Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any of the following questions by Shuguang Zhang:
- Why humans eat beef but do not become a cow, eat fish but do not become fish?
Proteins are made from the same 20 amino acids, and digestion breaks them down into individual amino acids, which are then reused to build proteins humans need, not cow or fish proteins. This relates to the idea that the information (DNA → proteins), not the source of amino acids, defines the organism.
- Why are there only 20 natural amino acids?
According to Derek Lowe the 20 standard amino acids are not random, but represent a near-ideal set covering essential chemical properties, such as size and charge, more efficiently than alternatives. The study suggests these amino acids developed in two phases—starting with simple, meteorite-abundant forms and expanding with complex, metabolically synthesized ones—to optimize functional diversity while minimizing biosynthetic costs.
- Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely existed before life. Evidence comes from meteorites, which contain both L- and D-amino acids. These findings support the theory that amino acids were formed in space or through prebiotic chemistry, contributing to the origin of life on Earth.
- If you make an α -helix using D-amino acids, what handedness (right or left) would you expect?
Natural proteins are made from L-amino acids and form right-handed helices, so, D-amino acids would form left-handed helices, by mirror symmetry.
- Why are most molecular helices right-handed?
Henry Rzepa’s blog indicates that right-handed protein helices are preferred because they are more thermodynamically stable, offering about 1 kcal/mol more stability per amino acid compared to left-handed versions. This structural preference, identified in 1991, arises because right-handed coils minimize steric clashing between side chains and maximize efficient hydrogen bonding for L-amino acids.
- Why do β -sheets tend to aggregate?
β -sheets are very stable due to hydrogen bonding and hydrophobic interactions. So once they are formed, they are difficult to untangle, and so tend to stack and aggregate.
- What is the driving force for β -sheet aggregation?
The driving forces are hydrophobic interactions and alternating patterns of hydrophilic/hydrophobic residues.
- Why many amyloid diseases form β -sheet?
Alzheimer, Parkinson, type II Diabetes are examples of amyloid diseases, these involve protein misfolding into ordered, insoluble fibrils rich in cross beta-sheets structures, which then deposit in tissues and cause dysfunctions. These structures are highly stable, low-energy aggregates that stack tightly maximizing hydrogen bonds and are hard to clear from the body.
- Can you use amyloid β -sheets as materials?
The same stability makes them useful: β -sheet nanofibers can form hydrogels used in tissue engineering, cosmetics, and biomaterials.
- Design a β -sheet motif that forms a well-ordered structure.
A simple rule is to alternate hydrophobic and hydrophilic residues (e.g., alanine–arginine–alanine–arginine…). This pattern promotes beta-sheet formation with distinct sides: one hydrophobic, one hydrophilic. This arrangement supports ordered stacking and material formation.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
Green Fluorescent Protein (GFP)
- Briefly describe the protein you selected and why you selected it.
- Discovered in jellyfish (Aequorea victoria), widely used in biology as a fluorescent marker.
- Easy to visualize and well-documented.
- Cool beta-barrel structure which protects a central chromophore.
- Has both helices and beta-sheets.
I selected GFP as it has become a powerful tool in molecular and cellular biology. Scientists use it as a genetic reporter, tagging it to other proteins, to visualize their location, movement, and expression in real time within live cells or organisms. GFP has been engineered into many variants with different colors and improved brightness or stability.
- Identify the amino acid sequence of your protein.
1EMA_1|Chain A|GREEN FLUORESCENT PROTEIN|Aequorea victoria MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFTYGVQCFSRYPDHMKRHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
- How long is it? What is the most frequent amino acid?
Length: 238 amino acids Most frequent: G (22 times, 9.2%)
- How many protein sequence homologs are there for your protein? Hint: Use the Uniprot’s BLAST tool to search for homologs.
BLAST 201 results found in UniProtKB
- Does your protein belong to any protein family?
Yes, to the GFP family. These proteins are able to fluoresce different colours, some of which are non-fluorescent. These proteins are all essentially encoded by single genes, since both the substrate and the catalytic enzyme for pigment biosynthesis are provided within a single polypeptide chain. (InterPro, Pfam = PF01353).
- Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
1996-11-08 Initial release Resolution: 1.90 Å, which is considered high resolution (very high accuracy).
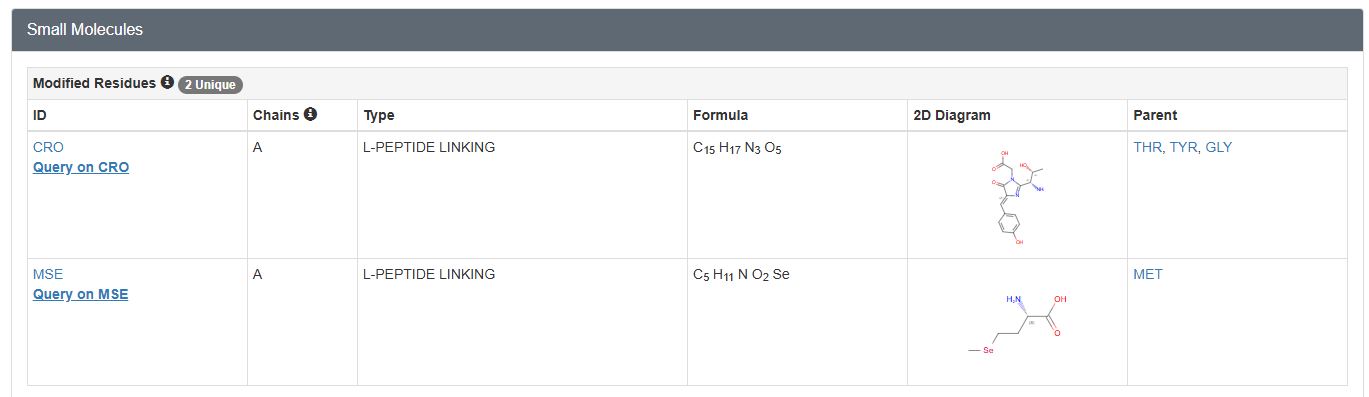
- Are there any other molecules in the solved structure apart from protein?
Yes, a peptide derived chromophore and a Selenomethionine ligand
- Does your protein belong to any structure classification family?
Superfamily: GFP-like Domain: 6OA8 A:4-238 SCOP ID: 8096046
- Open the structure of your protein in any 3D molecule visualization software:
- PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”
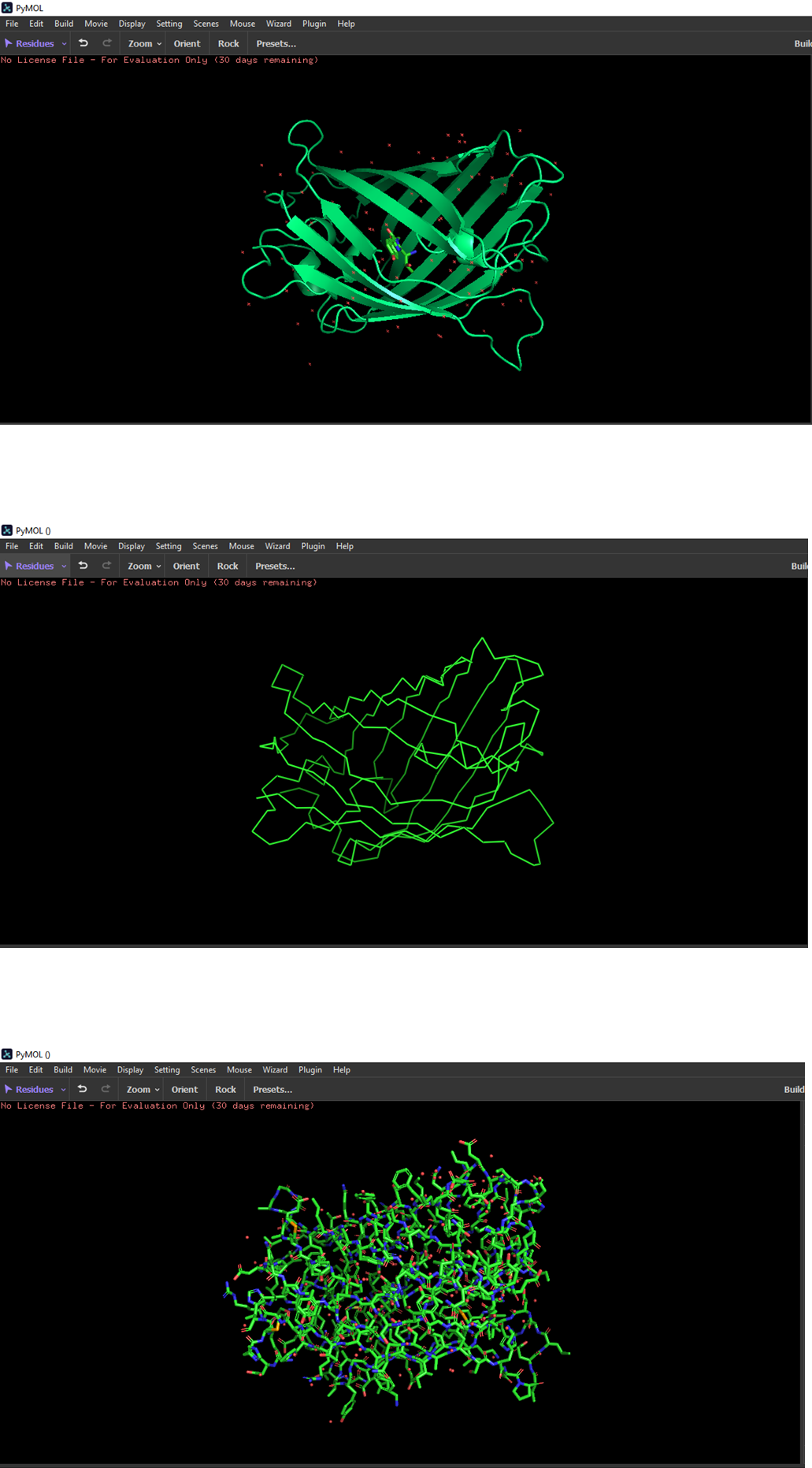
- Color the protein by secondary structure. Does it have more helices or sheets?
The structure has more sheets.
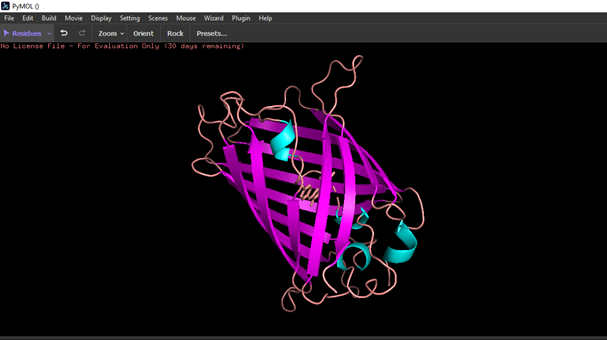
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophobic residues are mostly located in the core of the GFP structure, while hydrophilic residues are exposed on the surface, interacting with the aqueous environment.
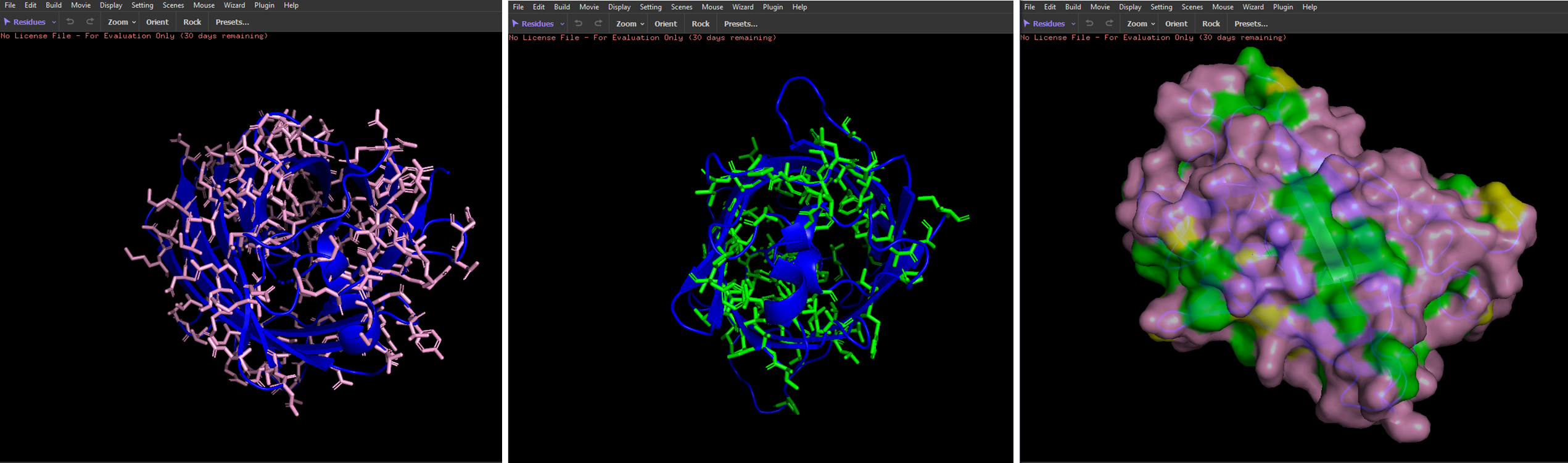
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
I couldn’t identify any.
Part C: Using ML-Based Protein Protein Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
Dispersin B
1YHT_1|Chain A|DspB|Aggregatibacter actinomycetemcomitans (714)
NCCVKGNSIYPQKTSTKQTGLMLDIARHFYSPEVIKSFIDTISLSGGNFLHLHFSDHENYAIESHLLNQRAENAVQGKDGIYINPYTGKPFLSYRQLDDIKAYAKAKGIELIPELDSPNHMTAIFKLVQKDRGVKYLQGLKSRQVDDEIDITNADSITFMQSLMSEVIDIFGDTSQHFHIGGDEFGYSVESNHEFITYANKLSYFLEKKGLKTRMWNDGLIKNTFEQINPNIEITYWSYDGDTQDKNEAAERRDMRVSLPELLAKGFTVLNYNSYYLYIVPKASPTFSQDAAFAAKDVIKNWDLGVWDGRNTKNRVQNTHEIAGAALSIWGEDAKALKDETIQKNTKSLLEAVIHKTNGDEHHHHHH
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
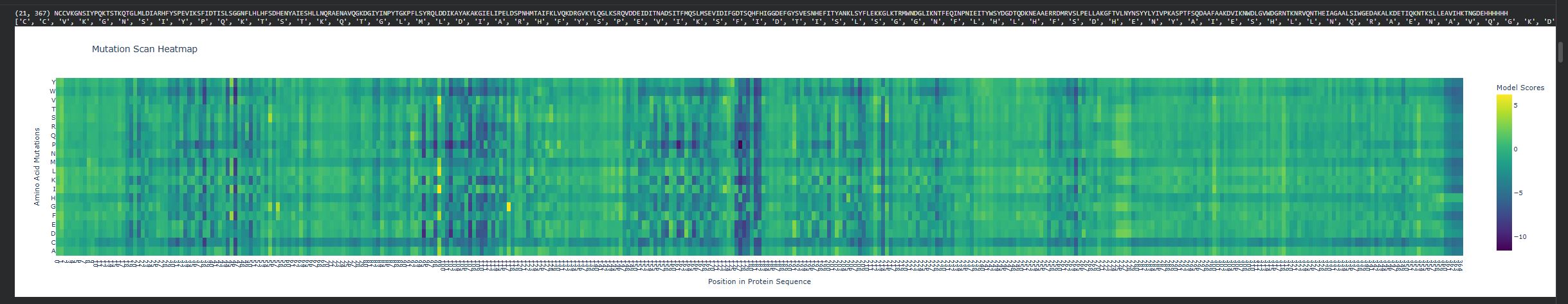
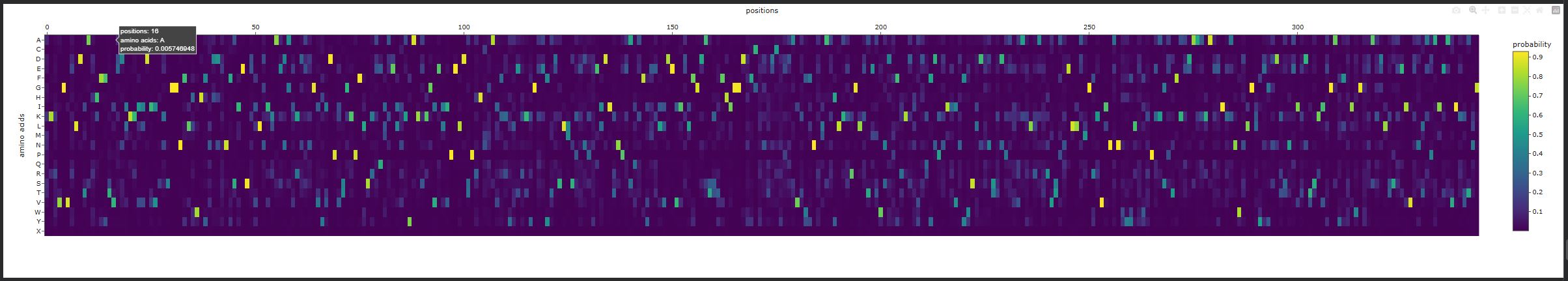
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
There are vertical columns that are blue or purple, which reveals positions that are not tolerant to mutations, meaning these residues are important structurally or functionally. The enzyme disperin B needs to have a stable structure to function correctly. In general there is a lot of green, which means that several positions do tolerate mutations.
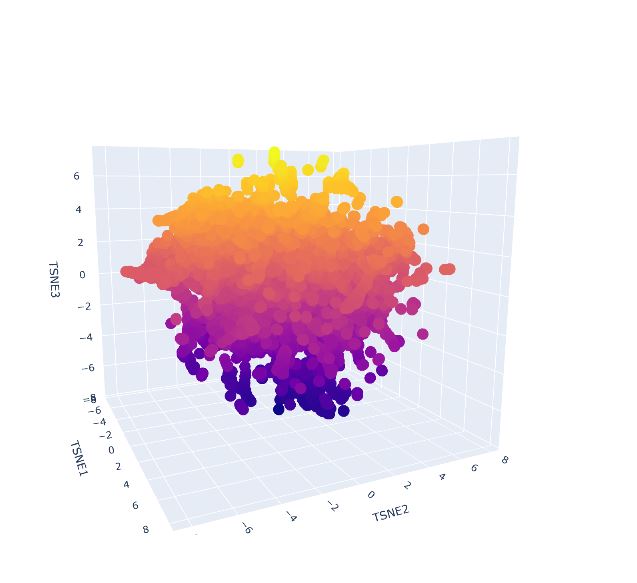
- Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
- Analyze the different formed neighborhoods: do they approximate similar proteins?
- Place your protein in the resulting map and explain its position and similarity to its neighbors
It was not possible to locate my protein, quite likely it is immersed within the protein clusters, which means it shares features with similar proteins. As it was not found on the external area or isolated, this supports its classification as it is not considered an outlier.
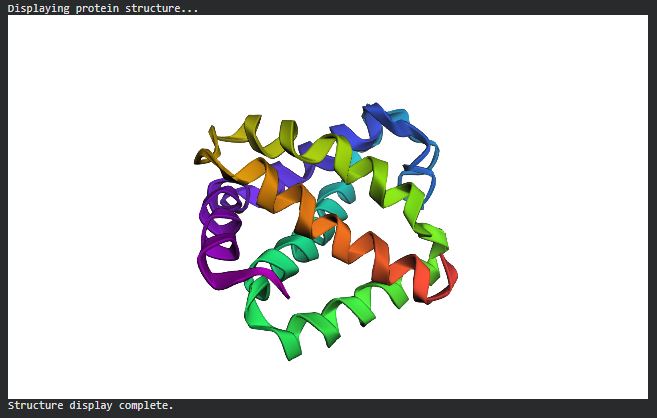
C2. Protein Folding
Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
{‘1YHT’: ([‘A’], [])} Length of chain A is 344
Generating sequences…
1YHT, score=1.4975, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020
TKQTGLMLDIARHFYSPEVIKSFIDTISLSGGNFLHLHFSDHENYAIESHLLNQRAENAVQGKDGIYINPYTGKPFLSYRQLDDIKAYAKAKGIELIPELDSPNHMTAIFKLVQKDRGVKYLQGLKSRQVDDEIDITNADSITFMQSLMSEVIDIFGDTSQHFHIGGDEFGYSVESNHEFITYANKLSYFLEKKGLKTRMWNDGLIKNTFEQINPNIEITYWSYDGDTQDKNEAAERRDMRVSLPELLAKGFTVLNYNSYYLYIVPKASPTFSQDAAFAAKDVIKNWDLGVWDGRNTKNRVQNTHEIAGAALSIWGEDAKALKDETIQKNTKSLLEAVIHKTNG
T=0.1, sample=0, score=0.8100, seq_recovery=0.4622
KKRVGVELDTARHFFTVEDIKKIIDIVAKAGGNFLHWHFNDDENFSIESEILNIKAEDAIKDENGVYIDPVSGKPFLSKEQIAELNKYAKEKGVELIPELDTPRHNMAIFKYLERKYGKEYTDSLMSPENPNEIDIKNPESQKFYEELVDEVIDLFGDATKTFHIGGDNFCYNTDCYEDAVKWFNRLAAHLKKRGLETETWNDPFRKDIVDQFDPNIIVNYWSLDENSTDPEVVAKLKATRVTANELLAMGRKVINANWYYLEWKPKKSPTFKEDAAKKAEEIKANWNLGLYEGDDKSNEITDTSKIEGAYFSIWGGEAKDLTSEEIVENLAPAIEAVIKTTNG
Generating sequences…
1YHT, score=1.4792, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020
TKQTGLMLDIARHFYSPEVIKSFIDTISLSGGNFLHLHFSDHENYAIESHLLNQRAENAVQGKDGIYINPYTGKPFLSYRQLDDIKAYAKAKGIELIPELDSPNHMTAIFKLVQKDRGVKYLQGLKSRQVDDEIDITNADSITFMQSLMSEVIDIFGDTSQHFHIGGDEFGYSVESNHEFITYANKLSYFLEKKGLKTRMWNDGLIKNTFEQINPNIEITYWSYDGDTQDKNEAAERRDMRVSLPELLAKGFTVLNYNSYYLYIVPKASPTFSQDAAFAAKDVIKNWDLGVWDGRNTKNRVQNTHEIAGAALSIWGEDAKALKDETIQKNTKSLLEAVIHKTNG
T=0.1, sample=0, score=0.8035, seq_recovery=0.4826
KKKTGVWLDTAHHFFSVENIKKIIDIVADAGGNFLLWHFNDDENFSIESEVLKILAEDGIKDENGVYIDPETGKPFLTREQIAELNAYAKAKGVEIIPELDSPRHMGAIFRYVERAKGKAYRDSLLSKEDPNELDITNPESQAFYKELLDEVIDLFGDATSTFHIGGDDFCWNTDCYDDAVKWFNDIAAYLKEKGLKTMMWNDPFRKDNYAQFDKDIEVLFWSLDDNTTDPKVAAELRATRVTAAELLAAGRTVINANWYYLYWTPKAGPTFKADAAAHAEDVKANWNLGLYEGSDTSNAITDTSKIAGALFWIDGWNAANLSDKEIVDATAPAIEAVIKKTNG
New Sequence: KKKTGVWLDTAHHFFSVENIKKIIDIVADAGGNFLLWHFNDDENFSIESEVLKILAEDGIKDENGVYIDPETGKPFLTREQIAELNAYAKAKGVEIIPELDSPRHMGAIFRYVERAKGKAYRDSLLSKEDPNELDITNPESQAFYKELLDEVIDLFGDATSTFHIGGDDFCWNTDCYDDAVKWFNDIAAYLKEKGLKTMMWNDPFRKDNYAQFDKDIEVLFWSLDDNTTDPKVAAELRATRVTAAELLAAGRTVINANWYYLYWTPKAGPTFKADAAAHAEDVKANWNLGLYEGSDTSNAITDTSKIAGALFWIDGWNAANLSDKEIVDATAPAIEAVIKKTNG
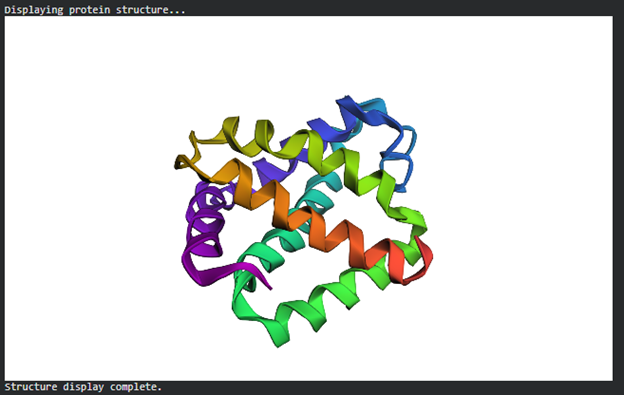
- Input this sequence into ESMFold and compare the predicted structure to your original.
References
- Greenwald, J., Riek, R., 2010. Biology of amyloid: Structure, function, and regulation. Structure. https://doi.org/10.1016/j.str.2010.08.009
- Lowe, D. (2023, February 27). Why these amino acids, anyway? | science | AAAS. Science. https://www.science.org/content/blog-post/why-these-amino-acids-anyway
- Rzepa, H. (2019, November 29). Why are α-helices in proteins mostly right handed? Henry Rzepa’s Blog. https://www.ch.ic.ac.uk/rzepa/blog/?p=3802#:~:text=We%20showed%20in%201991%5B6,0.9