Week 2 HW: DNA read, write and edit

Part 1: Benchling & In-silico Gel Art
- Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI
- Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
- You might find Ronan’s website a helpful tool for quickly iterating on designs!
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
I have chosen Glutamate Decarboxylase (GAD) from Lactobacillus reuteri because it is the key enzyme responsible for converting glutamate into GABA (gamma-aminobutyric acid). Since GABA plays a crucial role in reducing anxiety by calming the nervous system, engineering L. reuteri to enhance its GABA production could provide a natural, probiotic-based approach to mental health support. This selection aligns with my interest in the microbiome’s impact on mental health and the potential of synthetic biology to develop gut-based biosensors for anxiety management.
MAMLYGKHNHEAEEYLEPVFGAPSEQHDLPKYRLPKHSLSPREADRLVRDELLDEGNSRLNLATFCQTYMEPEAVELMKDTLAKNAIDKSEYPRTAEIENRCVNIIANLWHAPDDEHFTGTSTIGSSEACMLGGLAMKFAWRKRAQAAGLDLNAHRPNLVISAGYQVCWEKFWVYWDVDMHVVPMDEQHMALDVNHVLDYGDEYTIGIVGIMGITYTGQDDDLAALDKVVTHYNHQHPKLPVYIHVDAASGGFYTPFIEPQLIWDFRLANVVSINASGHKYGLVYPGVGWVVWRDRQFLPPELVFKVSYLGGELPKMAINFSHSAAQLIEQYYNFIRFGMDGYRKIQTKTHDVARYRATAPDKVGELKMINNGHQPPLICYQLPPREDRKGTLYDLSDRLLMNGWQVPTYPLPANLEQQVIQRIVVRADFGMNMAHDFMDDLTKAVHDLNHAHIVYHHDAAPKKYGFTH
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Employing the Reverse Translate tool from https://www.bioinformatics.org/sms2/rev_trans.html I chose this sequence, “Most Likely Codons” sequence, as it ensures efficient translation in Lactobacillus reuteri by selecting the most frequently used codons, minimizing translation delays caused by rare codons.
>reverse translation of Untitled to a 1407 base sequence of most likely codons. atggcgatgctgtatggcaaacataaccatgaagcggaagaatatctggaaccggtgttt ggcgcgccgagcgaacagcatgatctgccgaaatatcgcctgccgaaacatagcctgagc ccgcgcgaagcggatcgcctggtgcgcgatgaactgctggatgaaggcaacagccgcctg aacctggcgaccttttgccagacctatatggaaccggaagcggtggaactgatgaaagat accctggcgaaaaacgcgattgataaaagcgaatatccgcgcaccgcggaaattgaaaac cgctgcgtgaacattattgcgaacctgtggcatgcgccggatgatgaacattttaccggc accagcaccattggcagcagcgaagcgtgcatgctgggcggcctggcgatgaaatttgcg tggcgcaaacgcgcgcaggcggcgggcctggatctgaacgcgcatcgcccgaacctggtg attagcgcgggctatcaggtgtgctgggaaaaattttgggtgtattgggatgtggatatg catgtggtgccgatggatgaacagcatatggcgctggatgtgaaccatgtgctggattat ggcgatgaatataccattggcattgtgggcattatgggcattacctataccggccaggat gatgatctggcggcgctggataaagtggtgacccattataaccatcagcatccgaaactg ccggtgtatattcatgtggatgcggcgagcggcggcttttataccccgtttattgaaccg cagctgatttgggattttcgcctggcgaacgtggtgagcattaacgcgagcggccataaa tatggcctggtgtatccgggcgtgggctgggtggtgtggcgcgatcgccagtttctgccg ccggaactggtgtttaaagtgagctatctgggcggcgaactgccgaaaatggcgattaac tttagccatagcgcggcgcagctgattgaacagtattataactttattcgctttggcatg gatggctatcgcaaaattcagaccaaaacccatgatgtggcgcgctatcgcgcgaccgcg ccggataaagtgggcgaactgaaaatgattaacaacggccatcagccgccgctgatttgc tatcagctgccgccgcgcgaagatcgcaaaggcaccctgtatgatctgagcgatcgcctg ctgatgaacggctggcaggtgccgacctatccgctgccggcgaacctggaacagcaggtg attcagcgcattgtggtgcgcgcggattttggcatgaacatggcgcatgattttatggat gatctgaccaaagcggtgcatgatctgaaccatgcgcatattgtgtatcatcatgatgcg gcgccgaaaaaatatggctttacccat
>KF751352.2 Lactobacillus reuteri strain ATCC 23272 glutamate decarboxylase (gadB) gene, complete cds ATGGCAATGTTATACGGTAAACACAATCATGAAGCTGAAGAATACTTGGAACCAGTCTTTGGTGCGCCTT CTGAACAACATGATCTTCCTAAGTATCGGTTACCAAAGCATTCATTATCCCCTCGAGAAGCCGATCGCTT AGTTCGTGATGAATTATTAGATGAAGGCAATTCACGACTGAACTTGGCAACTTTTTGTCAGACCTATATG GAACCCGAAGCCGTTGAATTGATGAAGGATACGCTGGCTAAGAATGCCATCGACAAATCTGAGTACCCCC GCACGGCCGAGATTGAAAATCGGTGTGTGAACATTATTGCCAATCTGTGGCACGCACCTGATGACGAACA CTTTACGGGTACCTCTACGATTGGCTCCTCTGAAGCTTGTATGTTAGGCGGTTTAGCAATGAAATTCGCC TGGCGTAAACGCGCTCAAGCGGCAGGTTTAGATCTGAATGCCCATCGACCTAACCTCGTTATTTCGGCTG GCTATCAAGTTTGCTGGGAAAAGTTTTGGGTCTACTGGGACGTTGACATGCACGTGGTCCCAATGGATGA GCAACACATGGCCCTTGACGTTAACCACGTCTTAGACTACGGGGACGAATACACAATTGGTATCGTCGGT ATCATGGGCATCACTTATACCGGTCAAGATGACGACCTAGCCGCACTCGATAAGGTCGTTACTCACTACA ATCATCAGCATCCCAAATTACCAGTCTACATTCACGTTGACGCAGCGTCAGGTGGCTTCTATACCCCATT TATTGAGCCGCAACTCATCTGGGACTTCCGGTTGGCTAACGTCGTTTCAATCAACGCCTCCGGACACAAG TACGGTTTAGTTTATCCCGGGGTCGGCTGGGTCGTTTGGCGTGATCGTCAGTTTTTACCGCCAGAATTAG TCTTCAAAGTTAGTTATTTAGGGGGGGAATTGCCGAAAATGGCGATCAACTTCTCACATAGTGCAGCCCA GCTCATTGAACAATACTATAATTTCATTCGCTTTGGTATGGACGGTTACCGCAAAATTCAAACAAAGACT CACGATGTTGCCCGCTACCGGGCAACCGCTCCGGATAAAGTTGGTGAATTAAAAATGATCAATAACGGAC ACCAACCCCCCCTGATTTGTTACCAACTACCCCCGCGCGAAGATCGTAAAGGGACCCTTTATGATTTATC GGATCGCCTATTAATGAACGGTTGGCAAGTACCAACGTATCCTTTACCTGCTAATCTGGAACAACAAGTC ATCCAACGAATCGTCGTTCGGGCTGACTTTGGCATGAATATGGCCCACGATTTCATGGATGACCTGACCA AGGCTGTCCATGACTTAAACCACGCCCACATTGTCTATCATCATGACGCGGCACCTAAGAAATACGGATT CACACACTGA
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Codon optimization involves redesigning a DNA sequence to use codons preferred by the host organism while preserving the same amino acid sequence. This improves translation efficiency, protein yield, and expression stability. Glutamate decarboxylase (gadB) gene with Codon-Optimization (E. coli)
ATGGCGATGCTGTATGGCAAACATAACCATGAAGCGGAAGAATATTTGGAACCGGTGTTTGGCGCGCCGAGCGAACAGCATGATCTGCCGAAGTATCGCCTGCCGAAACATAGCCTGAGCCCGCGTGAAGCGGATCGCCTGGTGCGTGATGAACTGCTGGATGAAGGCAACAGCCGCCTGAACTTGGCGACCTTTTGCCAGACCTATATGGAACCGGAAGCGGTGGAATTGATGAAAGATACGCTGGCGAAAAACGCGATTGATAAAAGCGAGTATCCGCGCACGGCGGAGATTGAAAACCGCTGCGTGAACATTATTGCGAACCTGTGGCATGCGCCGGATGACGAACATTTTACGGGCACCAGCACGATTGGCAGCAGCGAAGCGTGCATGCTGGGCGGCCTGGCGATGAAATTTGCGTGGCGTAAACGCGCGCAGGCGGCGGGCCTGGATCTGAACGCGCATCGCCCGAACCTCGTGATTTCGGCGGGCTATCAGGTGTGCTGGGAAAAATTTTGGGTGTATTGGGATGTGGATATGCATGTGGTGCCGATGGATGAGCAGCATATGGCGCTGGATGTGAACCATGTGCTGGATTATGGCGATGAATATACCATTGGCATTGTGGGCATTATGGGCATTACCTATACCGGCCAGGATGATGATCTGGCGGCGCTCGATAAAGTGGTGACCCATTATAACCATCAGCATCCGAAACTGCCGGTGTATATTCATGTGGATGCGGCGAGCGGCGGCTTCTATACCCCGTTTATTGAGCCGCAGCTCATTTGGGATTTTCGCTTGGCGAACGTGGTGAGCATTAACGCGAGCGGACATAAGTATGGCCTGGTGTATCCGGGCGTGGGCTGGGTGGTGTGGCGTGATCGTCAGTTTCTGCCGCCGGAACTGGTGTTTAAAGTGAGTTATCTGGGCGGCGAATTGCCGAAAATGGCGATTAACTTTAGCCATAGTGCGGCGCAGCTCATTGAACAGTATTATAACTTTATTCGCTTTGGCATGGATGGCTATCGCAAAATTCAGACCAAAACCCATGATGTGGCGCGCTATCGCGCGACCGCGCCGGATAAAGTGGGCGAACTGAAAATGATTAACAACGGACATCAGCCGCCGCTGATTTGCTATCAGCTGCCGCCGCGCGAAGATCGTAAAGGCACCCTGTATGATCTGTCGGATCGCCTGCTGATGAACGGCTGGCAGGTACCGACGTATCCGCTGCCGGCGAACCTGGAACAGCAGGTGATTCAGCGCATTGTGGTGCGCGCGGATTTTGGCATGAACATGGCGCATGATTTTATGGATGATCTGACCAAAGCGGTGCATGACCTGAACCATGCGCATATTGTGTATCATCATGATGCGGCGCCGAAAAAATATGGATTCACCCATTAA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
For producing Glutamate Decarboxylase (GAD) from Lactobacillus reuteri, I will first express it in E. coli as an initial step before moving to L. reuteri. To achieve this, I will clone the codon-optimized gadB gene into a plasmid with a strong bacterial promoter, such as pLac or T7, for efficient expression. I will then introduce the plasmid into competent E. coli cells using heat shock transformation. To ensure only transformed cells grow, I will use selective markers, such as an antibiotic resistance gene (ampicillin or kanamycin) on the plasmid. I may also include a reporter gene, such as GFP (Green Fluorescent Protein), to visually confirm successful transformation under UV light. The transformed E. coli colonies will be plated on selective agar containing the antibiotic, ensuring that only bacteria carrying the plasmid with the GAD insert survive.
Part 4: Prepare a Twist DNA Synthesis Order
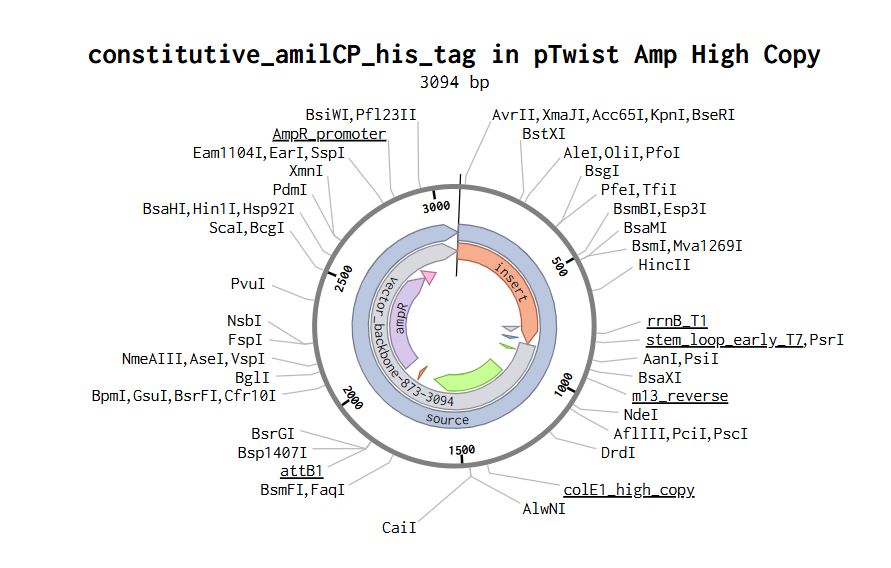
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would sequence the gadB gene from L. reuteri to ensure accurate expression and function in the engineered strain. Sequencing this DNA would confirm that no mutations occurred during cloning, preserving the enzyme’s ability to produce GABA efficiently. This is crucial for validating the stability and effectiveness of the modified strain before using it as part of a probiotic-based anxiety treatment.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Sanger sequencing to confirm the accuracy of the gadB gene after cloning. Sanger sequencing is first-generation sequencing, known for its high accuracy in small DNA fragments like single genes. It is cost-effective, widely accessible, and ideal for verifying cloned plasmids before moving to functional testing.
However, if larger-scale sequencing were needed (e.g., confirming genomic integration or long fragments), I could use Nanopore sequencing, as I have access to one in the lab. Nanopore is third-generation sequencing, capable of reading long DNA fragments without amplification, making it useful for analyzing structural variations.
Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
Sanger sequencing is First-generation (based on chain termination using fluorescently labeled nucleotides).
Nanopore sequencing is Third-generation (reads long DNA molecules directly without PCR).
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
Sanger sequencing: the input is a PCR-amplified fragment of the gadB gene. Preparation steps:
- PCR Amplification – Generate multiple copies of the gene.
- Purification – Remove excess primers and nucleotides.
- Cycle Sequencing – Incorporate fluorescently labeled dideoxynucleotides (ddNTPs).
- Capillary Electrophoresis – Separate DNA fragments by size for base calling.
Nanopore sequencing Preparation includes:
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
I chose Sanger sequencing because it is a first-generation method that provides high accuracy for sequencing small DNA fragments, such as the gadB gene. The process involves the following essential steps:
- PCR Amplification – The target DNA (gadB) is amplified using specific primers to generate multiple copies.
- Cycle Sequencing Reaction: DNA polymerase (to extend the strand), Regular dNTPs (to continue synthesis), and Fluorescently labeled ddNTPs (to randomly terminate elongation) is added.
- Chain Termination & Fragment Generation: When a ddNTP (dideoxynucleotide) is incorporated, DNA extension stops at that base. This results in fragments of different lengths, each ending with a labeled ddNTP.
- Capillary Electrophoresis: The fragments are separated by size in a capillary gel, with shorter fragments moving faster. A fluorescent detector reads the color signal of the last base in each fragment.
- Base Calling (Decoding the DNA Sequence): A computer analyzes the fluorescence signals from the gel. It assembles the fragment order based on size to reconstruct the full DNA sequence. The final output is a chromatogram, where each peak represents a nucleotide. This method is ideal for verifying inserted genes, such as my cloned gadB sequence, ensuring no mutations occurred during the cloning process.
- What is the output of your chosen sequencing technology?
The final output is a chromatogram, where each peak represents a nucleotide.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would synthesize the gadB gene from L. reuteri, optimized for efficient GABA production in a probiotic-based anxiety treatment. This gene encodes Glutamate Decarboxylase (GAD), an enzyme responsible for converting glutamate into GABA, a neurotransmitter known to reduce stress and anxiety.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use Twist Bioscience’s gene synthesis technology to synthesize the gadB gene because it provides highly accurate, scalable, and cost-effective de novo DNA synthesis. This technology enables the precise assembly of codon-optimized genes, ensuring efficient expression in Lactobacillus reuteri for enhanced GABA production.
Also answer the following questions:
- What are the essential steps of your chosen sequencing methods?
Essential steps of Sanger sequencing
a) DNA template preparation
b) Chain termination PCR
c) Separation of DNA fragments
d) Detection and analysis
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The limitation I find for Sanger sequencing could be the restriction regarding the length of the sequence, compared to Nanopore sequencing or Illumina.
5.3 DNA Edit.
(i) What DNA would you want to edit and why?
In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
It could be possible to edit the genome of L. reuteri to integrate the optimized gadB construct directly into the chromosome instead of relying on plasmids. This could help to increase the genetic stability, also avoid using antibiotic resistance markers, and also I wouldn´t have to worry about plasmid loss.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-based genome editing because it allows precise and targeted integration of DNA into the chromosome. This technology is efficient and used for bacterial genome engineering.
Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
CRISPR-Cas9 uses a guide RNA (gRNA) to recognize a specific DNA sequence in the genome. The Cas9 enzyme creates a double-strand break at that location. Then, the cell repairs the DNA using its natural repair mechanisms. By providing a donor DNA template with homology to the target region, the cell can integrate the gadB construct into the chromosome through homology directed repair (HDR).
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The main design and inputs include:
- Designing a guide RNA (gRNA) specific to the target genomic locus, which could be a neutral, non-essential chromosomal locus in L. reuteri
- A donor DNA template containing the optimized gadB gene with homology arms
- A plasmid-based Cas9 system, the plasmid could also include the donor DNA template
- The host cells would be L. reuteri
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- CRISPR may cut unintended regions
- Efficiency of HDR can be low in some bacteria
REFERENCES https://blog.addgene.org/crispr-101-homology-directed-repair
Gustafsson, C., Govindarajan, S., & Minshull, J. (2004). Codon bias and heterologous protein expression. Trends in biotechnology, 22(7), 346-353.
Icer, M. A., Sarikaya, B., Kocyigit, E., Atabilen, B., Çelik, M. N., Capasso, R., … & Budán, F. (2024). Contributions of gamma-aminobutyric acid (GABA) produced by lactic acid bacteria on food quality and human health: Current applications and future prospects. Foods, 13(15), 2437.
Week 2 Lecture Prep
Jacobson’s Questions
- Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error rate of polymerase: Natural DNA polymerase has an error rate of 1 in 10^6 bases.
Human genome length: The human genome is around 3.2 billion base pairs.
At an error rate of 1 in 10⁶, polymerase would introduce roughly:
3.2 x 109 / 1 x 106 = 3200 errors per genome
How biology deals with it: Despite this potential for errors, the cell uses multiple mechanisms to maintain genome stability:
Proofreading by DNA polymerase during replication.
Mismatch repair systems that fix errors after DNA synthesis.
Excision repair pathways for damaged or mismatched bases.
These systems reduce the final mutation rate to approximately 1 in 10⁹ bases, making DNA replication remarkably accurate.
- How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Because the genetic code is degenerate —meaning 61 different codons specify only 20 amino acids— a single protein sequence can be represented by countless, often billions or more, combinations of DNA sequences. For an average human protein of 400 amino acids, there are approximately 10^200 different DNA sequences that can encode the same protein.
Why not all work:
Codon bias (organisms prefer certain codons over others).
mRNA stability and folding may affect translation.
Regulatory sequences (e.g., hairpins, internal ribosome entry sites) may be unintentionally formed.
Toxic sequences might be formed (e.g., repeats, CpG motifs).