Class Assignment: Biosensors for Anxiety 1. Describe a biological engineering application or tool you want to develop and why. Currently as I am working with microbiome sequencing, I have developed an interest in the relationship between the microbiome and mental health, hence my interest in using genetically modified bacteria to create biosensors for anxiety. Consider bacteria like Lactobacillus reuteri, which are specifically engineered to identify high quantities of cortisol, the hormone that human bodies release during stressful situations. GABA (gamma-aminobutyric acid), a naturally occurring substance that aids in nervous system relaxation, would be produced by the modified bacteria in response to an increase in these cortisol levels. Not everyone reacts to traditional anxiety medications equally, and they frequently have negative side effects. So my proposed strategy presents a viable substitute—a probiotic supplement that supports mental health in real time and functions organically with the body. The simplicity of this concept—using something as ubiquitous as gut microbes to create a significant impact—is what makes it beautiful and potentially innocuous.
Part 1: Benchling & In-silico Gel Art Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. You might find Ronan’s website a helpful tool for quickly iterating on designs! Part 3: DNA Design Challenge 3.1. Choose your protein.
Assignment: Python Script for Opentrons Artwork 1. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. I used the GUI for converting my rising phoenix image into the dot design, I loved the tool, very helpful, easy to use and to edit and perfectionate my design. The tool supplied the coordinates to add to my code, and the result looks perfect!
Part A. Conceptual Questions Answer any of the following questions by Shuguang Zhang: Why humans eat beef but do not become a cow, eat fish but do not become fish? Proteins are made from the same 20 amino acids, and digestion breaks them down into individual amino acids, which are then reused to build proteins humans need, not cow or fish proteins. This relates to the idea that the information (DNA → proteins), not the source of amino acids, defines the organism.
1. Describe a biological engineering application or tool you want to develop and why.
Currently as I am working with microbiome sequencing, I have developed an interest in the relationship between the microbiome and mental health, hence my interest in using genetically modified bacteria to create biosensors for anxiety. Consider bacteria like Lactobacillus reuteri, which are specifically engineered to identify high quantities of cortisol, the hormone that human bodies release during stressful situations. GABA (gamma-aminobutyric acid), a naturally occurring substance that aids in nervous system relaxation, would be produced by the modified bacteria in response to an increase in these cortisol levels.
Not everyone reacts to traditional anxiety medications equally, and they frequently have negative side effects. So my proposed strategy presents a viable substitute—a probiotic supplement that supports mental health in real time and functions organically with the body. The simplicity of this concept—using something as ubiquitous as gut microbes to create a significant impact—is what makes it beautiful and potentially innocuous.
2. Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
This technology needs to be developed ethically if it is to be genuinely helpful. The following are the main objectives that ought to direct its governance:
Promoting safety, preventing harm, and upholding ethical behaviors are the main objectives.
Sub-Goal 1: Biosafety - Reduce the possibility of accidentally releasing genetically modified bacteria into the environment. Make sure the modified strains don’t upset the human gut microbiome’s natural equilibrium.
Sub-Goal 2: Informed Consent and Public Awareness - Provide simple, understandable information to users regarding the biosensor’s operation. Encourage openness by making sure individuals are aware of the advantages as well as any possible risks.
Sub-Goal 3: Access Equity - Make sure that not only a select few have access to this technology. Ensuring extensive and cheap access is essential in Ecuador due to its various communities and healthcare inequities.
In Ecuador, genetically modified (GM) crops and seeds are banned, except in rare cases where the President and the National Assembly approve them for national interest. Although GM crops are not grown locally, some food products containing GM ingredients (like maize and soy) are imported. Additionally, research with GMOs is allowed under strict biosafety conditions. Food products with over 0.9% GM content must be labeled clearly. This regulatory context will guide how Sbiosensors are developed and used responsibly in Ecuador.
3. Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Governance Actions
Action
Purpose
Design
Assumptions
Risks of Failure & Success
1. Biosafety Certification
Confirm that the bacteria are safe for people and the environment.
Review by the National Biosafety Committee (CONABIO) and the Ministry of Public Health.
Belief that existing biosafety protocols are sufficient.
Potential for unforeseen health risks or environmental issues.
2. Public Education Campaigns
Help the public understand biosensors and their safety.
Awareness programs led by NGOs, universities, and health agencies.
Assuming public trust will naturally follow accurate information.
Misinformation could spread more rapidly than facts.
3. International Collaboration
Ensure Ecuador’s policies align with global biosafety standards.
Ecuador already collaborates with organizations such as FAO and WHO on biosafety issues. The goal is to extend these collaborations to cover new technologies, including biosensors for mental health applications.
Expectation that international guidelines fit Ecuador’s context.
Some global policies may not consider local Ecuadorian realities.
4. Equity Monitoring Program
Guarantee equal access to the technology across all regions.
Oversight by public health institutions focusing on affordability and distribution.
Assumes regulations will prevent unequal access.
Risk of high costs or limited availability in remote communities.
4. Score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the option:
Action 1: Biosafety Certification
Action 2: Public Education Campaigns
Action 3: International Collaboration
Action 4: Equity Monitoring Program
Enhance Biosecurity
• By preventing incidents
1
2
1
3
• By helping respond
2
1
2
2
Foster Lab Safety
• By preventing incidents
1
3
2
N/A
• By helping respond
2
1
2
N/A
Protect the environment
• By preventing incidents
1
N/A
2
3
• By helping respond
2
N/A
2
2
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
2
2
• Feasibility?
2
1
2
2
• Not impede research
1
1
1
N/A
• Promote constructive applications
1
1
1
1
5. Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Biosafety Certification is the most important factor when it comes to scoring because it guarantees that the product is safe for human use and reduces environmental hazards. Prior to any public deployment, this fundamental step is essential. International collaboration is also key, as it offers access to global biosafety standards, proven governance frameworks, and technical support. Ecuador already works with organizations such as the FAO and WHO, and expanding these collaborations can strengthen the effectiveness of national efforts across biosafety, education, and equity. The significance of public education campaigns, which promote understanding and trust among the general public and are essential for acceptance and appropriate use, comes next. Finally, the Equity Monitoring Program is also important since it will help guarantee that the technology is accessible to everyone who needs it, not just those in places with ample resources, especially in Ecuador where access to healthcare can differ greatly between urban and rural areas. By defining a balance between these criteria, biosensors for anxiety will develop into a useful, moral, and safe instrument that will benefit not only Ecuador but the entire world.
References
Bravo, J. A., Forsythe, P., Chew, M. V., Escaravage, E., Savignac, H. M., Dinan, T. G., … & Cryan, J. F. (2011). Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proceedings of the National Academy of Sciences, 108(38), 16050-16055.
Cryan, J. F., & Dinan, T. G. (2012). Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nature Reviews. Neuroscience, 13(10), 701–712. https://doi.org/10.1038/nrn3346
Santos, E., Sánchez, E., Hidalgo, L., Chávez, T., Villao, L., Pacheco, R., & Navarrete, O. (2014, August). Status and challenges of genetically modified crops and food in Ecuador. In XXIX International Horticultural Congress on Horticulture: Sustaining Lives, Livelihoods and Landscapes (IHC2014): 1110 (pp. 229-235).
The assistance of OpenAI’s ChatGPT was employed to help clarify ideas, revise language and grammar.
Week 2 HW: DNA read, write and edit
Part 1: Benchling & In-silico Gel Art
Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs!
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
I have chosen Glutamate Decarboxylase (GAD) from Lactobacillus reuteri because it is the key enzyme responsible for converting glutamate into GABA (gamma-aminobutyric acid). Since GABA plays a crucial role in reducing anxiety by calming the nervous system, engineering L. reuteri to enhance its GABA production could provide a natural, probiotic-based approach to mental health support. This selection aligns with my interest in the microbiome’s impact on mental health and the potential of synthetic biology to develop gut-based biosensors for anxiety management.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Employing the Reverse Translate tool from https://www.bioinformatics.org/sms2/rev_trans.html I chose this sequence, “Most Likely Codons” sequence, as it ensures efficient translation in Lactobacillus reuteri by selecting the most frequently used codons, minimizing translation delays caused by rare codons.
>reverse translation of Untitled to a 1407 base sequence of most likely codons.
atggcgatgctgtatggcaaacataaccatgaagcggaagaatatctggaaccggtgttt
ggcgcgccgagcgaacagcatgatctgccgaaatatcgcctgccgaaacatagcctgagc
ccgcgcgaagcggatcgcctggtgcgcgatgaactgctggatgaaggcaacagccgcctg
aacctggcgaccttttgccagacctatatggaaccggaagcggtggaactgatgaaagat
accctggcgaaaaacgcgattgataaaagcgaatatccgcgcaccgcggaaattgaaaac
cgctgcgtgaacattattgcgaacctgtggcatgcgccggatgatgaacattttaccggc
accagcaccattggcagcagcgaagcgtgcatgctgggcggcctggcgatgaaatttgcg
tggcgcaaacgcgcgcaggcggcgggcctggatctgaacgcgcatcgcccgaacctggtg
attagcgcgggctatcaggtgtgctgggaaaaattttgggtgtattgggatgtggatatg
catgtggtgccgatggatgaacagcatatggcgctggatgtgaaccatgtgctggattat
ggcgatgaatataccattggcattgtgggcattatgggcattacctataccggccaggat
gatgatctggcggcgctggataaagtggtgacccattataaccatcagcatccgaaactg
ccggtgtatattcatgtggatgcggcgagcggcggcttttataccccgtttattgaaccg
cagctgatttgggattttcgcctggcgaacgtggtgagcattaacgcgagcggccataaa
tatggcctggtgtatccgggcgtgggctgggtggtgtggcgcgatcgccagtttctgccg
ccggaactggtgtttaaagtgagctatctgggcggcgaactgccgaaaatggcgattaac
tttagccatagcgcggcgcagctgattgaacagtattataactttattcgctttggcatg
gatggctatcgcaaaattcagaccaaaacccatgatgtggcgcgctatcgcgcgaccgcg
ccggataaagtgggcgaactgaaaatgattaacaacggccatcagccgccgctgatttgc
tatcagctgccgccgcgcgaagatcgcaaaggcaccctgtatgatctgagcgatcgcctg
ctgatgaacggctggcaggtgccgacctatccgctgccggcgaacctggaacagcaggtg
attcagcgcattgtggtgcgcgcggattttggcatgaacatggcgcatgattttatggat
gatctgaccaaagcggtgcatgatctgaaccatgcgcatattgtgtatcatcatgatgcg
gcgccgaaaaaatatggctttacccat
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Codon optimization involves redesigning a DNA sequence to use codons preferred by the host organism while preserving the same amino acid sequence. This improves translation efficiency, protein yield, and expression stability.
Glutamate decarboxylase (gadB) gene with Codon-Optimization (E. coli)
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
For producing Glutamate Decarboxylase (GAD) from Lactobacillus reuteri, I will first express it in E. coli as an initial step before moving to L. reuteri.
To achieve this, I will clone the codon-optimized gadB gene into a plasmid with a strong bacterial promoter, such as pLac or T7, for efficient expression. I will then introduce the plasmid into competent E. coli cells using heat shock transformation.
To ensure only transformed cells grow, I will use selective markers, such as an antibiotic resistance gene (ampicillin or kanamycin) on the plasmid. I may also include a reporter gene, such as GFP (Green Fluorescent Protein), to visually confirm successful transformation under UV light. The transformed E. coli colonies will be plated on selective agar containing the antibiotic, ensuring that only bacteria carrying the plasmid with the GAD insert survive.
Part 4: Prepare a Twist DNA Synthesis Order
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would sequence the gadB gene from L. reuteri to ensure accurate expression and function in the engineered strain. Sequencing this DNA would confirm that no mutations occurred during cloning, preserving the enzyme’s ability to produce GABA efficiently. This is crucial for validating the stability and effectiveness of the modified strain before using it as part of a probiotic-based anxiety treatment.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Sanger sequencing to confirm the accuracy of the gadB gene after cloning. Sanger sequencing is first-generation sequencing, known for its high accuracy in small DNA fragments like single genes. It is cost-effective, widely accessible, and ideal for verifying cloned plasmids before moving to functional testing.
However, if larger-scale sequencing were needed (e.g., confirming genomic integration or long fragments), I could use Nanopore sequencing, as I have access to one in the lab. Nanopore is third-generation sequencing, capable of reading long DNA fragments without amplification, making it useful for analyzing structural variations.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
Sanger sequencing is First-generation (based on chain termination using fluorescently labeled nucleotides).
Nanopore sequencing is Third-generation (reads long DNA molecules directly without PCR).
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
Sanger sequencing: the input is a PCR-amplified fragment of the gadB gene.
Preparation steps:
PCR Amplification – Generate multiple copies of the gene.
Purification – Remove excess primers and nucleotides.
Capillary Electrophoresis – Separate DNA fragments by size for base calling.
Nanopore sequencing
Preparation includes:
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
I chose Sanger sequencing because it is a first-generation method that provides high accuracy for sequencing small DNA fragments, such as the gadB gene. The process involves the following essential steps:
PCR Amplification – The target DNA (gadB) is amplified using specific primers to generate multiple copies.
Cycle Sequencing Reaction: DNA polymerase (to extend the strand), Regular dNTPs (to continue synthesis), and Fluorescently labeled ddNTPs (to randomly terminate elongation) is added.
Chain Termination & Fragment Generation: When a ddNTP (dideoxynucleotide) is incorporated, DNA extension stops at that base. This results in fragments of different lengths, each ending with a labeled ddNTP.
Capillary Electrophoresis: The fragments are separated by size in a capillary gel, with shorter fragments moving faster. A fluorescent detector reads the color signal of the last base in each fragment.
Base Calling (Decoding the DNA Sequence): A computer analyzes the fluorescence signals from the gel. It assembles the fragment order based on size to reconstruct the full DNA sequence. The final output is a chromatogram, where each peak represents a nucleotide. This method is ideal for verifying inserted genes, such as my cloned gadB sequence, ensuring no mutations occurred during the cloning process.
What is the output of your chosen sequencing technology?
The final output is a chromatogram, where each peak represents a nucleotide.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would synthesize the gadB gene from L. reuteri, optimized for efficient GABA production in a probiotic-based anxiety treatment. This gene encodes Glutamate Decarboxylase (GAD), an enzyme responsible for converting glutamate into GABA, a neurotransmitter known to reduce stress and anxiety.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use Twist Bioscience’s gene synthesis technology to synthesize the gadB gene because it provides highly accurate, scalable, and cost-effective de novo DNA synthesis. This technology enables the precise assembly of codon-optimized genes, ensuring efficient expression in Lactobacillus reuteri for enhanced GABA production.
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
Essential steps of Sanger sequencing
a) DNA template preparation
b) Chain termination PCR
c) Separation of DNA fragments
d) Detection and analysis
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The limitation I find for Sanger sequencing could be the restriction regarding the length of the sequence, compared to Nanopore sequencing or Illumina.
5.3 DNA Edit.
(i) What DNA would you want to edit and why?
In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
It could be possible to edit the genome of L. reuteri to integrate the optimized gadB construct directly into the chromosome instead of relying on plasmids. This could help to increase the genetic stability, also avoid using antibiotic resistance markers, and also I wouldn´t have to worry about plasmid loss.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-based genome editing because it allows precise and targeted integration of DNA into the chromosome. This technology is efficient and used for bacterial genome engineering.
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
CRISPR-Cas9 uses a guide RNA (gRNA) to recognize a specific DNA sequence in the genome. The Cas9 enzyme creates a double-strand break at that location. Then, the cell repairs the DNA using its natural repair mechanisms. By providing a donor DNA template with homology to the target region, the cell can integrate the gadB construct into the chromosome through homology directed repair (HDR).
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The main design and inputs include:
Designing a guide RNA (gRNA) specific to the target genomic locus, which could be a neutral, non-essential chromosomal locus in L. reuteri
A donor DNA template containing the optimized gadB gene with homology arms
A plasmid-based Cas9 system, the plasmid could also include the donor DNA template
The host cells would be L. reuteri
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Gustafsson, C., Govindarajan, S., & Minshull, J. (2004). Codon bias and heterologous protein expression. Trends in biotechnology, 22(7), 346-353.
Icer, M. A., Sarikaya, B., Kocyigit, E., Atabilen, B., Çelik, M. N., Capasso, R., … & Budán, F. (2024). Contributions of gamma-aminobutyric acid (GABA) produced by lactic acid bacteria on food quality and human health: Current applications and future prospects. Foods, 13(15), 2437.
Week 2 Lecture Prep
Jacobson’s Questions
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error rate of polymerase: Natural DNA polymerase has an error rate of 1 in 10^6 bases.
Human genome length: The human genome is around 3.2 billion base pairs.
At an error rate of 1 in 10⁶, polymerase would introduce roughly:
3.2 x 109 / 1 x 106 = 3200 errors per genome
How biology deals with it:
Despite this potential for errors, the cell uses multiple mechanisms to maintain genome stability:
Proofreading by DNA polymerase during replication.
Mismatch repair systems that fix errors after DNA synthesis.
Excision repair pathways for damaged or mismatched bases.
These systems reduce the final mutation rate to approximately 1 in 10⁹ bases, making DNA replication remarkably accurate.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Because the genetic code is degenerate —meaning 61 different codons specify only 20 amino acids— a single protein sequence can be represented by countless, often billions or more, combinations of DNA sequences. For an average human protein of 400 amino acids, there are approximately 10^200 different DNA sequences that can encode the same protein.
Why not all work:
Codon bias (organisms prefer certain codons over others).
mRNA stability and folding may affect translation.
Regulatory sequences (e.g., hairpins, internal ribosome entry sites) may be unintentionally formed.
Toxic sequences might be formed (e.g., repeats, CpG motifs).
Week 3 HW: Lab Automation
Assignment: Python Script for Opentrons Artwork
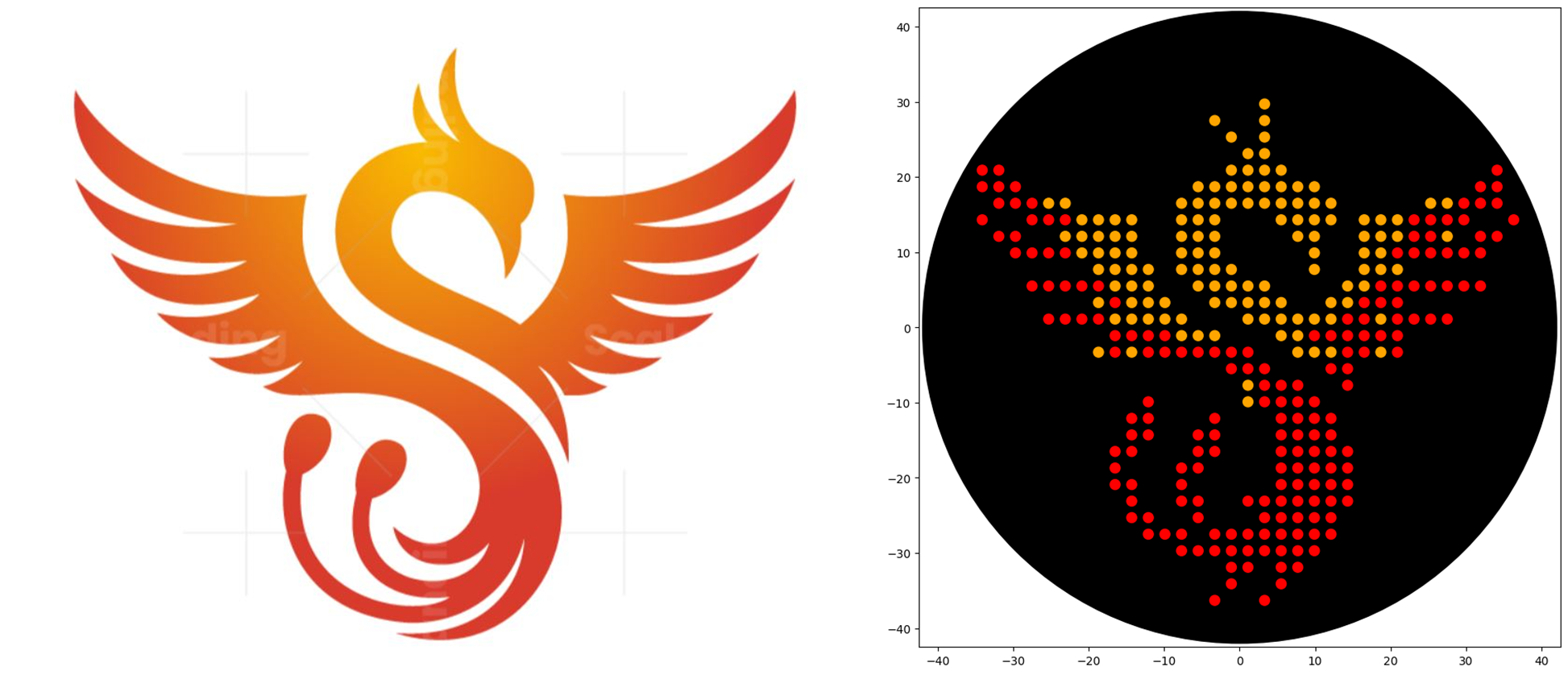
1. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
I used the GUI for converting my rising phoenix image into the dot design, I loved the tool, very helpful, easy to use and to edit and perfectionate my design. The tool supplied the coordinates to add to my code, and the result looks perfect!
I selected the rising phoenix as it represents transformation, resilience, and renewal. It reminds me that growth often comes from challenges, and I have the capacity to transform and emerge stronger.
2. Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
Ready in Google Colab
I used ChatGPT to polish written explanations, to clarify concepts and clean and debug Opentrons Python code. All final decisions and interpretations were reviewed and confirmed by me.
Post-Lab Questions
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
“Cell-free biosensor with automated acoustic liquid handling for rapid and scalable characterization of cellobiohydrolases on microcrystalline cellulose”
This study presents a cell-free biosensor designed to rapidly detect the activity of enzymes that break down crystalline cellulose. Instead of relying on traditional, slow assays or cell-based systems, the authors built a transcription factor-based biosensor that produces a fluorescent signal when cellobiose is generated. By integrating this system with the Echo 525 acoustic liquid handler, they demonstrated highly precise, optimized the reactions for small-volumes, and automated reactions that match manual performance while increasing a lot the throughput. Overall, the work shows how combining cell-free systems with automation can significantly accelerate enzyme screening and improve the efficiency of the complete experiment cycle in synthetic biology.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
For my project on biosensors for anxiety, I plan to use automation tools like Opentrons to support the genetic engineering of bacteria. My goal is to modify Lactobacillus reuteri so that it can sense elevated cortisol levels and respond by producing GABA, a calming neurotransmitter. By automating key molecular cloning steps, I hope to make the strain engineering process more consistent, efficient, and scalable while reducing human error during repetitive lab work.
Molecular cloning is a central step in building this biosensor system. Automating these procedures will help ensure precision and reproducibility, allowing the design-build-test cycle to move faster and more reliably.
2.1.DNA Fragment Preparation
Use Opentrons liquid handling to accurately pipette reagents for plasmid extraction and PCR amplification of gene fragments.
Automate purification steps (e.g., magnetic bead-based purification or column-based cleanup) to standardize DNA yields.
2.2.Restriction Digestion & Ligation
Automate enzymatic digestion using restriction enzymes to cut plasmids and insert fragments under precise conditions.
Program ligase reactions for efficient DNA assembly, ensuring consistent reaction times and temperatures.
2.3.Transformation of Engineered Constructs
Use Opentrons to pipette competent cells, mix them with recombinant DNA, and automate the heat shock or electroporation process.
Plate transformed cells onto selective agar using a robotic liquid handler to ensure even distribution.
2.4.Screening and Colony Picking
Automate pipetting of selection media for colony growth.
Use robotic colony pickers (if available) or automate PCR setup for rapid screening of positive clones.
2.5.PCR & Validation
Automate PCR setup for screening transformed colonies, ensuring each reaction is prepared with high accuracy.
If sequencing is needed, automate DNA preparation for submission to sequencing facilities.
REFERENCES
Taeok Kim, Eun Jung Jeon, Kil Koang Kwon, Minji Ko, Ha-Neul Kim, Seong Keun Kim, Eugene Rha, Jonghyeok Shin, Haseong Kim, Dae-Hee Lee, Bong Hyun Sung, Soo-Jung Kim, Hyewon Lee, Seung-Goo Lee, Cell-free biosensor with automated acoustic liquid handling for rapid and scalable characterization of cellobiohydrolases on microcrystalline cellulose, Synthetic Biology, Volume 10, Issue 1, 2025, ysaf005, https://doi.org/10.1093/synbio/ysaf005
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any of the following questions by Shuguang Zhang:
Why humans eat beef but do not become a cow, eat fish but do not become fish?
Proteins are made from the same 20 amino acids, and digestion breaks them down into individual amino acids, which are then reused to build proteins humans need, not cow or fish proteins. This relates to the idea that the information (DNA → proteins), not the source of amino acids, defines the organism.
Why are there only 20 natural amino acids?
According to Derek Lowe the 20 standard amino acids are not random, but represent a near-ideal set covering essential chemical properties, such as size and charge, more efficiently than alternatives. The study suggests these amino acids developed in two phases—starting with simple, meteorite-abundant forms and expanding with complex, metabolically synthesized ones—to optimize functional diversity while minimizing biosynthetic costs.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely existed before life. Evidence comes from meteorites, which contain both L- and D-amino acids. These findings support the theory that amino acids were formed in space or through prebiotic chemistry, contributing to the origin of life on Earth.
If you make an α -helix using D-amino acids, what handedness (right or left) would you expect?
Natural proteins are made from L-amino acids and form right-handed helices, so, D-amino acids would form left-handed helices, by mirror symmetry.
Why are most molecular helices right-handed?
Henry Rzepa’s blog indicates that right-handed protein helices are preferred because they are more thermodynamically stable, offering about 1 kcal/mol more stability per amino acid compared to left-handed versions. This structural preference, identified in 1991, arises because right-handed coils minimize steric clashing between side chains and maximize efficient hydrogen bonding for L-amino acids.
Why do β -sheets tend to aggregate?
β -sheets are very stable due to hydrogen bonding and hydrophobic interactions. So once they are formed, they are difficult to untangle, and so tend to stack and aggregate.
What is the driving force for β -sheet aggregation?
The driving forces are hydrophobic interactions and alternating patterns of hydrophilic/hydrophobic residues.
Why many amyloid diseases form β -sheet?
Alzheimer, Parkinson, type II Diabetes are examples of amyloid diseases, these involve protein misfolding into ordered, insoluble fibrils rich in cross beta-sheets structures, which then deposit in tissues and cause dysfunctions. These structures are highly stable, low-energy aggregates that stack tightly maximizing hydrogen bonds and are hard to clear from the body.
Can you use amyloid β -sheets as materials?
The same stability makes them useful: β -sheet nanofibers can form hydrogels used in tissue engineering, cosmetics, and biomaterials.
Design a β -sheet motif that forms a well-ordered structure.
A simple rule is to alternate hydrophobic and hydrophilic residues (e.g., alanine–arginine–alanine–arginine…). This pattern promotes beta-sheet formation with distinct sides: one hydrophobic, one hydrophilic. This arrangement supports ordered stacking and material formation.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
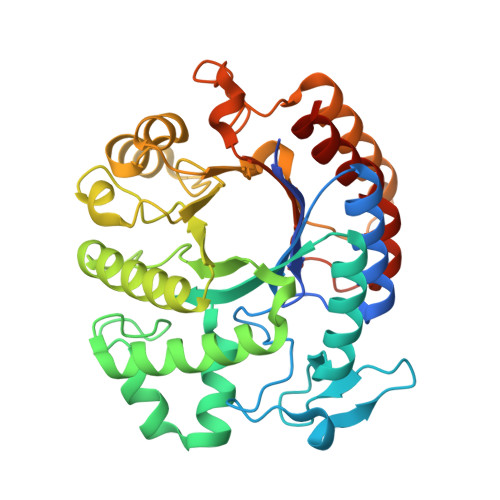
Green Fluorescent Protein (GFP)
Briefly describe the protein you selected and why you selected it.
Discovered in jellyfish (Aequorea victoria), widely used in biology as a fluorescent marker.
Easy to visualize and well-documented.
Cool beta-barrel structure which protects a central chromophore.
Has both helices and beta-sheets.
I selected GFP as it has become a powerful tool in molecular and cellular biology. Scientists use it as a genetic reporter, tagging it to other proteins, to visualize their location, movement, and expression in real time within live cells or organisms. GFP has been engineered into many variants with different colors and improved brightness or stability.
Identify the amino acid sequence of your protein.
1EMA_1|Chain A|GREEN FLUORESCENT PROTEIN|Aequorea victoria
MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFTYGVQCFSRYPDHMKRHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
How long is it? What is the most frequent amino acid?
Length: 238 amino acids
Most frequent: G (22 times, 9.2%)
How many protein sequence homologs are there for your protein? Hint: Use the Uniprot’s BLAST tool to search for homologs.
BLAST 201 results found in UniProtKB
Does your protein belong to any protein family?
Yes, to the GFP family. These proteins are able to fluoresce different colours, some of which are non-fluorescent. These proteins are all essentially encoded by single genes, since both the substrate and the catalytic enzyme for pigment biosynthesis are provided within a single polypeptide chain. (InterPro, Pfam = PF01353).
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
1996-11-08 Initial release
Resolution: 1.90 Å, which is considered high resolution (very high accuracy).
Are there any other molecules in the solved structure apart from protein?
Yes, a peptide derived chromophore and a Selenomethionine ligand
Does your protein belong to any structure classification family?
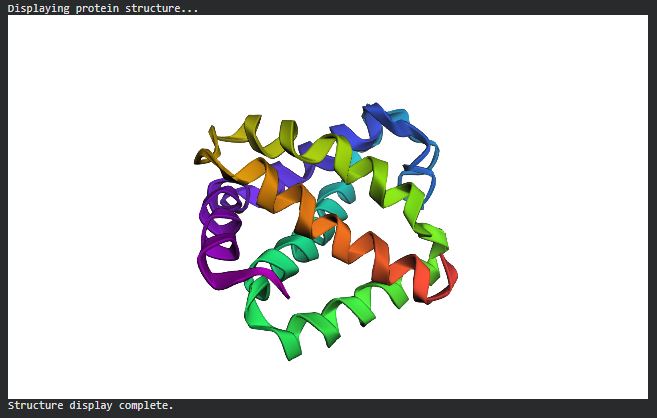
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”
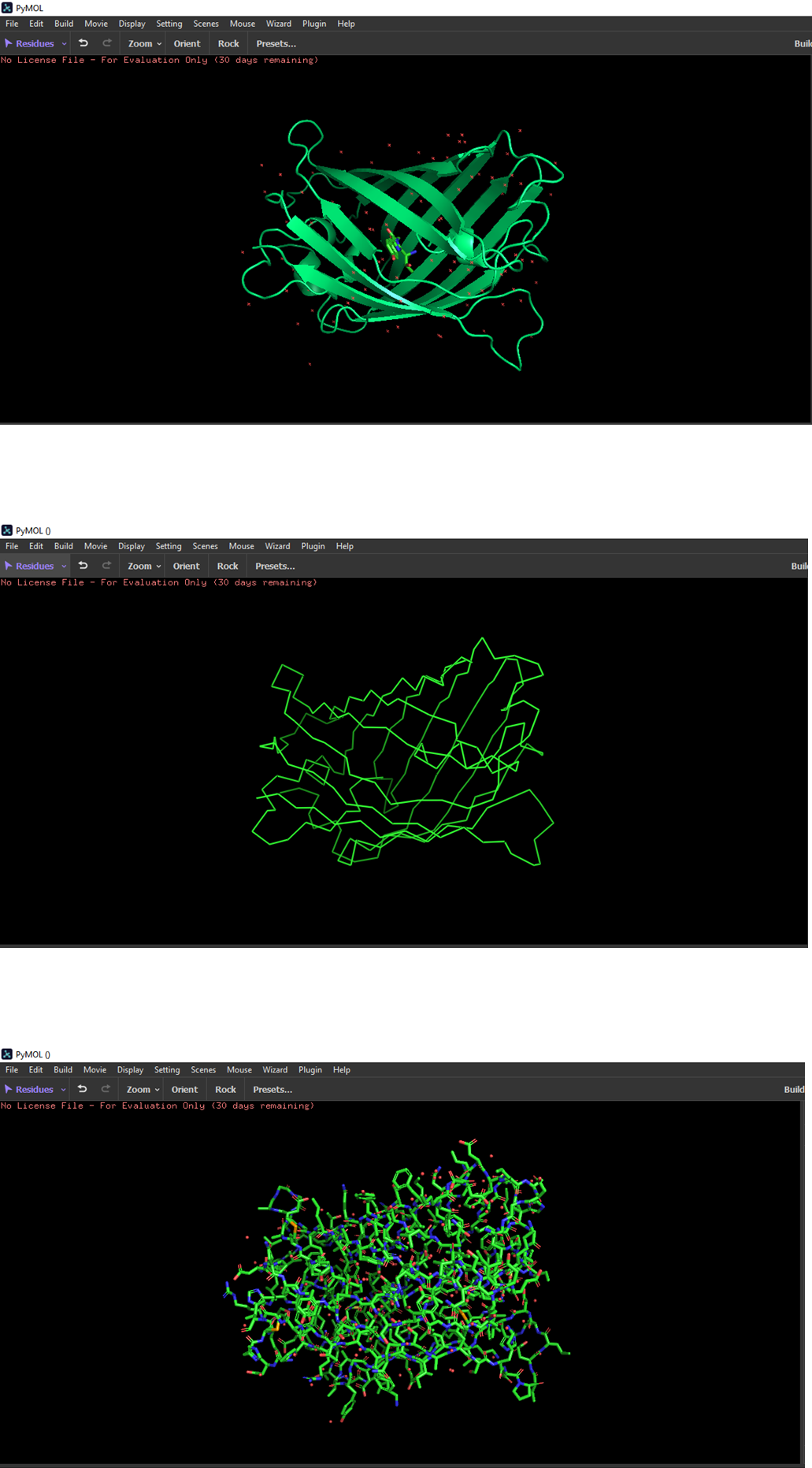
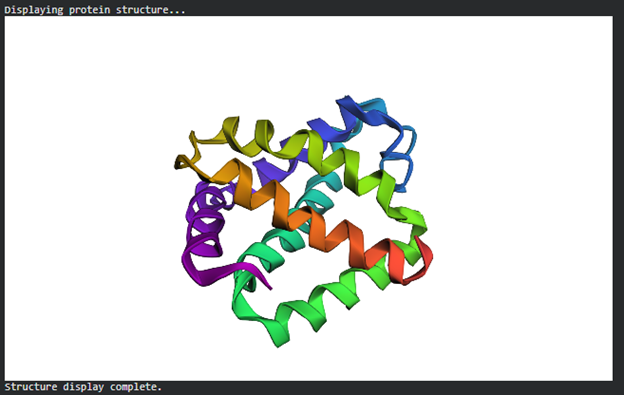
Color the protein by secondary structure. Does it have more helices or sheets?
The structure has more sheets.
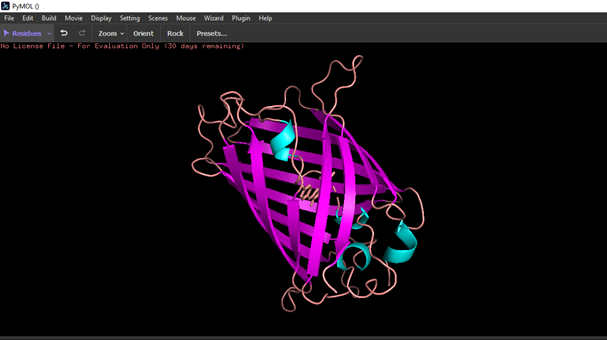
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophobic residues are mostly located in the core of the GFP structure, while hydrophilic residues are exposed on the surface, interacting with the aqueous environment.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
I couldn’t identify any.
Part C: Using ML-Based Protein Protein Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Deep Mutational Scans
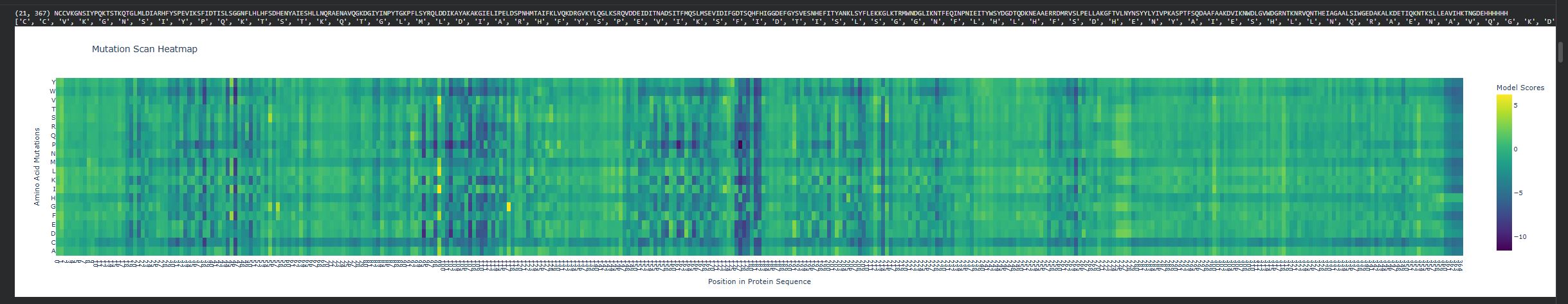
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
There are vertical columns that are blue or purple, which reveals positions that are not tolerant to mutations, meaning these residues are important structurally or functionally. The enzyme disperin B needs to have a stable structure to function correctly. In general there is a lot of green, which means that several positions do tolerate mutations.
Latent Space Analysis
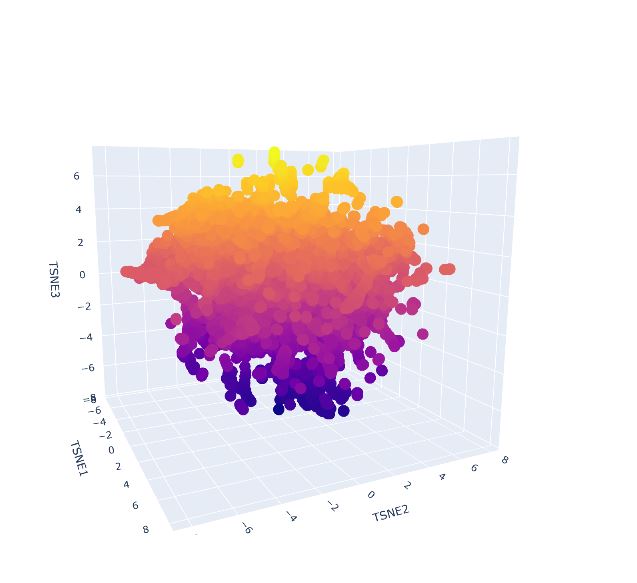
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors
It was not possible to locate my protein, quite likely it is immersed within the protein clusters, which means it shares features with similar proteins. As it was not found on the external area or isolated, this supports its classification as it is not considered an outlier.
C2. Protein Folding
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
C3. Protein Generation
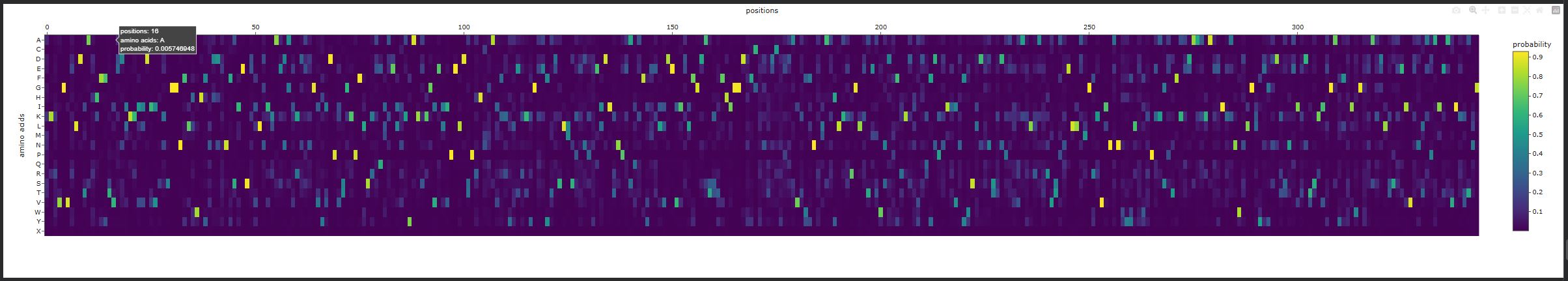
Inverse-Folding a protein:Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
New Sequence:
KKKTGVWLDTAHHFFSVENIKKIIDIVADAGGNFLLWHFNDDENFSIESEVLKILAEDGIKDENGVYIDPETGKPFLTREQIAELNAYAKAKGVEIIPELDSPRHMGAIFRYVERAKGKAYRDSLLSKEDPNELDITNPESQAFYKELLDEVIDLFGDATSTFHIGGDDFCWNTDCYDDAVKWFNDIAAYLKEKGLKTMMWNDPFRKDNYAQFDKDIEVLFWSLDDNTTDPKVAAELRATRVTAAELLAAGRTVINANWYYLYWTPKAGPTFKADAAAHAEDVKANWNLGLYEGSDTSNAITDTSKIAGALFWIDGWNAANLSDKEIVDATAPAIEAVIKKTNG
Input this sequence into ESMFold and compare the predicted structure to your original.