genes with strong T5 promoters significantly improved β-carotene production in engineered E. coli strains. Strong promoters can enhance precursor flux and increase carotenoid biosynthesis, although excessive overexpression may also generate metabolic burden and reduce cellular fitness (Liu et al., 2026).
Origin of Replication
1. What is an origin of replication?
The origin of replication (Ori) is the DNA sequence that allows a plasmid to replicate independently inside the host cell. The selected origin strongly influences plasmid copy number, stability, and metabolic burden.
(Cooper, 2000)
2. Types of origins
Origins of replication can be classified as high-copy-number, medium-copy-number, or low-copy-number origins. High-copy plasmids typically produce larger amounts of recombinant proteins or metabolites, whereas low-copy plasmids reduce metabolic burden and improve cellular stability.
(Ding & Koren, 2020)
3. Compatibility groups
If the goal is to suppress transcription in response to a metabolite, repressible promoters would be useful because they decrease gene expression after sensing a specific molecule. Conversely, inducible promoters are useful when gene expression should increase in the presence of a metabolite or inducer. Dynamic promoter regulation is important in metabolic engineering because excessive expression of biosynthetic genes can impose a metabolic burden and reduce cell growth.
(Ding & Koren, 2020)
Best origin for this construct
For this construct, a medium-copy-number origin such as p15A ori would be appropriate because it provides a balance between carotenoid production and cellular fitness. High-copy plasmids may increase carotenoid synthesis but can also generate significant metabolic burden and stress on the host cell.
(Liu et al., 2026)
Other bioparts
RBS
Ribosome binding sites (RBSs) are short regulatory sequences that recruit ribosomes and initiate translation. RBS strength strongly influences protein expression levels and can be tuned to balance metabolic flux in engineered biosynthetic pathways.
(Wu et al., 2018)
Terminators
Terminators are DNA sequences that stop transcription and stabilize RNA transcripts. Proper terminator selection reduces transcriptional readthrough and improves the stability of engineered genetic circuits.
(Torella et al., 2013; Deaner & Alper, 2016)
Operators
Operators are regulatory DNA elements recognized by transcription factors or repressors. They help regulate promoter activity and allow dynamic control of gene expression in response to environmental or metabolic signals.
(Addgene, 2019)
Aptamers and riboswitches
Aptamers are nucleic acid structures capable of binding specific target molecules, whereas riboswitches are regulatory RNA elements that alter gene expression in response to metabolites. In metabolic engineering, riboswitches can dynamically regulate biosynthetic pathways to improve metabolic balance, reduce toxicity, and optimize carotenoid production.
(Zhang et al., 2019)
Joining DNA parts together
The plasmid construct could be assembled using restriction enzyme cloning through the Benchling assembly wizard. Restriction enzymes can generate compatible sticky ends between the plasmid backbone and the insert containing the promoter, RBS, crtY coding sequence, and terminator. This approach allows modular insertion of the carotenoid expression cassette into an existing plasmid backbone.
Dream biosynthetic pathway
One interesting dream biosynthetic pathway would involve engineering Escherichia coli to detect early biofilm-associated physiological states on food-contact surfaces and activate a fluorescent or antimicrobial response before mature biofilm formation occurs. This system could combine biosensing circuits with metabolic pathways capable of producing detectable pigments, antimicrobial peptides, or protective metabolites in response to quorum sensing or stress-associated signals. Such a platform could contribute to early contamination monitoring and improved food safety strategies while reducing the risk of persistent biofilm-associated infections.
1. Mockup Plasmid Creation
A conceptual crtY expression plasmid was designed in Benchling using a pAC-BETA-inspired backbone for simulated carotenoid bioproduction in Escherichia coli. The objective was to create a simplified synthetic biology construct containing a promoter, ribosome binding site, crtY coding sequence, and transcriptional terminator assembled into a plasmid backbone carrying an origin of replication and chloramphenicol resistance marker.
DNA Sequence | Optimize with Cusabio. Online
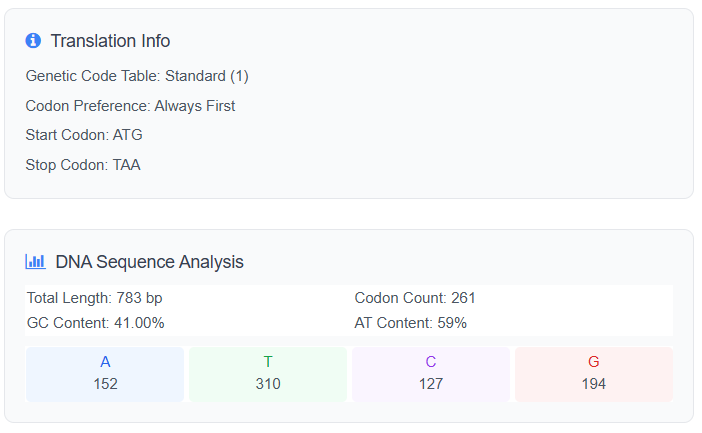
ATGATGCATTATGATAATGTTGGTGCTGGTGCTAATGGTAATGCTCGTGATATGCGTAATAATGATGCTGCTGCTGGTGGTAATCATACTTGGTCTCATCATGATGATACTTCTCATCGTTGGAATGCTGTTGTTCATCATTGGGATTATGTTCGTACTCGTCGTCGTAAAAATTCTGGTTATTGTAATACTTCTCGTGCTGTTCGTGGTCATTGGATGGATACTGCTGTTGCTGTTAATGCTTCTGTTCGTAAAAAAGGTGTTAATGGTGCTCGTGCTGTTAATGATGGTCGTGGTTATGCTGCTAATTCTGCTTCTGTTGGTGCTAATGGTTGGCGTTCTCATCATGGTTCTTCTAATAATATGGATGCTACTGTTGATAATGGTTATCGTGTTTATTCTTCTACTCGTAATGATACTCATTATAATGATAATGCTACTGATTGTGCTCGTAATAATTGTGATTATGCTGCTGGTTGGACTCGTGGTGCTAATACTTCTGGTAATGCTGATGCTTGGCGTGCTTGTTCTGGTCGTGCTGGTCATACTACTGGTTATTCTGCTGTTGCTGTTGCTGATCGTTCTGCTGATGTTACTTCTGCTTCTAATCATCATGCTAATACTCATGCTCGTCGTTGGGGTCGTATGAATCGTATGGCTGGTGCTGATTCTCGTTGGCGTGTTATGCGTTATGGTGATAATGCTCGTTATGCTGGTAAAACTACTGATCGTCGTAATTCTGGTAAAGTTGTTGCTGCTGCTAATATGACTACTCATCGTTAA
Analyze DNA Sequence from Cusabio:
Link of the Reverse Translate Protein to DNA: Cusabio.online
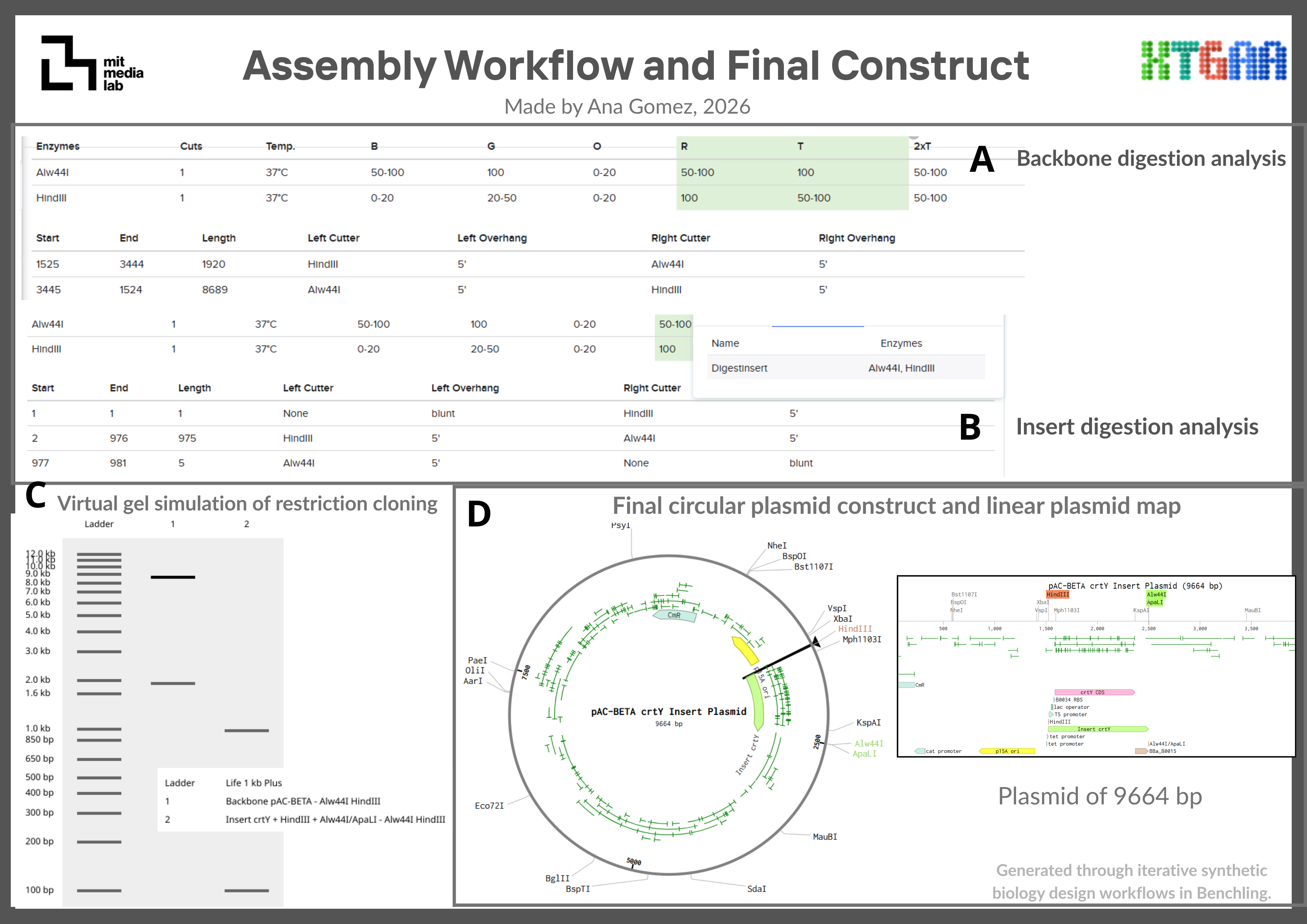
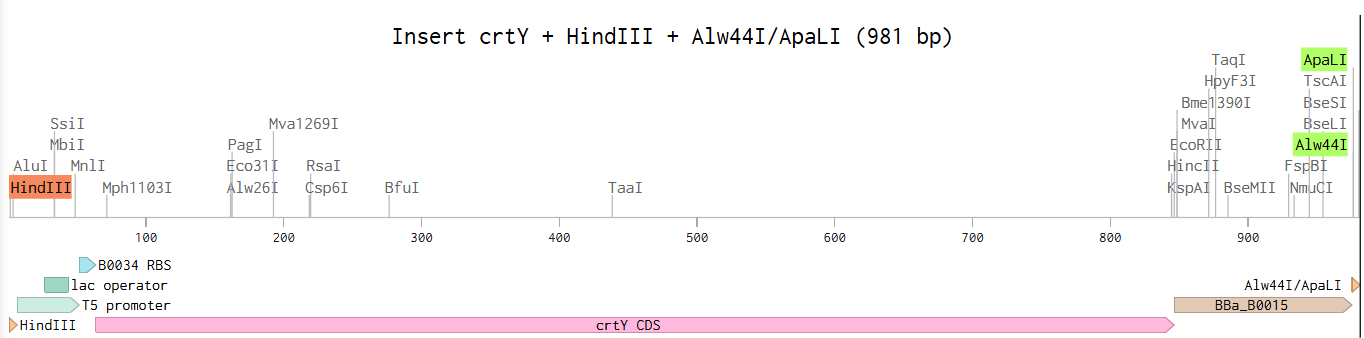
Restriction cloning was simulated using HindIII and Alw44I/ApaLI restriction enzymes. These enzymes were selected because they cut once within the pAC-BETA backbone while avoiding internal cuts within the crtY insert sequence, allowing directional insertion of the expression cassette into the plasmid backbone.
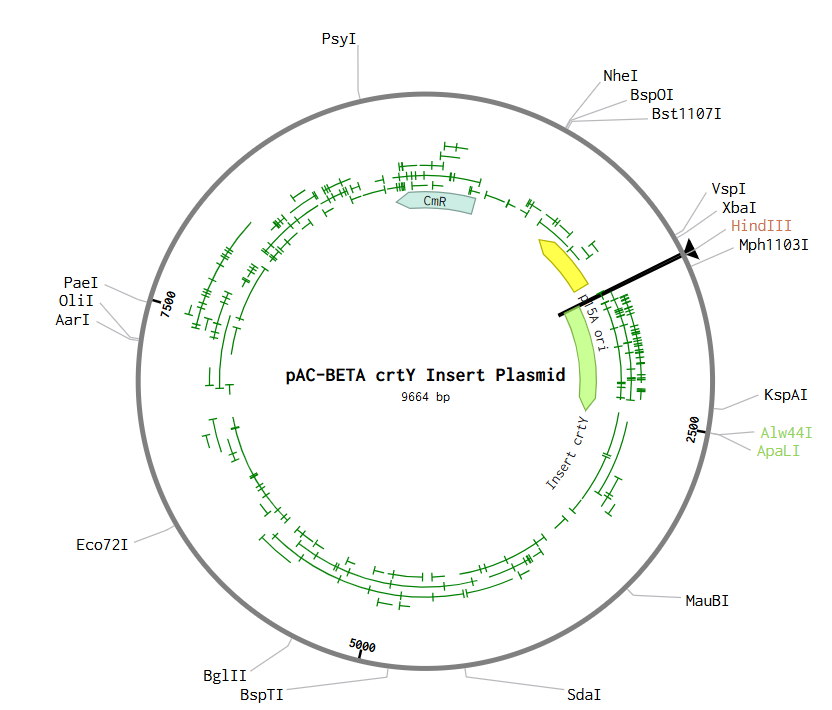
pAC-BETA crtY Insert Plasmid
Enzyme
Position
Cut site sequence
Strand
HindIII
1525-1525
AAGCTT
Forward
Alw44I/ApaLI
2499-2499
GTGCAC
Forward
Insert crtY
Enzyme
Position
Cut site sequence
Strand
HindIII
1-6
AAGCTT
Forward
Alw44I/ApaLI
976-981
GTGCAC
Forward
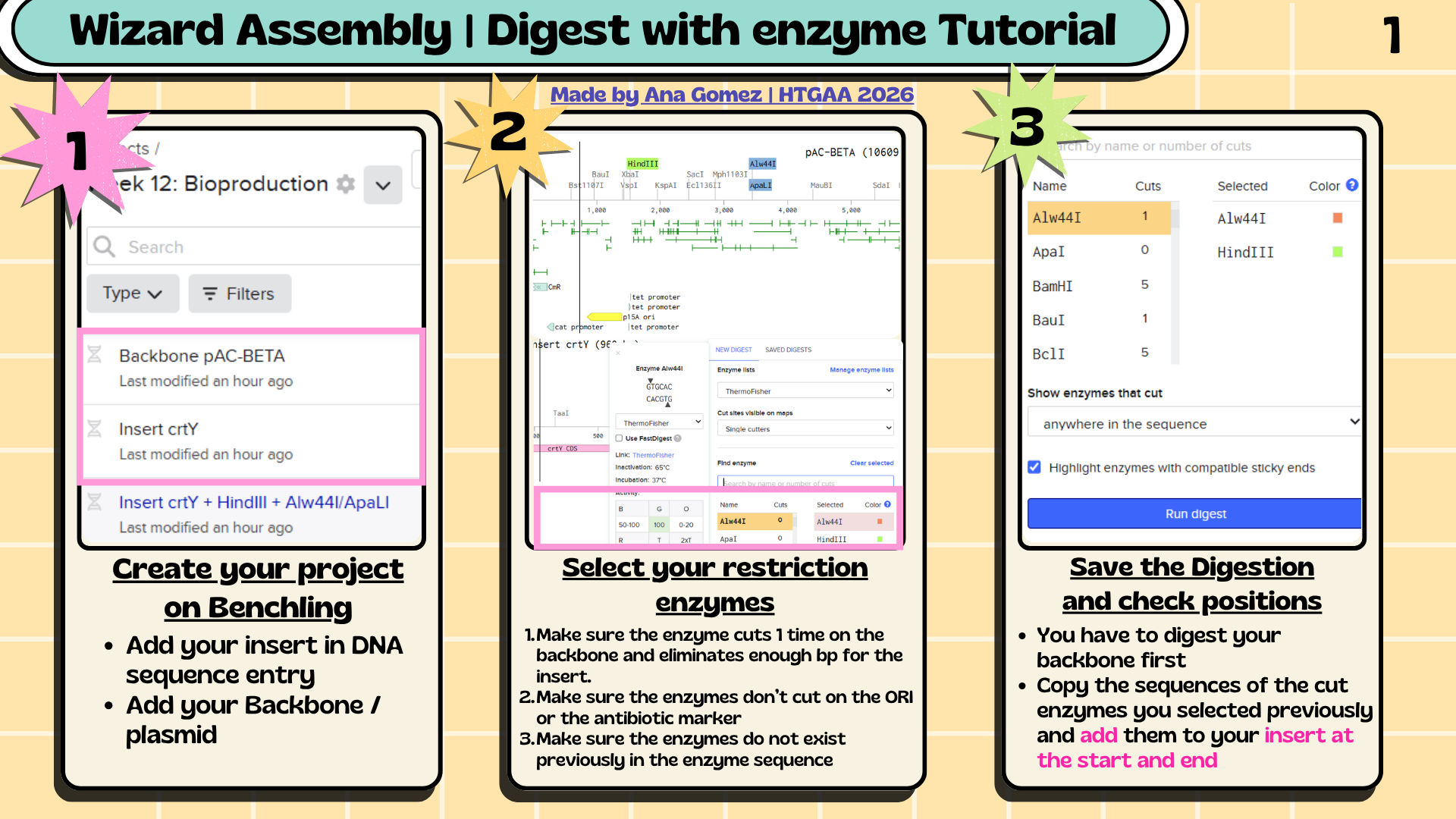
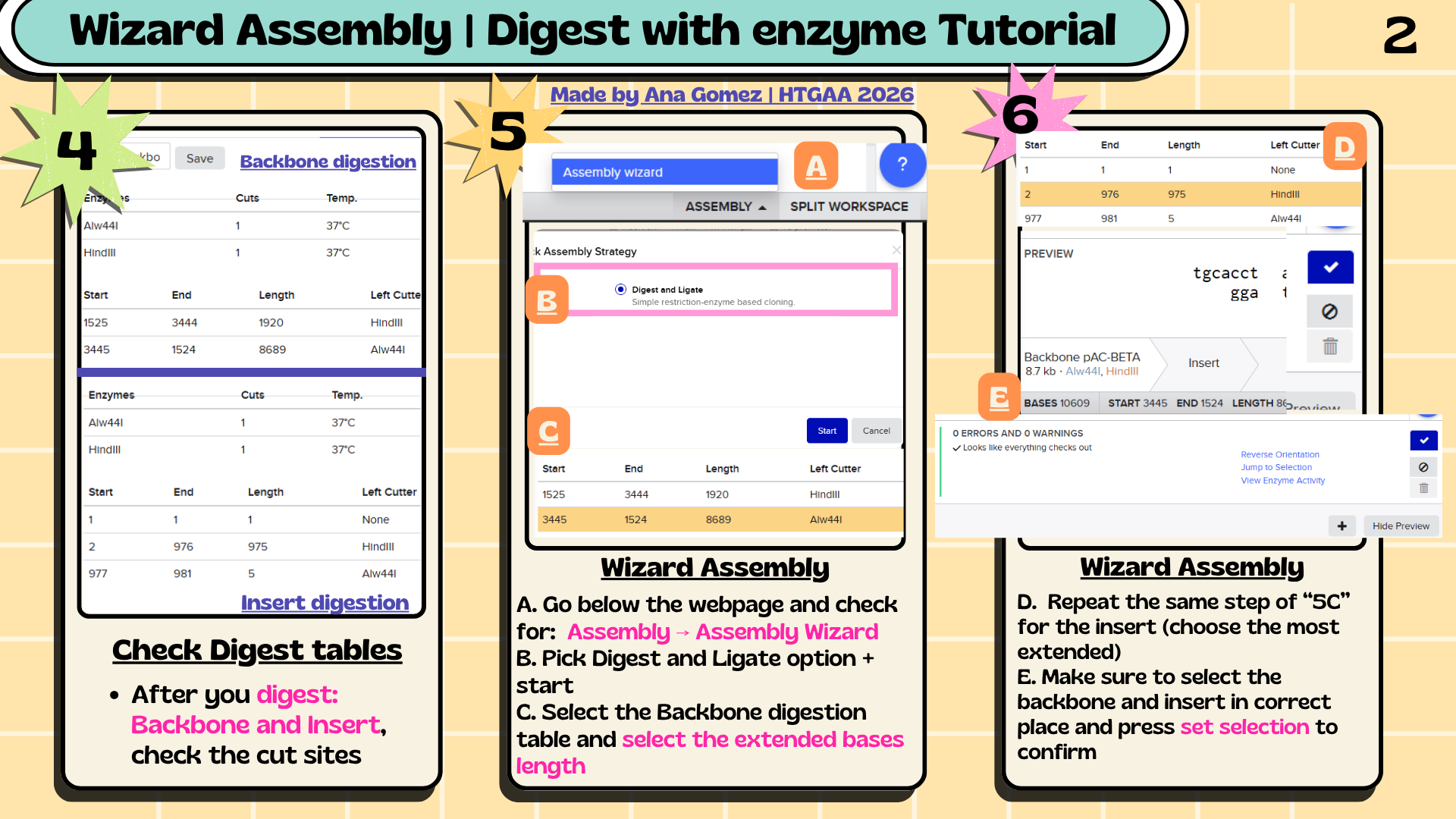
6. Assembly Workflow
Figure 1. Overview of the restriction cloning workflow performed in Benchling for the assembly of the crtY expression cassette into the pAC-BETA-inspired backbone.
6.1 The assembly workflow consisted of:
Analyze restriction sites in the plasmid backbone.
Select restriction enzymes compatible with the insert sequence.
Add restriction sites to the crtY insert.
Simulate backbone digestion in Benchling.
Assemble the insert into the backbone using the Benchling Assembly Wizard.
A conceptual crtY expression plasmid was designed in Benchling using a pAC-BETA-inspired backbone containing the p15A origin of replication and chloramphenicol resistance marker. The crtY expression cassette included a T5 promoter, lac operator, B0034 ribosome binding site, crtY coding sequence, and B0015 terminator. Restriction cloning was simulated using HindIII and Alw44I restriction sites to assemble the insert into the plasmid backbone. It’s summarized in Figures 2 & 3 for the insert and the final plasmid.
Figure 2. Conceptual crtY expression cassette containing the T5 promoter, lac operator, B0034 ribosome binding site, crtY coding sequence, and B0015 terminator flanked by HindIII and Alw44I/ApaLI restriction sites.
Deaner, M., & Alper, H. S. (2016). Promoter and terminator Discovery and Engineering. Advances in Biochemical Engineering, Biotechnology, 162, 21–44. https://doi.org/10.1007/10_2016_8
Ding, Q., & Koren, A. (2020). Positive and Negative Regulation of DNA Replication Initiation. Trends in genetics : TIG, 36(11), 868–879. https://doi.org/10.1016/j.tig.2020.06.020
Huang, G., Lan, Y., Duan, C., & Yan, G. (2025). Engineering microbial cell factories for the production of lycopene: Advances and perspectives. Food Research International, 227, 118270. https://doi.org/10.1016/j.foodres.2025.118270
Jing, Y., Guo, F., Zhang, S., Dong, W., Zhou, J., Xin, F., Zhang, W., & Jiang, M. (2021). Recent advances on biological synthesis of lycopene by using industrial yeast. Industrial & Engineering Chemistry Research, 60(9), 3485–3494. https://doi.org/10.1021/acs.iecr.0c05228
Liu, J., Shi, Y., Zhao, D., Lin, M., Wang, P., Zhou, Y., & Yan, X. (2026). Strategies for Metabolic Engineering of Escherichia coli for β-Carotene Biosynthesis. Molecules (Basel, Switzerland), 31(4), 611. https://doi.org/10.3390/molecules31040611
Sandmann, G., & Misawa, N. (1992). New functional assignment of the carotenogenic genescrtBandcrtEwith constructs of these genes fromErwiniaspecies. FEMS Microbiology Letters, 90(3), 253–258. https://doi.org/10.1111/j.1574-6968.1992.tb05162.x
Torella, J. P., Boehm, C. R., Lienert, F., Chen, J., Way, J. C., & Silver, P. A. (2013). Rapid construction of insulated genetic circuits via synthetic sequence-guided isothermal assembly. Nucleic Acids Research, 42(1), 681–689. https://doi.org/10.1093/nar/gkt860
Wang, C., Zhao, S., Shao, X., Park, J., Jeong, S., Park, H., Kwak, W., Wei, G., & Kim, S. (2019). Challenges and tackles in metabolic engineering for microbial production of carotenoids. Microbial Cell Factories, 18(1), 55. https://doi.org/10.1186/s12934-019-1105-1
Wang, Z., Sun, J., Yang, Q., & Yang, J. (2020). Metabolic Engineering Escherichia coli for the Production of Lycopene. Molecules, 25(14), 3136. https://doi.org/10.3390/molecules25143136
Wu, Y., Yan, P., Li, Y., Liu, X., Wang, Z., Chen, T., & Zhao, X. (2020). Enhancing β-Carotene Production in Escherichia coli by Perturbing Central Carbon Metabolism and Improving the NADPH Supply. Frontiers in Bioengineering and Biotechnology, 8, 585. https://doi.org/10.3389/fbioe.2020.00585
Wu, F., Zhang, Q., & Wang, X. (2018). Design of adjacent transcriptional regions to tune gene expression and facilitate circuit construction. Cell Systems, 6(2), 206-215.e6. https://doi.org/10.1016/j.cels.2018.01.010
Zhang, Y., Lai, B. S., & Juhas, M. (2019). Recent Advances in Aptamer Discovery and Applications. Molecules (Basel, Switzerland), 24(5), 941. https://doi.org/10.3390/molecules24050941