Week 5 — Protein Design Part II
✨ Part A. SOD1 Binder Peptide Design ✨
Part 1: Generate Binders with PepMLM
I searched for the SOD1 amino acid sequence in the UniProt database (P00441) and found that the protein has 154 amino acids:
>MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
After that, I inserted the sequence into the Colab notebook. I set the parameters to generate 4 peptide binders, each with a length of 12 amino acids. The model then generated the following peptides:
| Binder | Pseudo Perplexity |
|---|---|
| WHSYAAGVAWKX | 12.096901 |
| HHYPAVVVAHKE | 12.724163 |
| WHVGVVVVRHKX | 18.296936 |
| WLYGATVVRLKE | 20.406855 |
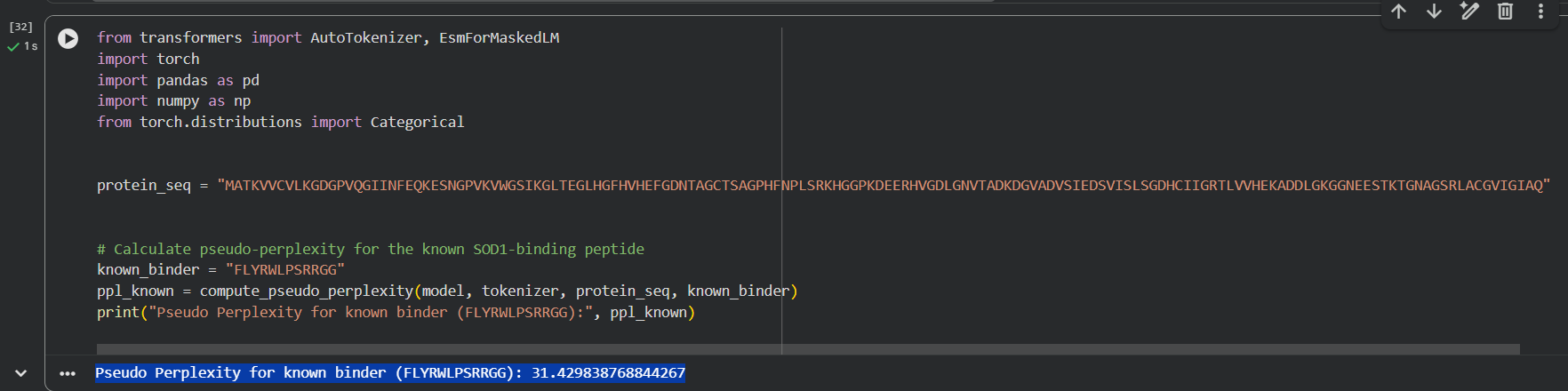
I added a new block in the Colab notebook where I used a code to calculate the pseudo-perplexity for the known SOD1-binding peptide. The sequence FLYRWLPSRRGG was used as input, and the model returned a pseudo-perplexity of 31.43.
I recorded the pseudo-perplexity scores for all the generated peptide binders, as well as for the known SOD1-binding peptide FLYRWLPSRRGG. These scores indicate PepMLM’s confidence in each peptide: a lower pseudo-perplexity means the model predicts the peptide is more likely to bind the target protein, while a higher value indicates less confidence.
Part 2: Evaluate Binders with AlphaFold3
I first input the SOD1 sequence together with the peptide binder into the model. Initially, the prediction failed because SOD1 is a homodimer, so I corrected the input by including two chains with the same sequence of 154 amino acids, followed by the peptide binder. Two of the previously generated binders could not be used because they contained an “X”, which represents an unknown amino acid.
I first tested the known SOD1-binding peptide FLYRWLPSRRGG (pseudo-perplexity pp = 31). The predicted complex produced an ipTM score of 0.88, which indicates a reasonably confident interaction. In the structure visualization, the two SOD1 chains appear in blue, while the binder appears in orange, corresponding to very low confidence (pLDDT < 50). This may suggest that the position of the peptide is not very stable in the predicted structure. The peptide does not localize near the N-terminus where the A4V mutation is located. Instead, it appears close to residues around 90–100, near several β-sheets, indicating that it engages the β-barrel region of the protein.
Next, I tested two of the generated binders. For HHYPAVVVAHKE (pp = 12.72), the predicted complex had an ipTM score of 0.87, and the visualization showed a similar pattern, with the peptide again appearing orange, indicating low confidence. For WLYGATVVRLKE (pp = 20.40), the ipTM score was 0.82, with the same color distribution observed.
In all cases, the peptide does not appear tightly attached to the protein surface. Instead, it seems to float near the chain, suggesting that the peptide is surface-bound rather than deeply buried within the structure.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
After evaluating both HHYPAVVVAHKE and WLYGATVVRLKE in PeptiVerse, we observed the following:
- HHYPAVVVAHKE shows high solubility (probability = 1.0) and is non-hemolytic (probability = 0.02), suggesting it is safe and easy to formulate. Its predicted binding affinity is classified as weak (pKd/pKi = 4.968), but in AlphaFold it had a relatively high ipTM score (0.87), indicating a reasonably confident structural interaction with the A4V mutant SOD1. The peptide has a neutral net charge (0.03 at pH 7), a molecular weight of 1386.6 Da, an isoelectric point of 7.03, and slight negative hydrophobicity (GRAVY = -0.31), all suggesting favorable biophysical properties.
- WLYGATVVRLKE also shows high solubility (probability = 1.0) and is non-hemolytic (probability = 0.074). Its predicted binding affinity is weaker (pKd/pKi = 6.159), and the ipTM from AlphaFold is lower (0.82), indicating a slightly less confident structural interaction. The peptide has a net charge of 0.77 at pH 7, molecular weight of 1434.7 Da, an isoelectric point of 8.59, and slight positive hydrophobicity (GRAVY = 0.22).
| Property | HHYPAVVVAHKE | WLYGATVVRLKE |
|---|---|---|
| ipTM | 0.87 | 0.82 |
| Solubility 💧 | 1.000 | 1.000 |
| Hemolysis 🩸 | 0.020 | 0.074 |
| Binding Affinity 🔗 | 4.971 | 6.163 |
| Length 📏 | 12 | 12 |
| Molecular Weight ⚖️ | 1386.6 | 1434.7 |
| Net Charge ⚡ | 0.03 | 0.77 |
| Isoelectric Point 🎯 | 7.03 | 8.59 |
| Hydrophobicity 💦 | -0.31 | 0.22 |
Based on the data and analysis, HHYPAVVVAHKE is the better peptide. The reasons are:
- Stronger predicted binding: Its pKd/pKi = 4.971, which is lower than WLYGATVVRLKE (6.163), indicating stronger predicted affinity for the A4V mutant SOD1.
- Higher structural confidence: ipTM = 0.87 vs 0.82, meaning AlphaFold predicts a more stable interaction.
- Favorable therapeutic properties: Both peptides are soluble and non-hemolytic, but HHYPAVVVAHKE has a neutral net charge and slightly more hydrophilic character, which is advantageous for formulation and bioavailability.
- Better overall balance: Combines reasonably strong binding with safe and favorable physicochemical properties, making it the most promising candidate to advance.
Part 4: Generate Optimized Peptides with moPPIt
In the peptide generation tool, I first pasted the A4V mutant SOD1 sequence. Then I set the peptide length to 12 amino acids. After that, I enabled the options “Enable motif and affinity guidance” (as well as solubility/hemolysis guidance if available). After running the tool, three peptide motifs were generated: RKMICGRYRYYI, SCFLYYYYTIIL, and SARRQKCVRYYT.
Then, I introduced the generated peptides into PeptiVerse, as in the previous step, in order to compare the peptides generated by PepMLM and moPPit and evaluate their physicochemical properties.
| Peptide | Solubility | Hemolysis | Binding Affinity (pKd/pKi) | Net Charge | GRAVY |
|---|---|---|---|---|---|
| RKMICGRYRYYI | Soluble | Non-hemolytic (0.085) | 8.512 | 3.75 | -0.69 |
| SCFLYYYYTIIL | Soluble | Non-hemolytic (0.205) | 9.137 | -0.55 | 1.27 |
| SARRQKCVRYYT | Soluble | Non-hemolytic (0.050) | 7.301 | 3.45 | -1.38 |
All three moPPIt peptides were predicted to be soluble and non-hemolytic, which indicates a favorable safety profile. Among them, SCFLYYYYTIIL shows the highest predicted binding affinity (9.137 pKd/pKi), suggesting stronger interaction with potential targets, although it is also the most hydrophobic. In contrast, SARRQKCVRYYT has the lowest hemolysis probability and the highest hydrophilicity, indicating potentially better biological compatibility.
Compared with the best peptide generated by PepMLM, the top moPPit peptide shows improved predicted properties such as stronger binding affinity and favorable solubility, suggesting that motif-guided design may produce peptides with more optimized functional characteristics.
✨ Part B: BRD4 Drug Discovery Platform Tutorial ✨
Text
✨ Part C: Final Project: L-Protein Mutants ✨
For this part of the assignment, I chose Option 3: Random Mutagenesis. I generated three mutations in Colab using a Python script (code provided below). I prompted ChatGPT with the original Colab code and the instructions from the course, and this is what I obtained.
>import random
def generate_random_mutations(sequence, possible_positions, n_mutations=2):
amino_acids = "ACDEFGHIKLMNPQRSTVWY"
# Select n_mutations random positions from the list of possible positions
positions_to_mutate = random.sample(possible_positions, n_mutations)
mutated_sequence = list(sequence)
mutations = []
for pos in positions_to_mutate:
# Choose a new amino acid different from the original one
new_aa = random.choice([aa for aa in amino_acids if aa != sequence[pos]])
mutated_sequence[pos] = new_aa
# Store mutation info as (position, original AA, new AA), using 1-based indexing
mutations.append((pos+1, sequence[pos], new_aa))
return "".join(mutated_sequence), mutations
# Example usage
sequence = "METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT"
possible_positions = [9, 25, 30, 39, 50, 53] # safe positions to mutate
mut_seq, mut_info = generate_random_mutations(sequence, possible_positions, n_mutations=3)
print(mut_seq)
print(mut_info)The three mutations generated are as follows:
- Mutation 1: Y → V at position 40
- Mutation 2: F → T at position 51
- Mutation 3: Q → M at position 54
After applying these mutations, the resulting protein sequence is:
>METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLY**V**LIFLAIFLSK**T**TN**M**LLLSLLEAVIRTVTTLQQLLT
WT-L-protein
Mutated-L-protein