Week 4 — Protein Design Part I
✨ Part A. Conceptual Questions ✨
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Using an online converter ( https://www.unitconverters.net/weight-and-mass/gram-to-dalton.htm ), I calculated that 100 Daltons (1 amino acid) corresponds to approximately 1.66 × 10⁻²² g. After dividing the mass of a 500 g piece of meat by this value, I found the total number of amino acid molecules:
1.66 × 10⁻²² g/molecule
≈ 3.01 × 10²⁴ molecules
This means there are ~ 3.01 × 10²⁴ amino acid molecules in 500 grams of meat.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans eat beef or fish, but we do not become cows or fish because each species has its own unique genome. Eating proteins from another species does not change our DNA; our body simply digests the proteins into amino acids and uses them to build its own proteins. We only take the building blocks, not the instructions for making another species.
3. Why are there only 20 natural amino acids?
There are only 20 natural amino acids because the genetic code in DNA and mRNA is built to encode only these 20. Although there are 64 codons, many codons code for the same amino acid (redundancy in the genetic code). Scientists are experimenting with creating non-natural amino acids to expand the range of possible proteins, but in nature, only 20 are used.
4. Can you make other non-natural amino acids? Design some new amino acids.
I’m not completely sure how it works, but I remember from George Church’s slides that scientists can create new non-natural nucleobases. I guess that by using these artificial bases in the genetic code, it might be possible to produce new non-natural amino acids, although I don’t know the exact method.
5. Where did amino acids come from before enzymes that make them, and before life started?
Based on the information I found in this article https://doi.org/10.1002/chem.202201419 , amino acids existed before life and before enzymes, formed through non-enzymatic chemistry. Enzymes appeared later as proteins that accelerated chemical reactions, including the synthesis of other proteins. Experimental evidence for prebiotic amino acid formation comes from the Miller–Urey experiment, from 1953, in which gases such as CH₄, NH₃, H₂O, and H₂ were exposed to electrical energy, producing amino acids. ( https://www.britannica.com/science/Miller-Urey-experiment )
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
D-amino acids (D = dextro, right) are enantiomers, meaning they are mirror versions of L-amino acids (the natural amino acids in proteins, L = levo, left). If you make an α-helix using D-amino acids, the helix will be left-handed. Even though L-amino acids are “left” in configuration, when they form a helix they twist to the right because this is the most stable arrangement for hydrogen bonds and steric interactions. D-amino acids are mirror images, so their α-helix twists in the opposite direction.
7. Can you discover additional helices in proteins?
I’m not sure, but I guess it might be possible to discover additional or unusual helices in proteins that we don’t know yet. Methods like X-ray crystallography or NMR might reveal new structures, but I don’t know the details.
8. Why most molecular helices are right-handed?
Most molecular helices are right-handed because the natural amino acids in proteins are L-amino acids (left-handed in configuration). When L-amino acids fold into a helix, the right-handed α-helix is the most stable arrangement due to optimal hydrogen bonding and minimal steric strain.
9. Why do β-sheets tend to aggregate? -What is the driving force for β-sheet aggregation?
10. Why do many amyloid diseases form β-sheets? -Can you use amyloid β-sheets as materials? Many amyloid diseases form β-sheets because misfolded proteins adopt β-sheet–rich structures that can form extensive hydrogen bonds between different molecules. This leads to stable aggregates called amyloid fibrils, which accumulate in tissues and cause disease. Yes, amyloid β-sheets can be used as materials because they form highly stable and mechanically strong fibrils. Scientists are studying them as biomaterials for nanotechnology and medical applications.
11. Design a β-sheet motif that forms a well-ordered structure. I am not completely sure how to design a specific β-sheet motif, but I would use amino acids that favor β-sheet formation, like valine and isoleucine. These amino acids are hydrophobic and have side chains that fit well in the extended β-strand structure, which helps the sheet stay stable. I would also alternate hydrophobic and polar residues, because in β-strands the side chains point up and down, so this pattern allows the sheet to interact with water on one side and form a stable hydrophobic core on the other. Together with hydrogen bonds between the strands, this could make a well-ordered and stable β-sheet.
✨ Part B. Protein Analysis and Visualization ✨
1. Briefly describe the protein you selected and why you selected it. I selected the Cry1A.105 protein from Bacillus thuringiensis. Cry1A.105 is a chimeric δ-endotoxin used in genetically modified Bt crops for insect pest control. The structure available corresponds to its tryptic core, which represents the active form of the toxin.
I chose this protein because it has a well-resolved 3D crystal structure and a clearly defined three-domain organization, making it ideal for structural analysis. Additionally, it is biologically and biotechnologically relevant, as it contributes to sustainable agriculture by reducing the need for chemical insecticides.
2. Identify the amino acid sequence of your protein. From the RCSB Protein Data Bank ( https://www.rcsb.org/structure/6DJ4 ), I downloaded the FASTA sequence:
>>6DJ4_1|Chain A|Cry1A.105|Bacillus thuringiensis IETGYTPIDISLSLTQFLLSEFVPGAGFVLGLVDIIWGIFGPSQWDAFLVQIEQLINQRIEEFARNQAISRLEGLSNLYQIYAESFREWEADPTNPALREEMRIQFNDMNSALTTAIPLFAVQNYQVPLLSVYVQAANLHLSVLRDVSVFGQRWGFDAATINSRYNDLTRLIGNYTDHAVRWYNTGLERVWGPDSRDWIRYNQFRRELTLTVLDIVSLFPNYDSRTYPIRTVSQLTREIYTNPVLENFDGSFRGSAQGIEGSIRSPHLMDILNSITIYTDAHRGEYYWSGHQIMASPVGFSGPEFTFPLYGTMGNAAPQQRIVAQLGQGVYRTLSSTLYRRPFNIGINNQQLSVLDGTEFAYGTSSNLPSAVYRKSGTVDSLDEIPPQNNNVPPRQGFSHRLSHVSMFRSGFSNSSVSIIRAPMFSWIHRSAEFNNIIASDSITQIPLVKAHTLQSGTTVVRGPGFTGGDILRRTSGGPFAYTIVNINGQLPQRYRARIRYASTTNLRIYVTVAGERIFAGQFNKTMDTGDPLTFQSFSYATINTAFTFPMSQSSFTVGADTFSSGNEVYIDRFELIPVTATLEAEYNLER
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. The Cry1A.105 protein consists of 591 amino acids, with serine (S) being the most frequent, appearing 54 times in the sequence.
.png)
3. Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å) The Cry1A.105 structure was initially deposited on 24 May 2018 and released on 12 September 2018. It was solved using X-ray diffraction and has a resolution of 3.01 Å, which indicates a good quality structure suitable for detailed structural analysis.
- Are there any other molecules in the solved structure apart from protein? Yes, apart from the protein polymer, the solved structure contains water molecules and crystallographic information, including the unit cell (C 1 2 1). No other ligands or cofactors are present.
- Does your protein belong to any structure classification family? Yes, Cry1A.105 belongs to the delta-endotoxin family, C-terminal domain, according to SCOP classification. The classification is based on chain A, residues 477–609, from Bacillus thuringiensis serovar aizawai.
4. Open the structure of your protein in any 3D molecule visualization software:
.png)
.png)
When the protein is colored by secondary structure, it shows both α-helices and β-sheets. In Cry1A.105, I would say that β-sheets are more abundant than α-helices, forming the main structural framework of the protein.
.png)
.png)
I visualized the solvent-accessible surface of Cry1A.105 and observed several cavities. I highlighted one region that seems to be a pocket, but I am not very confident in identifying binding pockets, so I am not sure if this is correct.
.png)
✨ Part C. Using ML-Based Protein Design Tools ✨
C1. Protein Language Modeling
- Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
I used ESM2 to generate a deep mutational scan for Cry1A.105 (PDB ID: 6DJ4). The results are shown as a heatmap, where each position in the sequence is tested with different possible mutations.
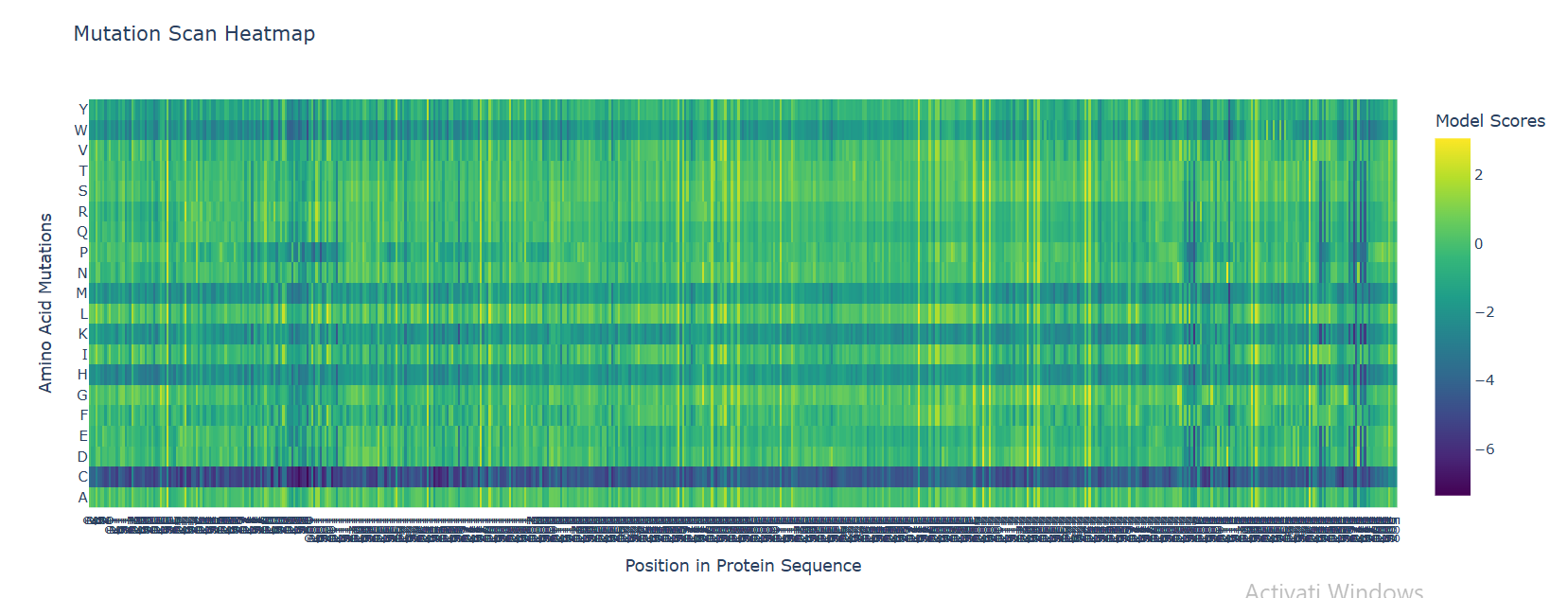
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
In the heatmap, yellow represents beneficial or tolerated mutations, while dark blue represents unfavorable mutations. I noticed that some positions are mostly dark blue, which suggests they are important for the protein structure and do not tolerate changes well.
For the positive mutation, at position 373, changing the residue to L (Leucine) gives a high score (+2.80). This basically means the model thinks leucine fits well there. The position is probably flexible or not very important structurally, so swapping in a hydrophobic residue like leucine doesn’t cause problems. In other words, the protein seems totally fine with this mutation.
c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I searched for experimental mutational scans (such as Deep Mutational Scanning datasets) for Cry1A.105, but no DMS data were available for this protein. Therefore, a comparison between language model predictions and experimental results (e.g., using a heatmap) could not be performed.
- Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
- I used the provided protein sequence dataset and generated embeddings with a pretrained model (ESM2). These embeddings are numerical representations of each protein.
- Then, I reduced their dimensionality using t-SNE and visualized them in a 3D interactive plot. Each point represents one protein, and similar proteins tend to cluster together.
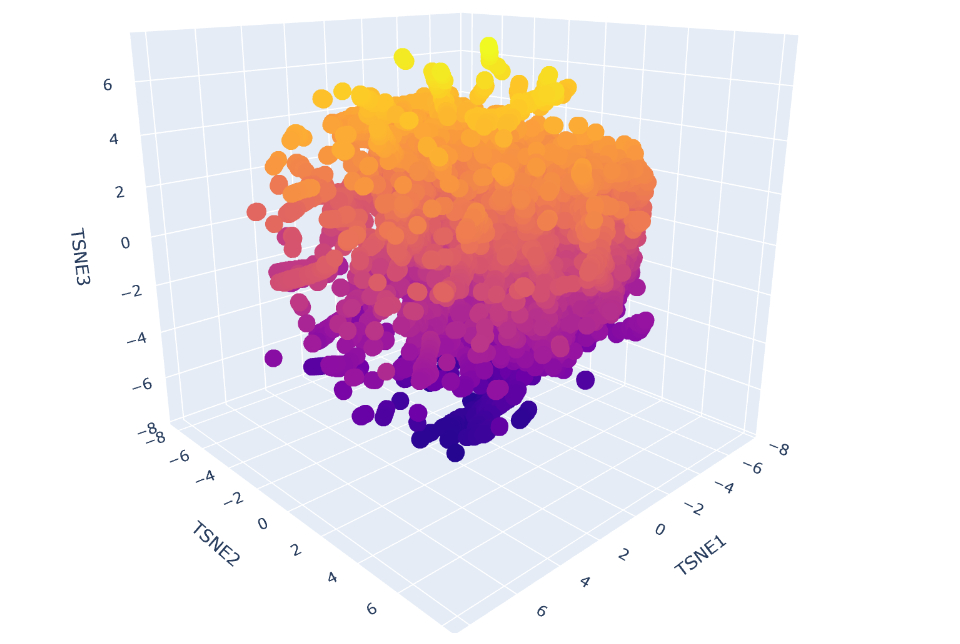
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
In the t‑SNE embedding, proteins that appear close to each other form neighborhoods that reflect real structural similarity. For example, one of the clusters I inspected contained proteins from very different species (PDB IDs 3UKN, 4D7S, and 5J3U), but all of them share the same type of domain: a cyclic‑nucleotide binding domain (CNBD/CNBHD). Even though they come from zebrafish, a thermophilic bacterium, and Toxoplasma gondii, their 3D structures belong to the same SCOPe fold family (b.82.x.x). This explains why the model places them close together.
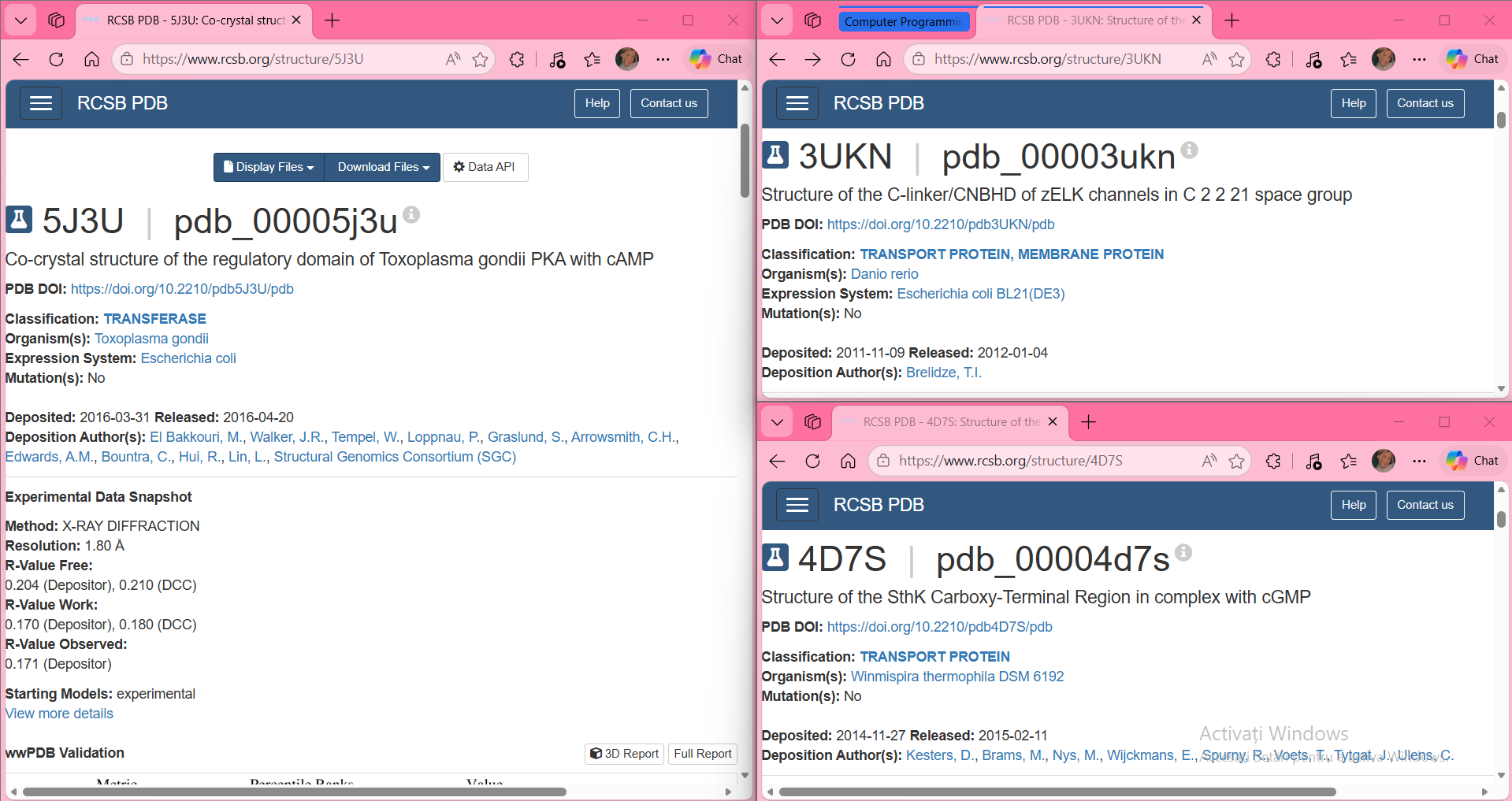
Conclusion: the neighborhoods approximate similar proteins — the embedding groups proteins by structural fold, not by species.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
How I highlighted my protein in the t‑SNE map
To clearly identify my protein in the t‑SNE embedding, I added a small piece of code that colors all SCOPe proteins in blue and my protein in green. I also increased the marker size for my protein so it stands out from the rest of the points in the map. This allowed me to easily locate it in the 3D projection and inspect its closest neighbors. By highlighting it in green, I could immediately see which proteins cluster around it and analyze their structural similarity to mine.
In the t-SNE map, 6DJ4 is located close to the following neighbors:
- 2BVC – glutamine synthetase
- 5Z37 – Abrin A chain
- 3MVG – IRIP (ribosome-inactivating protein)
- 2BU9 – Isopenicillin N synthase
Although these proteins have different biological roles (metabolic enzyme, toxin, oxidase), they share important structural similarities. All are relatively large, soluble, globular proteins with a mixed α/β architecture and compact catalytic-like domains. The fact that 6DJ4 clusters with these neighbors suggests that the embedding captures similarities in overall fold, secondary structure composition, and global 3D organization rather than strict functional similarity.
C2. Protein Folding
Folding a protein
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I folded my protein using ESMFold in Google Colab and obtained a predicted 3D structure. In the comparison image, the predicted model is shown on the left and the original PDB structure on the right.
The two structures look quite similar overall. The general shape of the protein and the arrangement of the main secondary structure elements (α-helices and β-sheets) are preserved. This means that ESMFold was able to correctly predict the overall fold of the protein.
There are small differences in some regions, probably in flexible loops, which is normal for structure prediction.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To test whether my protein structure is resilient to mutations, I introduced a single conservative point mutation in the middle region of the sequence (approximately positions 250–300), since central regions are generally more structurally stable than terminal ends.
Specifically, I replaced an isoleucine (I) with valine (V), changing the sequence fragment from:
MDILNSITIYTDA to: MDVLNSITIYTDA
The mutated structure (Figure A) appears very similar to both the original ESMFold prediction (Figure B) and the experimental PDB structure (Figure C). The global fold and overall 3D organization are preserved.
This suggests that the protein is resilient to small, conservative point mutations, as a single amino acid substitution did not significantly alter the overall structure.
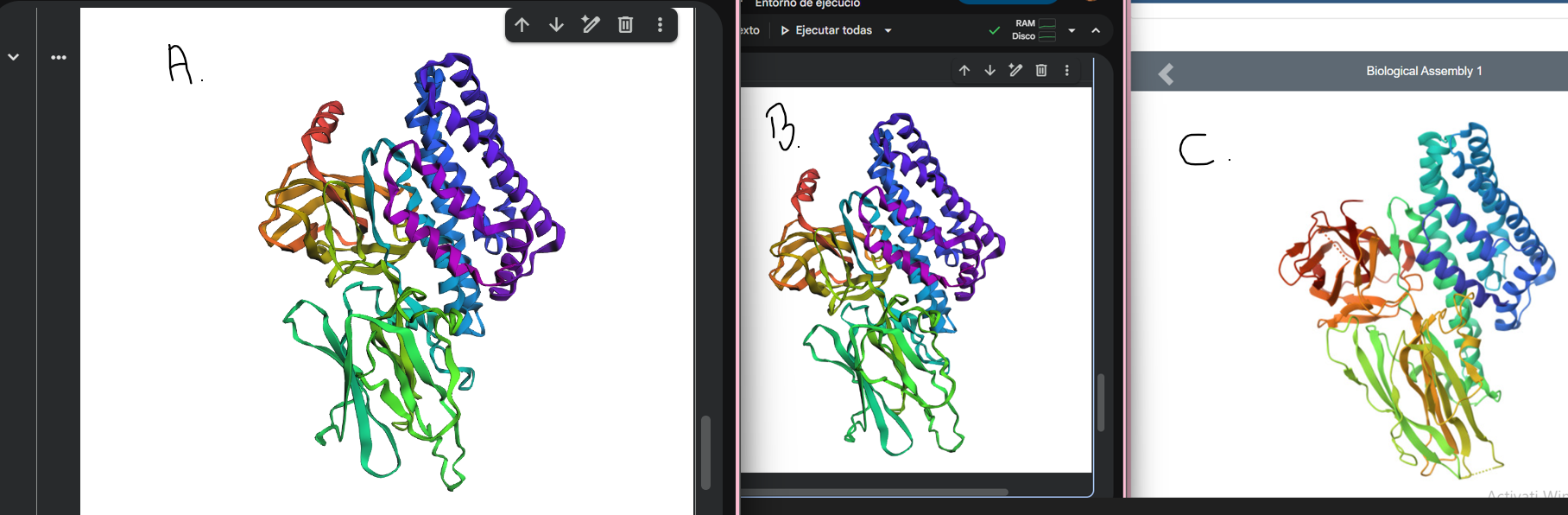
The larger segment mutation (MDILNSI → AAAAAAA) in Figure A causes a clear structural change compared to the original predicted (Figure B) and experimental PDB (Figure C) structures, showing that the protein fold is disrupted by extensive mutations.
C3. Protein Generation
Inverse-Folding a protein
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
I performed inverse folding using the backbone coordinates from the PDB structure (6DJ4) and obtained a new sequence predicted to be compatible with the 3D structure. The predicted sequence is:
>ASPETVALRLTRFLIEDNELGAKFAKGLVDIVWGAEGPALWREWIAAVEDLIGRAVPEAVRDAAIAAAEELNKLFKEFVATVEAWEADPTDPALRKAMIEAFERLIAALEEALPLYRPPGWAIPLLPLYVLVSILYLYVLSYVSKFGKKWGFSEEKIEEYKKKLKESIISFTEFVLKHYHEGLAAVRGPTEADFVRYLRYERVMTITCLDLVALFEKFDTDLFPIKVRFQLTRVLYFNPVLEARDEDLPGSAEAIRAALPAPALVRNLVSTTFYLGSVNGKTFISGLVNTSRPAGGPLPETTDPLRGVAETALPTGFTVTSRGDGIVNLKASVVYENPEPXXXKKLTLRTLDGITFEYGSTSSMAPDVVLKSGDVDTRDVFPPARTDVPYTAGFSFKLSDISMYYEGDLTGSDRIVRSPIFGFRHRSATDNNDVYPDQITIIPLTRATRLYPGVTVVKGPGFLGGDLLKITSPGNLARLSLRLKXXXGVTYQFRVRYSANADFTVWVTVDGTRTLSTNCSKTFNAGEPLTPKSFKYCTIPESFTFEKPTFTLDVGASNFPSGNTFYVDYVELVPTSL
2. Input this sequence into ESMFold and compare the predicted structure to your original.
Next, I input the predicted sequence from ProteinMPNN into ESMFold, following the same procedure as before. The resulting 3D structure is very similar to the original PDB structure, with the overall α/β fold and domain organization preserved.
This indicates that the inverse folding approach was successful: the predicted sequence is almost compatible with the original fold and can reproduce the protein’s global 3D structure.
✨ Part A. Conceptual Questions ✨
Engineering the MS2 L Protein: Disrupting Interaction with E. coli DnaJ
Goal
- Disrupt the interaction between MS2 L protein and E. coli DnaJ by truncating the N-terminal domain (aa 1–35) while keeping the transmembrane domain intact.
Tools
- AlphaFold Server – structure prediction for WT and truncated L protein, complex prediction with DnaJ.
- ColabFold (AlphaFold-Multimer) – interactive 3D structure prediction and visualization of WT and truncated complexes.
Pipeline
WT-L-protein
Truncated-L-protein
WT-L-protein + DnaJ
Truncated-L-protein + DnaJ
responsible for DnaJ interaction
Tools: AlphaFold Server
Tools: AlphaFold-Multimer / ColabFold
- WT: N-terminal tail contacts DnaJ
- Truncated: helix alone, no contact
Results
| Variant | Observation | pTM | ipTM |
|---|---|---|---|
| WT L-protein + DnaJ | N-terminal tail interacts with DnaJ | 0.54 | 0.12 |
| Truncated L-protein (1–35) + DnaJ | Helix alone, no contact | 0.59 | 0.11 |
- WT structure shows the N-terminal tail of L protein near DnaJ, confirming potential interaction.
- Truncated variant loses contact with DnaJ, while the transmembrane helix remains folded.
- ColabFold visualizations provide interactive 3D structures to illustrate the difference.
Potential Pitfalls
- AlphaFold predictions may not fully capture membrane environment effects.
- Low ipTM scores indicate uncertain interface prediction, experimental validation would be needed.
- Flexible regions in DnaJ and the N-terminal domain may differ in reality from predicted models.
Conclusion
- Truncating the N-terminal domain of MS2 L protein disrupts interaction with DnaJ without destabilizing the transmembrane domain.
- This approach supports the goal of modifying L protein for DnaJ-independent activity.