Group Final Project- Computational Engineering of the MS2 Lysis Protein to Modulate Lysis Timing and Improve Viral Yield

Computational Engineering of the MS2 Lysis Protein to Modulate Lysis Timing and Improve Viral Yield (Mini-documentation)
Abstract
Bacteriophage lysis timing plays a critical role in virion assembly efficiency and viral yield. In the MS2 bacteriophage system, the small membrane-associated L protein is responsible for host lysis and may be influenced by structural alterations caused by amino acid substitutions. This mini computational study evaluated previously reported MS2 L-protein mutations associated with altered lysis phenotypes to determine whether these variants preserved structurally plausible membrane-associated conformations. Mutant variants (L44P, F47Y, and R30L) were computationally generated from the WT MS2 L-protein sequence and analyzed using Benchling Boltz-2 and AlphaFold2 structural prediction approaches. Comparative structural analysis revealed that all variants preserved alpha-helical membrane-associated regions to varying degrees, although mutations produced distinct local conformational perturbations. Among the evaluated candidates, R30L displayed the closest structural similarity to the WT prediction, whereas L44P showed stronger local structural alterations consistent with the helix-disrupting properties of proline residues. These results suggest that selected MS2-L mutations may preserve structural plausibility while potentially altering local structural dynamics relevant to lysis-associated behavior. This work provides a preliminary computational framework for future experimental phage-engineering studies focused on lysis timing modulation and viral yield optimization.
Keywords: MS2 bacteriophage, lysis protein, protein engineering, structural prediction, AlphaFold2, membrane-associated proteins, phage engineering, computational biology
3. Objectives
The objective of this mini computational study is to evaluate previously reported MS2 L-protein mutations associated with altered lysis phenotypes and assess whether these variants preserve plausible structural integrity for future phage engineering applications.
Aim 1
Select and computationally evaluate previously reported MS2-L mutations associated with altered lysis phenotypes to identify structurally plausible candidates for future phage engineering applications.
Aim 2
Experimentally evaluate selected MS2-L variants in E. coli systems to determine whether altered lysis timing improves virion assembly efficiency and increases viral yield.
Aim 3
Develop computationally guided phage-engineering strategies capable of improving bacteriophage adaptability and robustness against bacterial resistance mechanisms for future phage therapy applications.
4. Pipeline / Workflow
- Retrieve the wild-type MS2 L-protein sequence from UniProt.
- Select previously reported mutations associated with altered lysis phenotypes from the MIT HTGAA dataset.
- Generate mutant protein variants computationally.
- Predict WT and mutant protein structures using ESMFold.
- Compare predicted structural integrity and membrane-associated features between WT and mutant variants.
- Identify structurally plausible candidates for future experimental evaluation related to lysis timing and viral yield modulation.
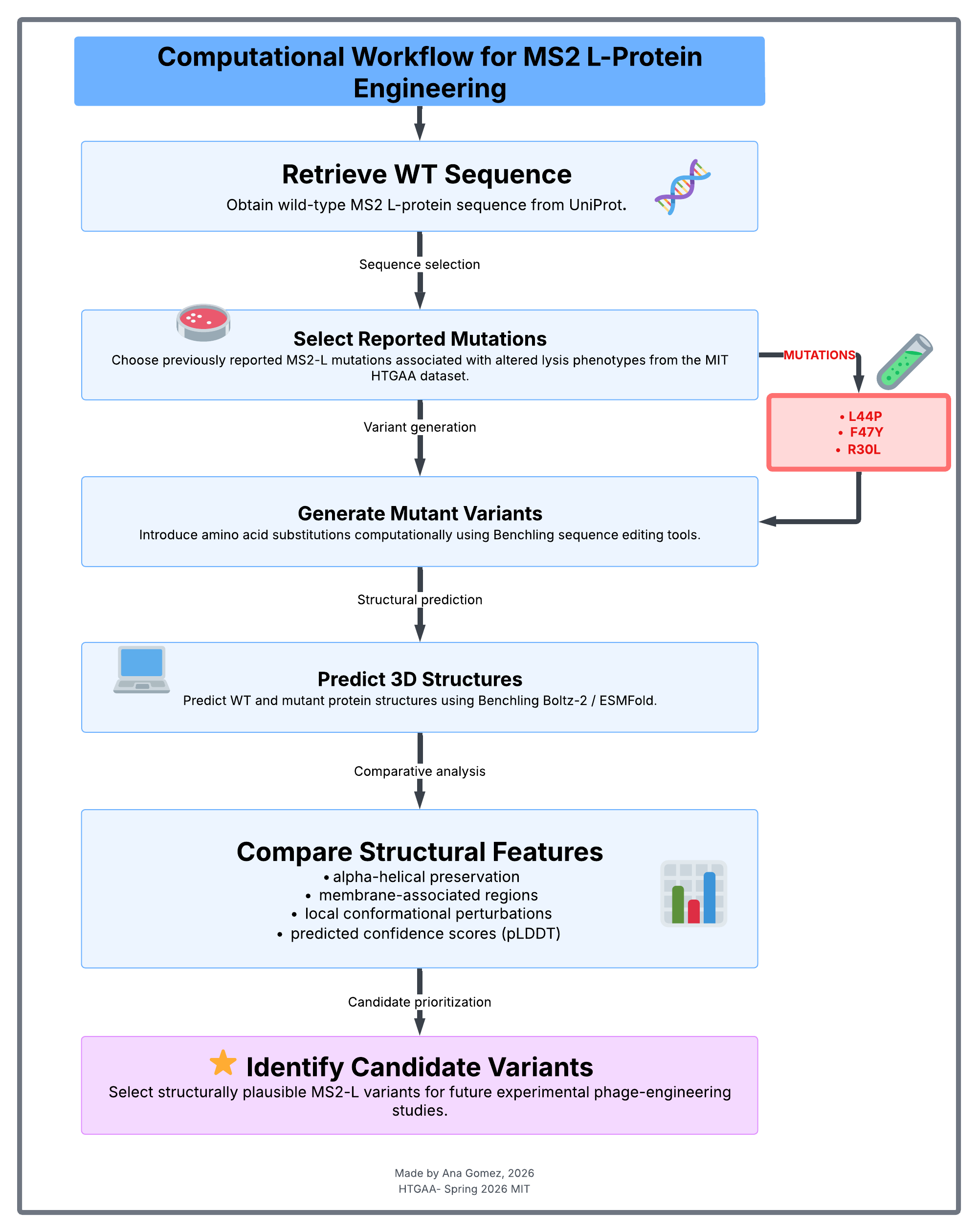
Figure 1. Computational Workflow for MS2 L-Protein Engineering Computational workflow used for the evaluation of previously reported MS2 L-protein mutations associated with altered lysis phenotypes. Wild-type and mutant variants were computationally generated and structurally analyzed using Benchling Boltz-2/ESMFold to identify structurally plausible candidates for future phage-engineering studies.
5. Possible Mutations / Strategy
5.1 Mutations
Previously reported MS2 L-protein mutations associated with altered lysis phenotypes were selected as candidate variants for computational structural evaluation. Mutations were prioritized based on their potential impact on membrane-associated alpha-helical regions, local conformational stability, or physicochemical residue properties.
| Mutation | Lysis phenotype | Reason for selection |
|---|---|---|
| L44P | altered | Potential helix-disrupting mutation |
| F47Y | altered | Conservative aromatic substitution |
| R30L | altered | Charge-to-hydrophobic substitution potentially affecting membrane interaction |
Previously reported mutations with altered lysis behavior were selected for computational structural evaluation.
5.1.1 Genetic parts:
1. MS2 lysis protein sequence: (WT)
- Uniprot code: P03609 · LYS_BPMS2
link: https://www.uniprot.org/uniprotkb/P03609/entry
2. L44P Mutation protein sequence:
3. F47Y Mutation protein sequence:
4. R30L Mutation protein sequence:
5.2 Strategy and computational assays
A computational protein-engineering workflow was implemented to evaluate whether previously reported MS2-L mutations preserved structurally plausible membrane-associated conformations. Structural prediction approaches were used as preliminary computational filters to compare WT and mutant variants and identify candidate mutations potentially suitable for future phage-engineering studies.
5.2.1 Structural Prediction Using Benchling Boltz-2
Wild-type and mutant MS2-L protein variants were analyzed using the Benchling 3D Structure Prediction platform with the Boltz-2 model. Predicted structures were compared qualitatively to assess preservation of membrane-associated structural features and identify mutations potentially associated with local conformational perturbations. Structural predictions were used as a preliminary computational filter for identifying candidate variants for future experimental evaluation.
Project folder in the next link: MS2 PROJECT ANA GOMEZ BENCHLING FOLDER
5.2.2 AlphaFold 2 Predictions
The WT and mutant MS2 L-protein sequences were additionally analyzed using AlphaFold2 through the ColabFold implementation. Predicted structures and pLDDT confidence profiles were compared to evaluate the preservation of alpha-helical membrane-associated regions and identify mutations associated with local conformational perturbations. Cross-model comparisons between AlphaFold2 and Benchling Boltz-2 predictions were used to strengthen structural consistency assessment across computational approaches.
Colab Notebook used: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb
5.2.3 Sequence Similarity Analysis
BLAST analysis of the WT MS2 L-protein sequence identified homologous lysis-associated proteins across related bacteriophages, supporting the evolutionary conservation of membrane-associated lysis systems. Sequence similarity analysis was used as an additional computational approach to contextualize the structural conservation of MS2-like lysis proteins. It’s shown in Figure 2.
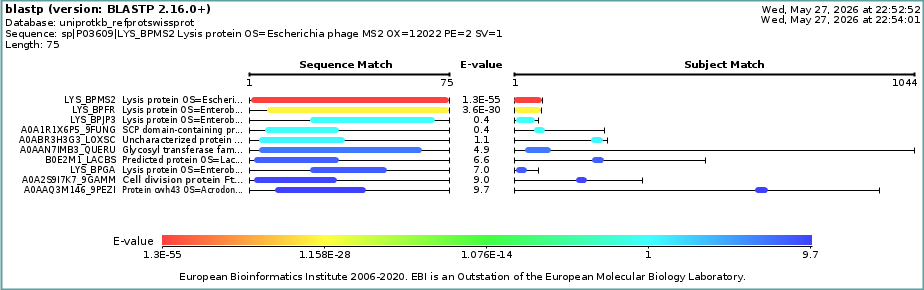
Figure 2. BLAST Similarity Analysis of the WT MS2 L-Protein Sequence BLAST similarity analysis of the WT MS2 L-protein sequence showing homologous lysis-associated proteins across related bacteriophages and supporting sequence conservation among membrane-associated phage lysis systems.
5.3 Computational Analysis
Structural predictions revealed that all mutant variants preserved membrane-associated alpha-helical features to varying degrees. Among the evaluated mutations, R30L showed the closest structural similarity to the WT prediction, suggesting that the mutation may be structurally tolerated while potentially altering local membrane-associated interactions. F47Y preserved the overall global fold with moderate local perturbations, whereas L44P exhibited more noticeable conformational alterations consistent with the known helix-disrupting effects of proline residues. These results suggest that certain MS2-L mutations may preserve structural plausibility while modifying local structural dynamics relevant to lysis-associated behavior. Comparative structural predictions generated using Benchling Boltz-2 are summarized in Figure 3.
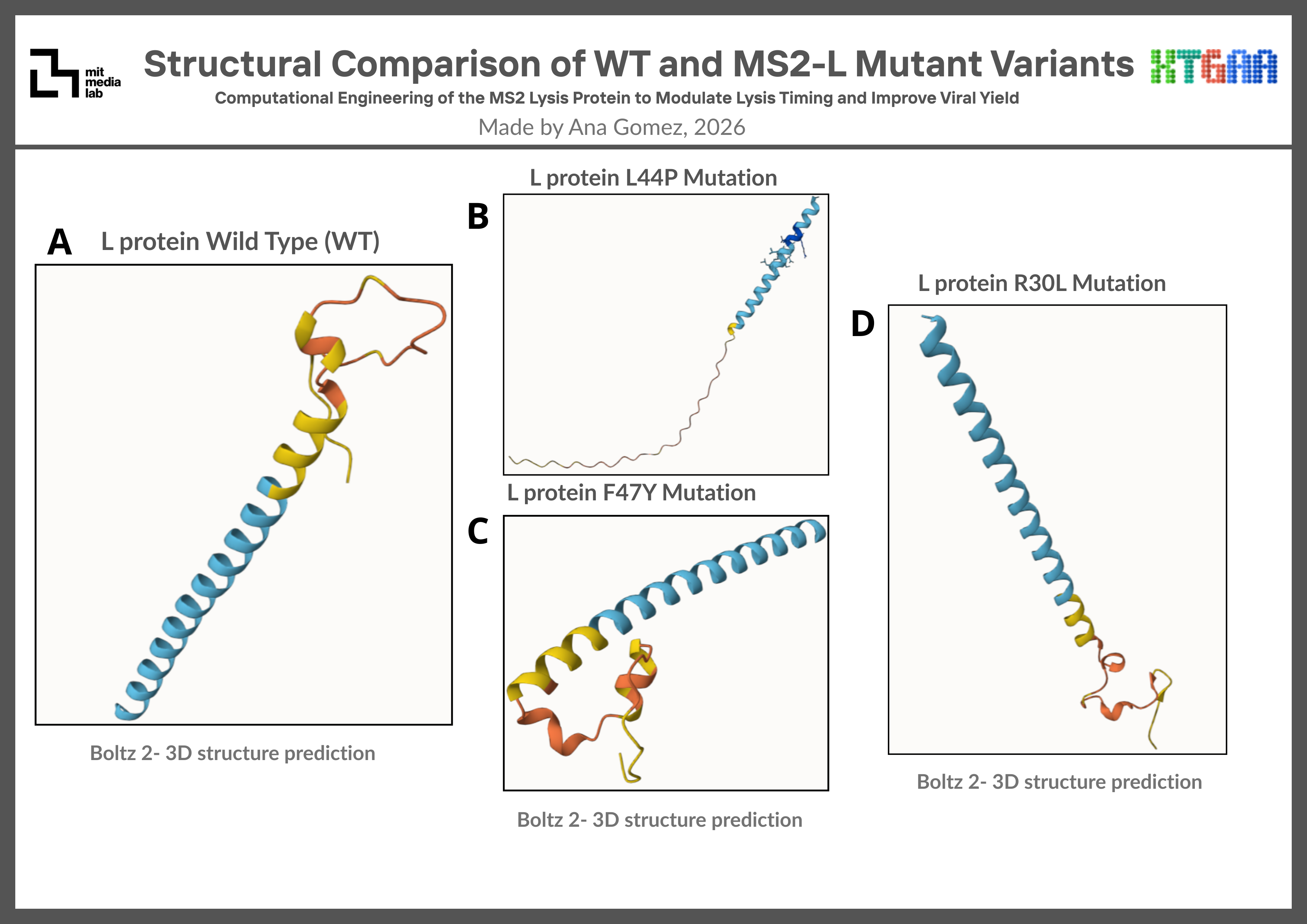
Figure 3. Structural Comparison of WT and MS2-L Mutant Variants Predicted structures of WT and mutant MS2 L-protein variants generated using Benchling Boltz-2. R30L preserved a fold most similar to the WT structure, whereas L44P showed stronger conformational perturbations potentially associated with local helix disruption. Mutations were selected from the MS2 L-protein mutational dataset provided during the MIT HTGAA Spring 2026 group project activities.
AlphaFold2 predictions were additionally used to compare confidence profiles and structural consistency among WT and mutant MS2-L variants. This is shown in Figure 4.
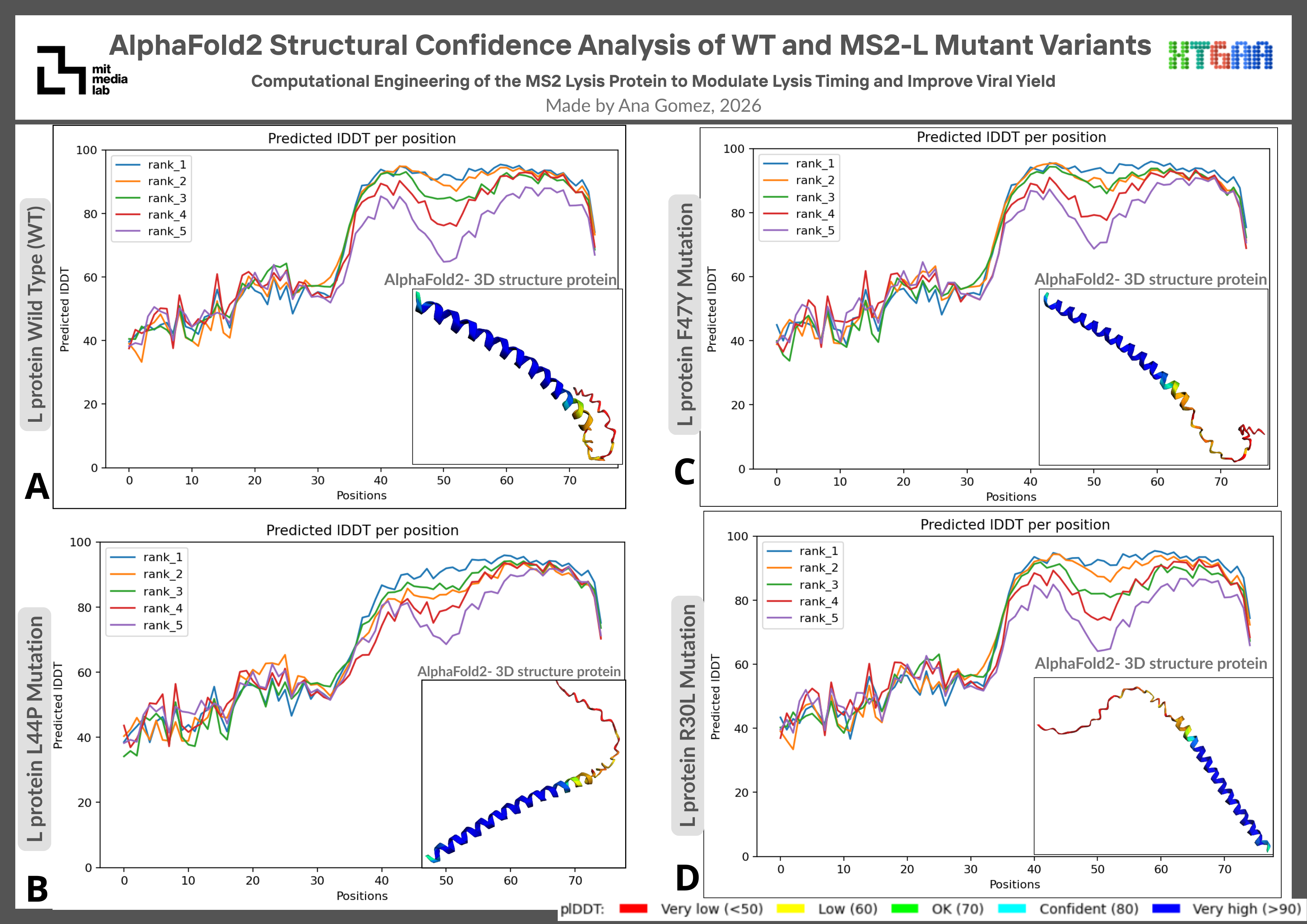
Figure 4. AlphaFold2 Structural Confidence Analysis of WT and MS2-L Mutant Variants AlphaFold2 structural predictions and pLDDT confidence profiles of WT and mutant MS2 L-protein variants. All variants preserved membrane-associated alpha-helical regions to varying degrees. R30L displayed the closest structural similarity to the WT prediction, whereas L44P exhibited stronger local conformational perturbations consistent with the helix-disrupting properties of proline residues. F47Y maintained an overall WT-like fold with moderate local structural variation.
Structural trends observed using Benchling Boltz-2 were generally consistent with AlphaFold2 predictions, particularly regarding preservation of membrane-associated alpha-helical regions and the relative structural similarity of the R30L variant to the WT structure. Cross-platform consistency strengthened confidence in the computational observations obtained during this preliminary analysis.
6. Potential Challenges
One limitation of this computational analysis is that structural plausibility does not directly predict biological performance. Although certain mutations may preserve overall folding, they could still negatively affect membrane insertion, oligomerization, host interactions, or dependence on bacterial chaperones such as DnaJ. Additionally, small membrane-associated proteins remain challenging targets for computational structure prediction, and experimental validation would ultimately be required to determine effects on lysis timing and viral yield.
7. Future Directions
Future work could experimentally evaluate selected MS2-L variants in E. coli systems to determine their effects on lysis timing, virion assembly efficiency, and viral titers. Additional studies may explore whether specific mutations reduce functional dependence on bacterial chaperones such as DnaJ or improve phage robustness against host resistance mechanisms. In the long term, this strategy could contribute to computationally guided phage engineering approaches for future phage therapy applications.
8. Conclusions
This mini computational study successfully fulfilled Aim 1 by selecting and structurally evaluating previously reported MS2 L-protein mutations associated with altered lysis phenotypes. Comparative predictions using Benchling Boltz-2 and AlphaFold2 revealed that all evaluated variants preserved membrane-associated alpha-helical regions to varying degrees, although mutations produced distinct local conformational perturbations.
Among the analyzed variants, R30L displayed the highest structural similarity to the WT prediction, suggesting that the mutation may represent a structurally tolerated candidate for future experimental evaluation. In contrast, L44P produced stronger local conformational perturbations consistent with the known helix-disrupting properties of proline residues.
Cross-model comparisons between Benchling Boltz-2 and AlphaFold2 predictions showed generally consistent structural trends, strengthening confidence in the computational observations obtained during this study.
Although biological effects on lysis timing and viral yield were not experimentally validated, this work establishes a preliminary computational framework for future phage-engineering studies focused on modulating lysis-associated dynamics and improving phage robustness.
Thanks for reading! You can find this information in my Notion webpage too! Notion MS2 Project Ana Gomez
9. Bibliography
UniProt Consortium. Lysis protein - Escherichia phage MS2 (P03609). UniProtKB. Available at: https://www.uniprot.org/uniprotkb/P03609/entry
Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589.
Mirdita M, Schütze K, Moriwaki Y, et al. ColabFold: making protein folding accessible to all. Nature Methods. 2022;19:679–682.
Chamakura, K. R., Tran, J. S., & Young, R. (2017). MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ. Journal of Bacteriology, 199(12). https://doi.org/10.1128/jb.00058-17
Chamakura KR, Young R. Phage single-gene lysis: Finding the weak spot in the bacterial cell wall. Journal of Biological Chemistry. 2019.
MIT HTGAA Spring 2026. MS2 L-protein mutational dataset and collaborative project resources.
10. Supplementary Material
Supplementary Figure S1. Early collaborative workflow drafts and brainstorming diagrams were developed during the HTGAA Spring 2026 group project phase. Workflow created by Viera, 2026. Group members: Cynthia Viera-Synbio USFQ , Paulina Flores - Synbio USFQ & Andrea Benítez- Synbio USFQ
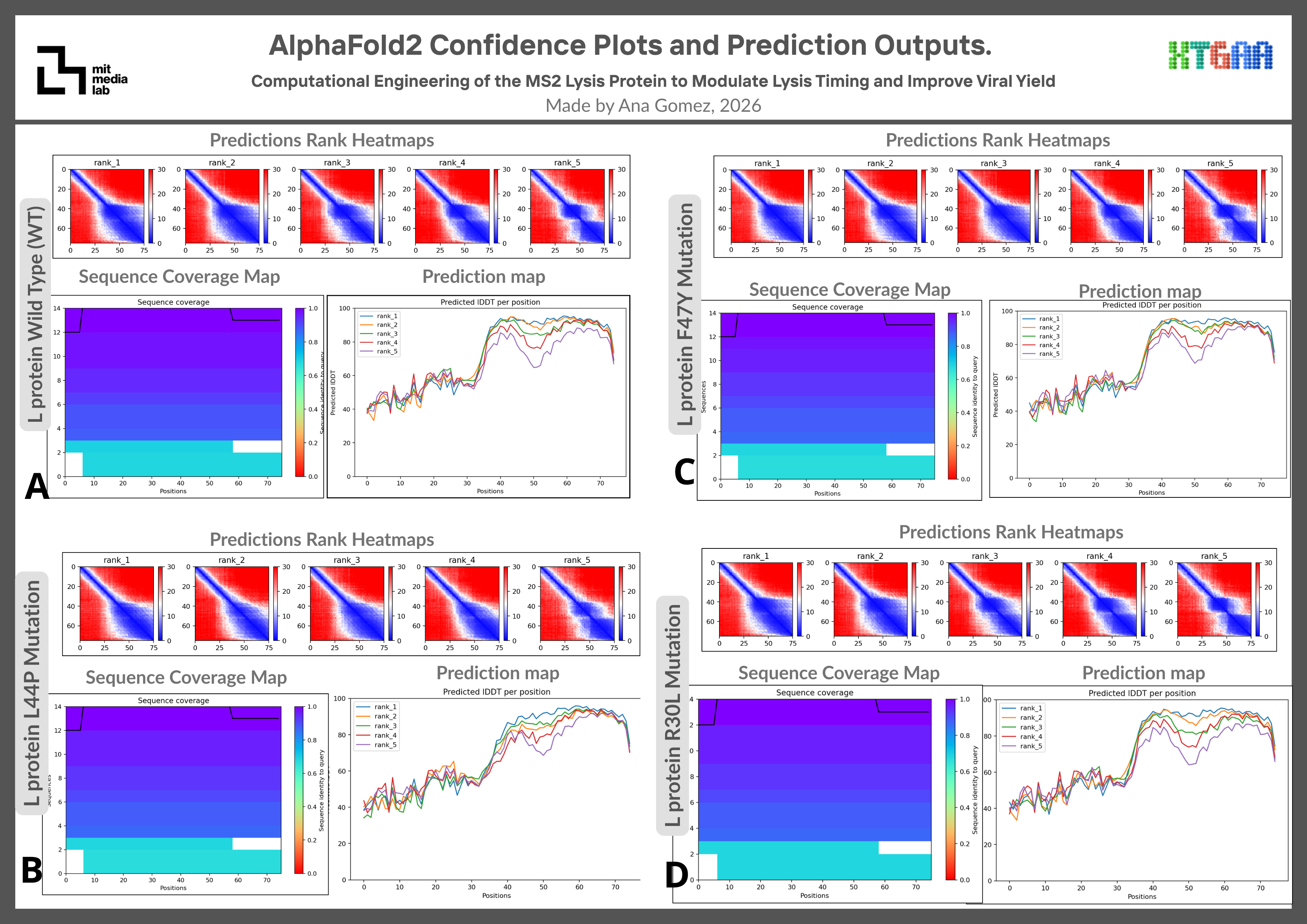
Supplementary Figure S2. Additional AlphaFold2 confidence plots and prediction outputs.
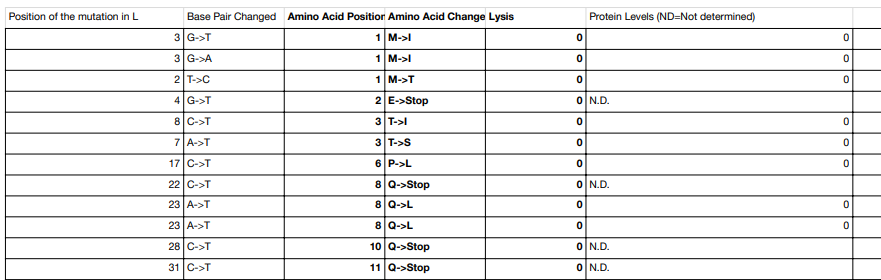
Supplementary Table S1. Mutation list extracted from the MIT HTGAA MS2-L mutational dataset.
Click here to download the document in PDF: mitmutantssheet.pdf