Week 2 hw: DNA, Read, Write & edit
Part 1: Benchling & In-silico Gel Art
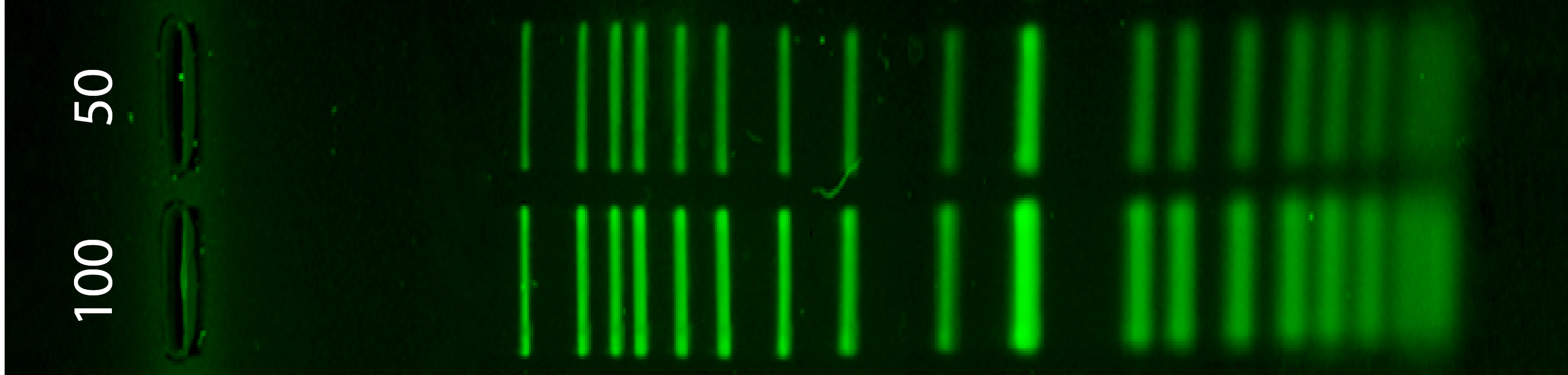
1.1 Simulation of the enzyme digestion with the following enzymes
Next to the ladder (Life 1kb Plus) there are the digestion from the enzymes:
- Search the genome “Escherichia phage Lambda” (for this I used NCBI)
- Download or export the complete genome as a genbank file
- Open Benchling and import new DNA/RNA sequence (import the .gb file and rename)
- Now we have a visualization of our genome. Next, go to the right side of the bar and click in digestion (the scisors symbol).
- Select the restriction enzimes required and run each one.
- Click in “run digest” and visualize the simulation.
- For each one, I recommend to save each process so you can consult the results anytime.
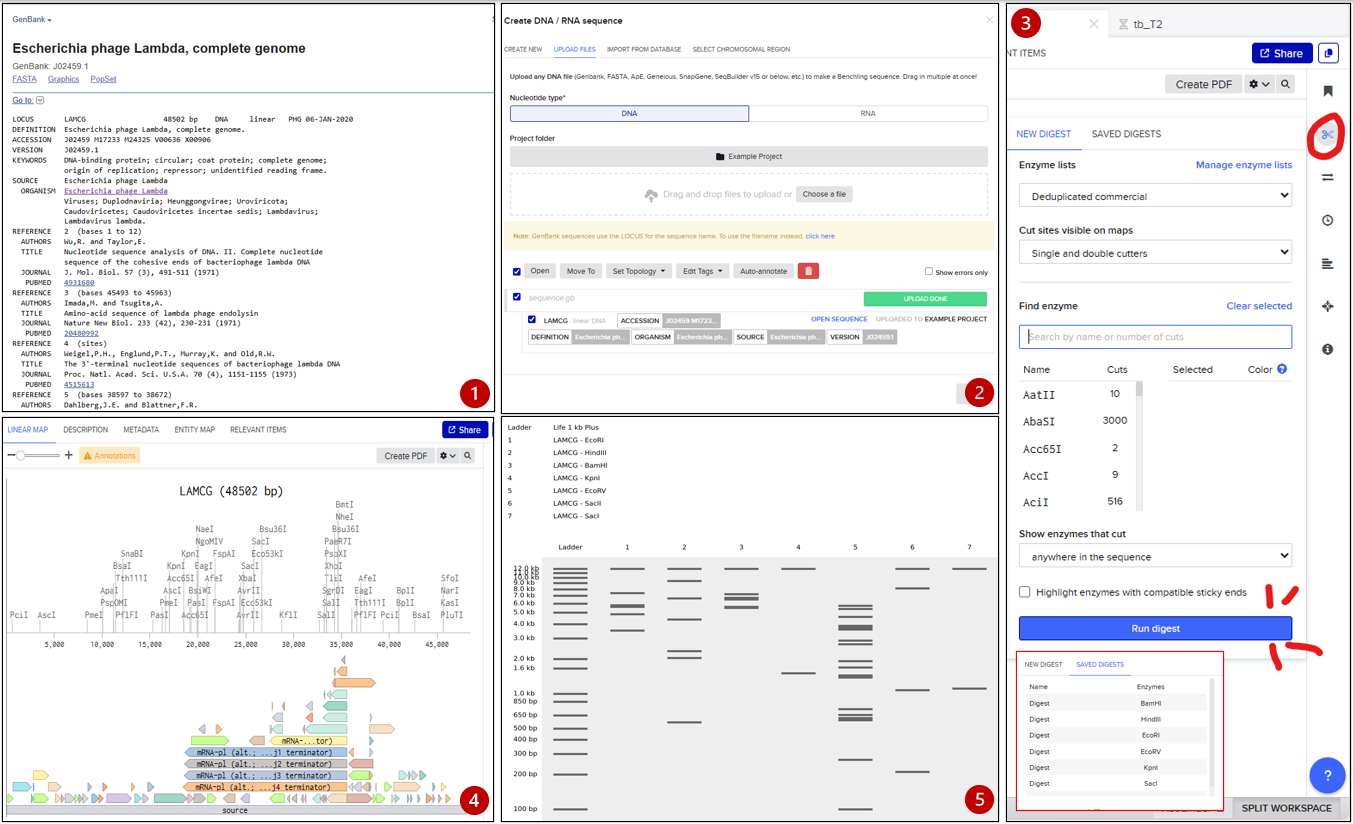
Benchling Workspace: https://benchling.com/s/seq-okBuD9NENrRXJIgK3yvh?m=slm-CAeF79q6rB0gWIBLHfkl
1.2 Pattern inspired by Paul Vanouse’s Artwork

All material was generated an inspired by Paul Vanouse’s Latent Figure Protocol artworks and designed with https://rcdonovan.com/gel-art
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Part 3: DNA Design Challenge
3.1. Choose your protein!!
I choose the tdTomato, is a basic (constitutively fluorescent) orange fluorescent protein published in 2004, derived from corals (Discosoma sp.). How it works? tbTomato absorbs light from an external source (such as blue-green light) then it re-emit it in a glowing red light. Due to the photostability that offers, it is primarily used in biomedical research as a biomarker to visualize tissues and cells inside living organisms. Personally, I like marine biology and how many things the sea can show us, so when I heard the origin of this protein while researching candidates I knew it. Also, my favorite group are mollusks but cnidarians (like corals) are quite special for their big potential in the production of innovative metabolites.

Comparing TdTomato glow with another fluorescent protein "mCherry" with same tone expresion. Image Extracted from paper "Optimizing the Protein Fluorescence Reporting System for Somatic Embryogenesis Regeneration Screening and Visual Labeling of Functional Genes in Cotton"
- Extracted from ncbi and FpBase (for reference)
AAV52169.1 tandem-dimer red fluorescent protein [synthetic construct] MVSKGEEVIKEFMRFKVRMEGSMNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGPLPFAWDILSPQFMYGS KAYVKHPADIPDYKKLSFPEGFKWERVMNFEDGGLVTVTQDSSLQDGTLIYKVKMRGTNFPPDGPVMQKK TMGWEASTERLYPRDGVLKGEIHQALKLKDGGHYLVEFKTIYMAKKPVQLPGYYYVDTKLDITSHNEDYT IVEQYERSEGRHHLFLGHGTGSTGSGSSGTASSEDNNMAVIKEFMRFKVRMEGSMNGHEFEIEGEGEGRP YEGTQTAKLKVTKGGPLPFAWDILSPQFMYGSKAYVKHPADIPDYKKLSFPEGFKWERVMNFEDGGLVTV TQDSSLQDGTLIYKVKMRGTNFPPDGPVMQKKTMGWEASTERLYPRDGVLKGEIHQALKLKDGGHYLVEF KTIYMAKKPVQLPGYYYVDTKLDITSHNEDYTIVEQYERSEGRHHLFLYGMDELYK
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
reverse translation of td_Tomato1 to a 1428 base sequence of most likely codons. atggtgagcaaaggcgaagaagtgattaaagaatttatgcgctttaaagtgcgcatggaa ggcagcatgaacggccatgaatttgaaattgaaggcgaaggcgaaggccgcccgtatgaa ggcacccagaccgcgaaactgaaagtgaccaaaggcggcccgctgccgtttgcgtgggat attctgagcccgcagtttatgtatggcagcaaagcgtatgtgaaacatccggcggatatt ccggattataaaaaactgagctttccggaaggctttaaatgggaacgcgtgatgaacttt gaagatggcggcctggtgaccgtgacccaggatagcagcctgcaggatggcaccctgatt tataaagtgaaaatgcgcggcaccaactttccgccggatggcccggtgatgcagaaaaaa accatgggctgggaagcgagcaccgaacgcctgtatccgcgcgatggcgtgctgaaaggc gaaattcatcaggcgctgaaactgaaagatggcggccattatctggtggaatttaaaacc atttatatggcgaaaaaaccggtgcagctgccgggctattattatgtggataccaaactg gatattaccagccataacgaagattataccattgtggaacagtatgaacgcagcgaaggc cgccatcatctgtttctgggccatggcaccggcagcaccggcagcggcagcagcggcacc gcgagcagcgaagataacaacatggcggtgattaaagaatttatgcgctttaaagtgcgc atggaaggcagcatgaacggccatgaatttgaaattgaaggcgaaggcgaaggccgcccg tatgaaggcacccagaccgcgaaactgaaagtgaccaaaggcggcccgctgccgtttgcg tgggatattctgagcccgcagtttatgtatggcagcaaagcgtatgtgaaacatccggcg gatattccggattataaaaaactgagctttccggaaggctttaaatgggaacgcgtgatg aactttgaagatggcggcctggtgaccgtgacccaggatagcagcctgcaggatggcacc ctgatttataaagtgaaaatgcgcggcaccaactttccgccggatggcccggtgatgcag aaaaaaaccatgggctgggaagcgagcaccgaacgcctgtatccgcgcgatggcgtgctg aaaggcgaaattcatcaggcgctgaaactgaaagatggcggccattatctggtggaattt aaaaccatttatatggcgaaaaaaccggtgcagctgccgggctattattatgtggatacc aaactggatattaccagccataacgaagattataccattgtggaacagtatgaacgcagc gaaggccgccatcatctgtttctgtatggcatggatgaactgtataaa
3.3 Codon Optimization
Why you need to optimize codon usage? The expression varies in species, sometimes one specie prefers the usage of X codon instead of Y. As it was presented in the slides, it can be demonstrated in both stability of the RNA Structure and percentages of efficiency in the expression: Arginine expression in e.coli works smoothly with an CGC (22.3%) codon instead of CGG (5.4%).
Which organism have you chosen to optimize the codon sequence for and why? For this activity I choose e. coli because is a well know bacterian model, also it is easy and simple to use in compare to other candidates I had (like zebra fishes).
1 ATGGTATCTA AGGGCGAGGA AGTCATCAAG GAGTTTATGC GCTTTAAAGT TCGTATGGAG GGTAGCATGA 71 ACGGCCACGA ATTCGAAATC GAAGGCGAAG GCGAAGGCCG CCCGTACGAA GGTACCCAGA CCGCTAAACT 141 GAAAGTTACC AAGGGCGGCC CGCTGCCGTT TGCTTGGGAT ATCCTGTCCC CGCAGTTCAT GTACGGCTCT 211 AAAGCGTACG TAAAACATCC GGCGGACATC CCGGACTACA AAAAGCTGTC CTTTCCGGAA GGCTTCAAGT 281 GGGAACGCGT GATGAACTTC GAAGACGGCG GCCTGGTTAC TGTCACCCAG GACTCCTCTC TGCAGGATGG 351 CACTCTGATC TACAAAGTGA AAATGCGCGG TACCAACTTT CCGCCAGATG GTCCGGTTAT GCAGAAGAAA 421 ACCATGGGTT GGGAAGCAAG CACCGAGCGT CTGTACCCAC GTGATGGTGT CCTGAAGGGC GAAATCCACC 491 AGGCACTGAA ACTGAAAGAT GGTGGTCATT ACCTGGTGGA GTTCAAAACC ATCTATATGG CCAAGAAACC 561 GGTGCAACTG CCGGGCTACT ACTATGTTGA CACCAAACTG GATATTACCT CTCACAATGA GGACTATACT 631 ATCGTGGAAC AGTATGAACG CTCTGAAGGC CGCCACCACC TGTTTCTGGG CCACGGTACT GGTTCTACTG 701 GCTCCGGTTC TTCCGGCACT GCATCTTCCG AGGACAACAA CATGGCGGTT ATCAAAGAAT TCATGCGCTT 771 CAAAGTACGC ATGGAAGGCT CTATGAACGG TCACGAATTC GAAATCGAGG GTGAAGGCGA AGGTCGCCCG 841 TACGAAGGTA CGCAAACTGC GAAGCTGAAA GTGACGAAGG GTGGTCCGCT GCCATTCGCG TGGGATATTC 911 TGTCTCCGCA GTTCATGTAT GGTAGCAAAG CATATGTCAA GCACCCTGCT GATATCCCGG ACTATAAAAA 981 GCTGAGCTTC CCGGAAGGCT TCAAATGGGA GCGTGTCATG AACTTCGAGG ACGGTGGCCT GGTAACCGTT 1051 ACCCAGGACT CTTCTCTGCA GGACGGCACT CTGATCTACA AAGTCAAAAT GCGTGGTACT AACTTCCCGC 1121 CAGACGGCCC GGTGATGCAG AAAAAGACCA TGGGCTGGGA AGCCTCCACC GAACGTCTGT ACCCGCGCGA 1191 CGGTGTCCTG AAAGGTGAGA TCCATCAGGC GCTGAAACTG AAAGACGGTG GCCACTATCT GGTTGAATTC 1261 AAAACCATCT ACATGGCAAA AAAACCGGTG CAGCTGCCAG GTTATTATTA TGTGGATACC AAACTGGATA 1331 TTACCAGCCA CAATGAGGAC TACACTATCG TCGAACAGTA CGAACGTTCC GAAGGCCGCC ACCACCTGTT 1401 CCTGTATGGT ATGGACGAAC TGTACAAA
Codon optimization was executed with https://www.novoprolabs.com/tools/codon-optimization
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? You may describe either cell-dependent or cell-free methods, or both. (DeepseekAI assisted) For this step, I would choose cell-dependent expression. I would insert the tdTomato gene into a compatible plasmid and introduce the recombinant plasmid into E. coli cells using heat shock or electroporation. Then, I would incubate them and monitor their growth until they reach an optimal density. I would induce protein expression (using IPTG, since E. coli can metabolize lactose-based inducers) and incubate again for a few hours to visualize gene expression as a red fluorescent color (meaning the protein folded correctly).
Part 5: DNA Read/Write/Edit
5.1 DNA Read
What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
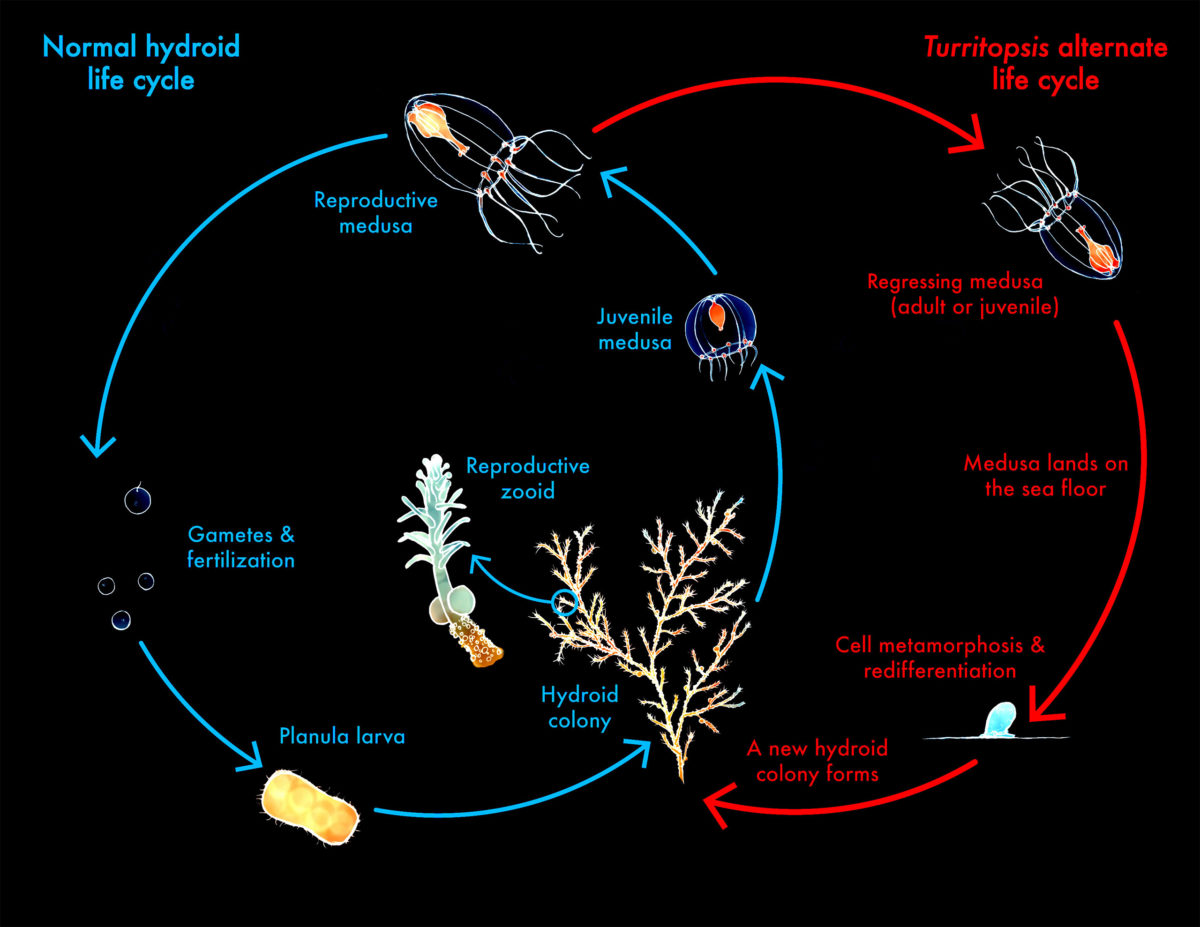
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so? The method I use is second-generation. I opted for Illumina given its accessibility and low cost compared to other methods1. Furthermore, since a reference genome already exists (provided by Hasegawa et al. (2023))2, the need for de novo sequencing and assembly is reduced, compare to a process that would require a hybrid method(PacBio x Illumina as Hasegawa et al used) if we were the pioneers in the study of this specie.
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps. INPUT: For DNA extraction, only one individual of T. dohrnii would be needed. Currently, modern Illumina kits have high sensitivity and require only 1–100 ng of DNA, thus we avoid having to clone or collect more specimens. The steps for the extraction are enlisted:
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)? (Assisted with deepseekAI)
Cluster generation: Fragments bind to the flow cell and are locally amplified to form “clusters” of identical copies. Sequencing by synthesis (SBS): Fluorescently labeled nucleotides (each base with a different color) with a reversible terminator are added. The polymerase incorporates one nucleotide per cycle. Base Calling: - A laser excites the fluorophores. - A camera records the color emitted by each cluster. - The software translates each color into a specific base (A, T, C, G) and assigns a quality score (Q-score) to each call. Repeat: The terminator and dye are removed, and the cycle is repeated (e.g., 150 cycles for 150 bp reads).
- What is the output of your chosen sequencing technology? (Assisted with deepseekAI) The direct output of the sequencing process would be FASTQ files containing millions of short reads (e.g., 150 bp) along with their corresponding quality scores (Q-scores) for each base call. After bioinformatic analysis, these reads are aligned against the T. dohrnii reference genome provided by Hasegawa et al. (2023)2, generating BAM files that contain the mapped reads. Finally, variant calling analysis produces a VCF file (Variant Call Format) listing all genetic variants found in the sequenced sample compared to the reference genome, along with coverage statistics and sequencing quality metrics2.
5.2 DNA write
5.2.1 What DNA would you want to synthesize (e.g., write) and why?
These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I choose the lactase enzime. This because there are studies related to Food_desensitization3 where the main methodology implies the ingestion of dosis of the alergen (in this case milk), but what if we could skip this step (a process that can be uncomfortable and time-consuming) and implement the lactase enzime to decrease the ingestion of the alergen. I propose producing recombinant lactase enzyme that could be administered directly as a supplement or incorporated into dairy products, allowing individuals to digest lactose without undergoing desensitization. Specifically, I want to focus on the evolved beta-galactosidase subunit alpha from Escherichia coli K-12. This enzyme catalyzes the hydrolysis of lactose into D-glucose and D-galactose (assimilable forms).
sp|P06864|BGA2_ECOLI Evolved beta-galactosidase subunit alpha MNRWENIQLTHENRLAPRAYFFSYDSVAQARTFARETSSLFLPLSGQWNFHFFDHPLQVPEAFTSELMADW GHITVPAMWQMEGHGKLQYTDEGFPFPIDVPFVPSDNPTGAYQRIFTLSDGWQGKQTLIKFDGVETYFEVYV NGQYVGFSKGSRLTAEFDISAMVKTGDNLLCVRVMQWADSTYVEDQDMWWSAGIFRDVYLVGKHLTHINDFT VRTDFDEAYCDATLSCEVVLENLAASPVVTTLEYTLFDGERVVHSSAIDHLAIEKLTSASFAFTVEQPQQWS AESPYLYHLVMTLKDANGNVLEVVPQRVGFRDIKVRDGLFWINNRYVMLHGVNRHDNDHRKGRAVGMDRVEK DLQLMKQHNINSVRTAHYPNDPRFYELCDIYGLFVMAETDVESHGFANVGDISRITDDPQWEKVYVERIVRH IHAQKNHPSIIIWSLGNESGYGCNIRAMYHAAKALDDTRLVHYEEDRDAEVVDIISTMYTRVPLMNEFGEYP HPKPRIICEYAHAMGNGPGGLTEYQNVFYKHDCIQGHYVWEWCDHGIQAQDDHGNVWYKFGGDYGDYPNNYN FCLDGLIYSDQTPGPGLKEYKQVIAPVKIHARDLTRGELKVENKLWFTTLDDYTLHAEVRAEGETLATQQIK LRDVAPNSEAPLQITLPQLDAREAFLNITVTKDSRTRYSEAGHPIATYQFPLKENTAQPVPFAPNNARPLTL EDDRLSCTVRGYNFAITFSKMSGKPTSWQVNGESLLTREPKINFFKPMIDNHKQEYEGLWQPNHLQIMQEHL RDFAVEQSDGEVLIISRTVIAPPVFDFGMRCTYIWRIAADGQVNVALSGERYGDYPHIIPCIGFTMGINGEY DQVAYYGRGPGENYADSQQANIIDIWRSTVDAMFENYPFPQNNGNRQHVRWTALTNRHGNGLLVVPQRPINF SAWHYTQENIHAAQHCNELQRSDDITLNLDHQLLGLGSNSWGSEVLDSWRVWFRDFSYGFTLLPVSGGEATA QSLASYEFGAGFFSTNLHSENKQ
5.2.2 What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions: a. What are the essential steps of your chosen sequencing methods? I would use oligonucleotide-based chemical synthesis (specifically, the phosphoramidite solid-phase synthesis method), which is the standard approach for custom gene synthesis offered by commercial providers like Twist Bioscience, IDT, or GenScript. b. What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
- The primary limitations of chemical DNA synthesis include an inherent error rate (typically 1 error per 500-1000 bases) that necessitates careful sequence verification and potential correction rounds, especially for longer genes like the ~3kb lactase sequence.
- Cost remains significant, with synthesis at $0.10-0.30 per base pair making a full-length gene $300-900 before cloning and validation.
- Sequence complexity can pose challenges, as highly repetitive regions or extreme GC content may be difficult to synthesize accurately. Turnaround time is relatively slow, with standard gene synthesis taking 2-4 weeks for delivery plus additional time for cloning and verification. Size constraints also apply, as genes beyond 3-5 kb may require special assembly strategies. Finally, codon optimization, while computationally guided, involves some uncertainty since the “optimal” codon set for a given expression host cannot always be perfectly predicted and may require empirical testing of multiple variants to achieve maximum expression yields.
5.3 DNA Edit
5.3.1 What DNA would you want to edit and why?
In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I personally choose the genes related to the pigmentation of human iris. Since I have partial heterochromia I’ve been always curious about how this mutation occured or what were the mechanisms related to the correct pigmentation of iris that mutated the moment I was just a cell!!. A particular region of chromosome 15 plays an important role in eye color. Within this region, there are two genes located very close together, OCA2 and HERC2. The protein produced from the OCA2 gene, known as the P protein, is involved in the maturation of melanosomes, cellular structures that produce and store melanin. Therefore, the P protein plays a crucial role in the amount and quality of melanin present in the iris. Several common variations (polymorphisms) in the OCA2 gene reduce the amount of functional P protein that is produced. Less P protein means there is less melanin present in the iris, leading to blue eyes instead of brown eyes in people with a polymorphism in this gene.4
The presence of other genes involved in pigmentation is known; however, the effects of these genes are likely combined with those of OCA2 and HERC2 to produce a continuum of eye colors in different people. My interest is to understand if its possible simulate this type of mutation, or induce it.
5.3.2 What technology or technologies would you use to perform these DNA edits and why? (Assisted with DeepseekAI)
Also answer the following questions: i. How does your technology of choice edit DNA? What are the essential steps? ii. What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? iii. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
To introduce or correct a mutation associated with heterochromia in the OCA2 or HERC2 genes, I would use CRISPR-Cas9 genome editing technology. This is the most suitable option due to its high precision, efficiency, and programmability. CRISPR-Cas9 edits DNA by creating a targeted double-strand break, which the cell then repairs through natural pathways.
The preparation begins with careful in silico design: selecting a unique 20bp target sequence in the OCA2 gene adjacent to a PAM motif, and designing a donor DNA template with homology arms if precise HDR editing is desired. The required inputs include the target cells (such as melanocytes or cell lines), Cas9 (provided as plasmid DNA, mRNA, or recombinant protein), a synthetic guide RNA designed for your specific gene sequence, a donor DNA template for precise edits, and a delivery reagent like lipofection reagents or electroporation devices to introduce these components into the cells.
Despite Crispr-CAS9 potential, it has some limits. The primary precision concern is off-target effects, where guide RNAs may bind to similar genomic sequences and introduce unintended mutations, potentially disrupting tumor suppressors or oncogenes. Regarding efficiency, Homology-Directed Repair (HDR) for precise edits remains inefficient compared to the dominant error-prone Non-Homologous End Joining (NHEJ) pathway, and HDR is largely restricted to dividing cells. Additional challenges include mosaicism in multicellular organisms, risk of large structural variants like deletions or translocations, delivery hurdles for in vivo applications, and potential immunogenicity from the bacterial-derived Cas9 protein.
References
Tamang, S. (2024, December 13). Illumina Sequencing: principle, steps, uses, diagram. Microbe Notes. https://microbenotes.com/illumina-sequencing/#limitations-of-illumina-sequencing ↩︎
Hasegawa, Y., Watanabe, T., Otsuka, R., Toné, S., Kubota, S., & Hirakawa, H. (2023). Genome assembly and transcriptomic analyses of the repeatedly rejuvenating jellyfish Turritopsis dohrnii. DNA research : an international journal for rapid publication of reports on genes and genomes, 30(1), dsac047. https://doi.org/10.1093/dnares/dsac047 ↩︎ ↩︎ ↩︎
Piraino Sosa, Pedro, & Ojeda Soley, Giovanni. (2023). Desensibilización con alimentos. Revista alergia México, 70(4), 284-292. Epub 29 de abril de 2024.https://doi.org/10.29262/ram.v70i4.1339 ↩︎
https://medlineplus.gov/spanish/genetica/entender/rasgos/colorojos/ ↩︎