Protein design part 1
Homework 4: Protein design part 1:
Part A:
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- 500g of meat => ~25% grams of protein => 125g of protein
- 125g of protein / 100 g per mole amino acids = 1.25 moles of AAs
- Nr of amino acids: 6.022 * 1023 * 1.25 = 7.5275 * 1023 amino acids in 500g of meat;
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- What does it even mean to become a cow??? To act like a cow? To look like a cow? Both? I guess it means that your genome is indistinguishable from the one of a cow. Everything that you ingest is broken down into the monomers of that thing (or very close), because otherwise your cells will struggle to obtain the nutrients that they need. The nutrients present in a cow are the same as the ones found in humans..so it’s not the molecules that make us what we are, but the way they are arranged and function in our organism. Cells do not invent new genes or proteins (only accidentally). They use the nutrients to follow the instructions already present in them. In conclusion, to become a cow, you would need to rewrite your genome, and we can’t do that JUST by eating cows.
Why are there only 20 natural amino acids? Why would there be more than 20 alpha-AAs? Life found a way to work with those 20 amino acids, adding a new amino acid means increasing complexity and causing a lot of new structural problems. “The simpler, the better” - it applies to any system, because it minimizes problems. There aren’t just 20 “standard amino acids” because it is physically impossible for life to exist in any other way, there are only 20 because it worked.
Can you make other non-natural amino acids? Design some new amino acids.
A side chain containing an azobenzene group, which changes shape (from straight to bent) when hit by specific wavelengths of light. (answer generated with gemini ai thinking mode)
Further research:
Where did amino acids come from before enzymes that make them, and before life started? - Probably they appeared through spontaneous processes that occurred under the conditions that existed at that time on Earth. Conditions that were pretty extreme.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? –>right-handed (L-amino acids are left-handed)
Can you discover additional helices in proteins? While the standard alpha helix is the most common, the sheer diversity of protein folding allows for variations that differ in their vertical rise and number of residues per rotation
Why are most molecular helices right-handed? Because to prevent overlapping, they form right-handed twists.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? beta-sheets tend to aggregate because they possess sticky exposed edges with unsatisfied hydrogen-bonding sites that seek to link with neighboring strands, creating a propitious environment for the formation of large, stable structures.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Design a β-sheet motif that forms a well-ordered structure.
Part B:
1.
Chosen protein: MAO B (Monoamine oxidase B)
- made of two identical subunits of 520 amino acids each; –> Further research:
- function: amine oxidation
- Because it creates ROS species, the overexpression of this protein damages surrounding tissue and is associated with Alzheimer’s and Parkinson’s;
2.
- Protein sequence: *
MSGDHLHNDS QIEADFRLND SHKHKDKHKD REHRHKEHKK EKDREKSKHS NSERDKERDK ERDKERDKER DREKDKDRER EREKEREKDS GKDRAKDKDR ERDRERDRER DREKDKEKDR DKDREKEKEK DREKDKEKDR DREKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR DKDKEKDRDK DKEKDRDKDK EKDRDKDKEK DRDKDKEKDR
- Leucine is the most frequent AA;
- 250 homologous proteins in other species;
- MAO-B is part of amine-oxidase family and is a mitochondrial membrane-bound protein;
3.
- Resolution: 3.00 Å => the resolution is not the best (<2 A)
- Released Date: 2001-11-29
- MAO-B is part of amine-oxidase family
4.
MAOB in pyMOL:
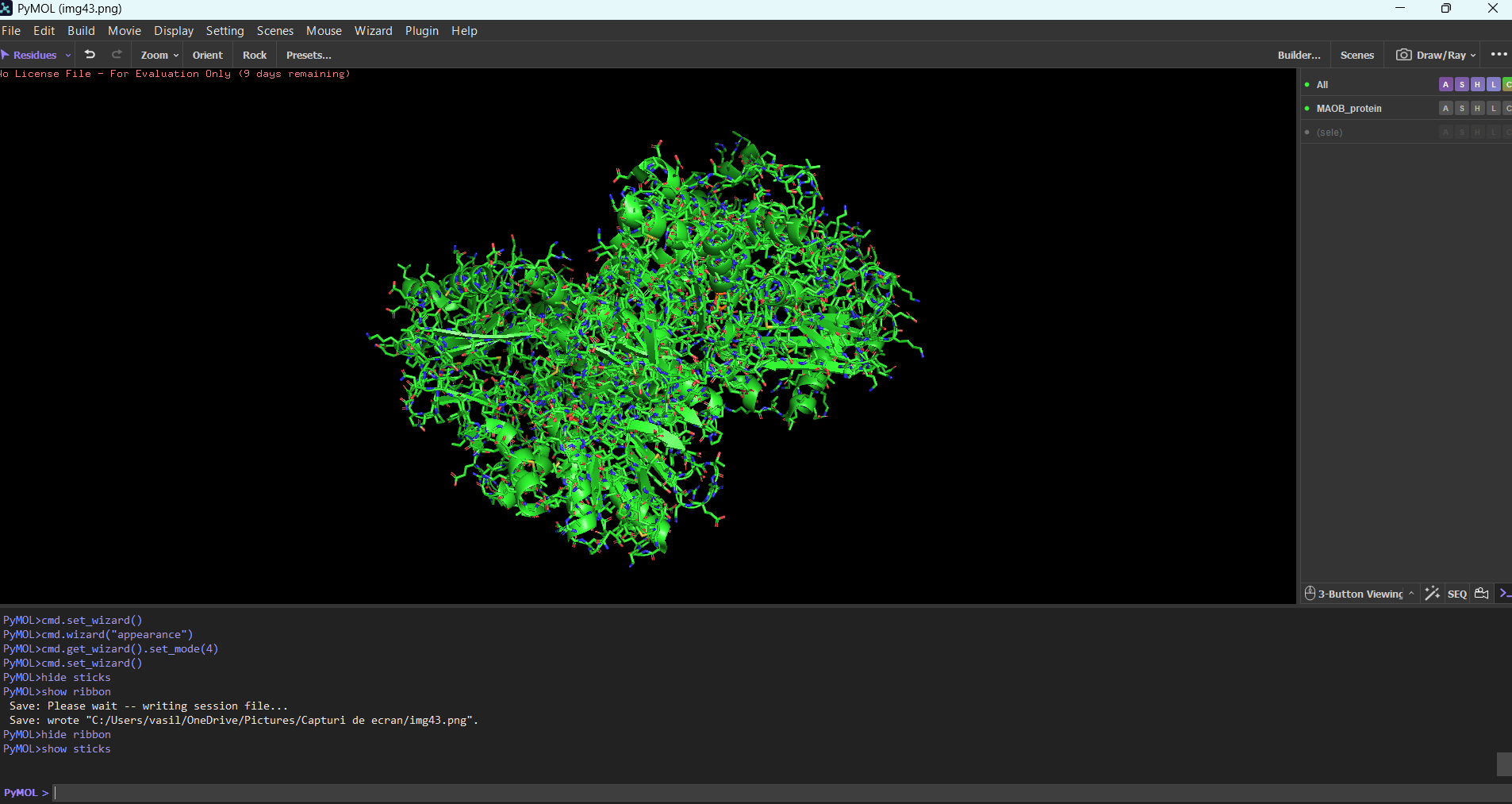
Ribbon mode:
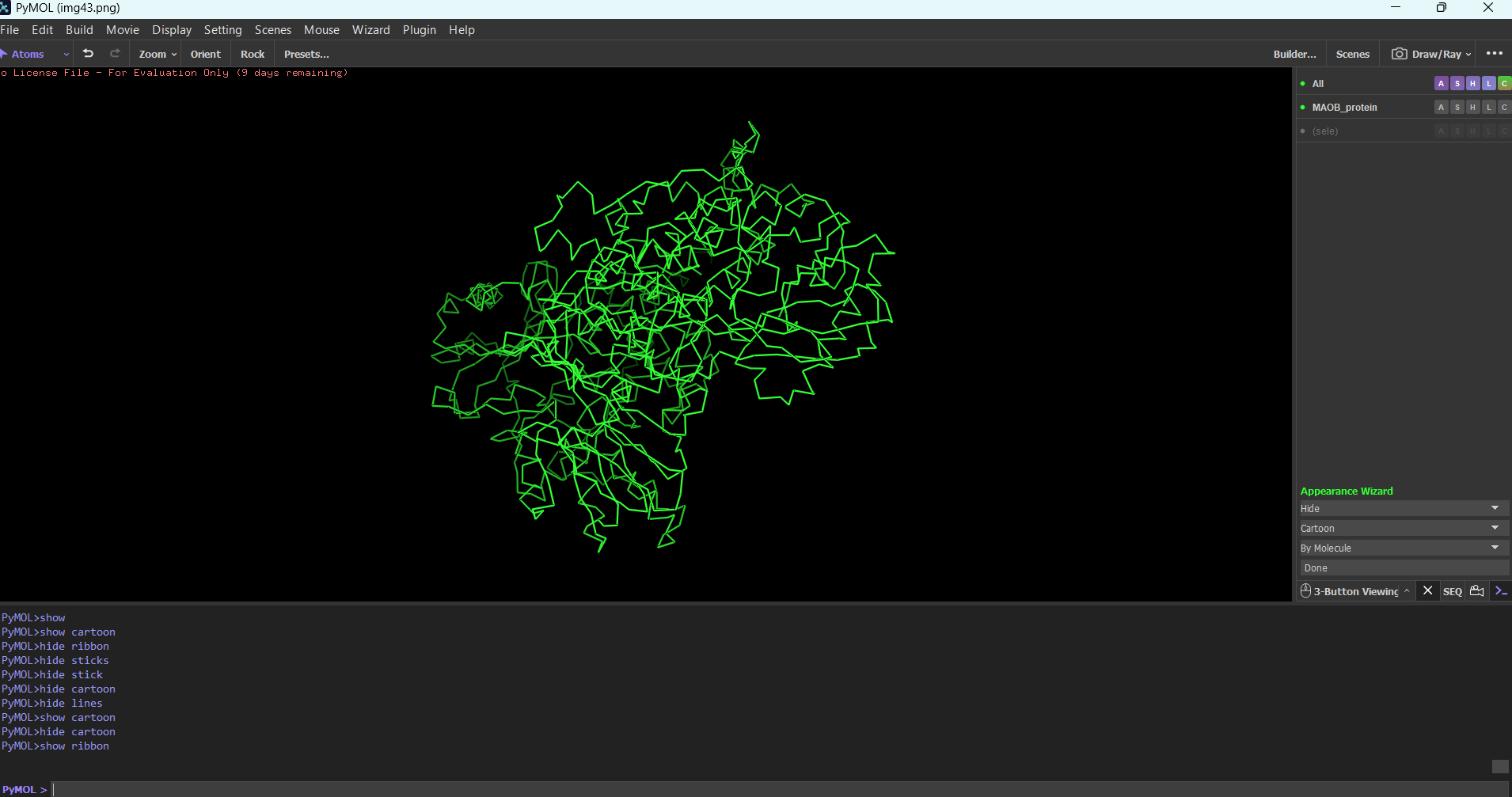
Sticks mdoe:
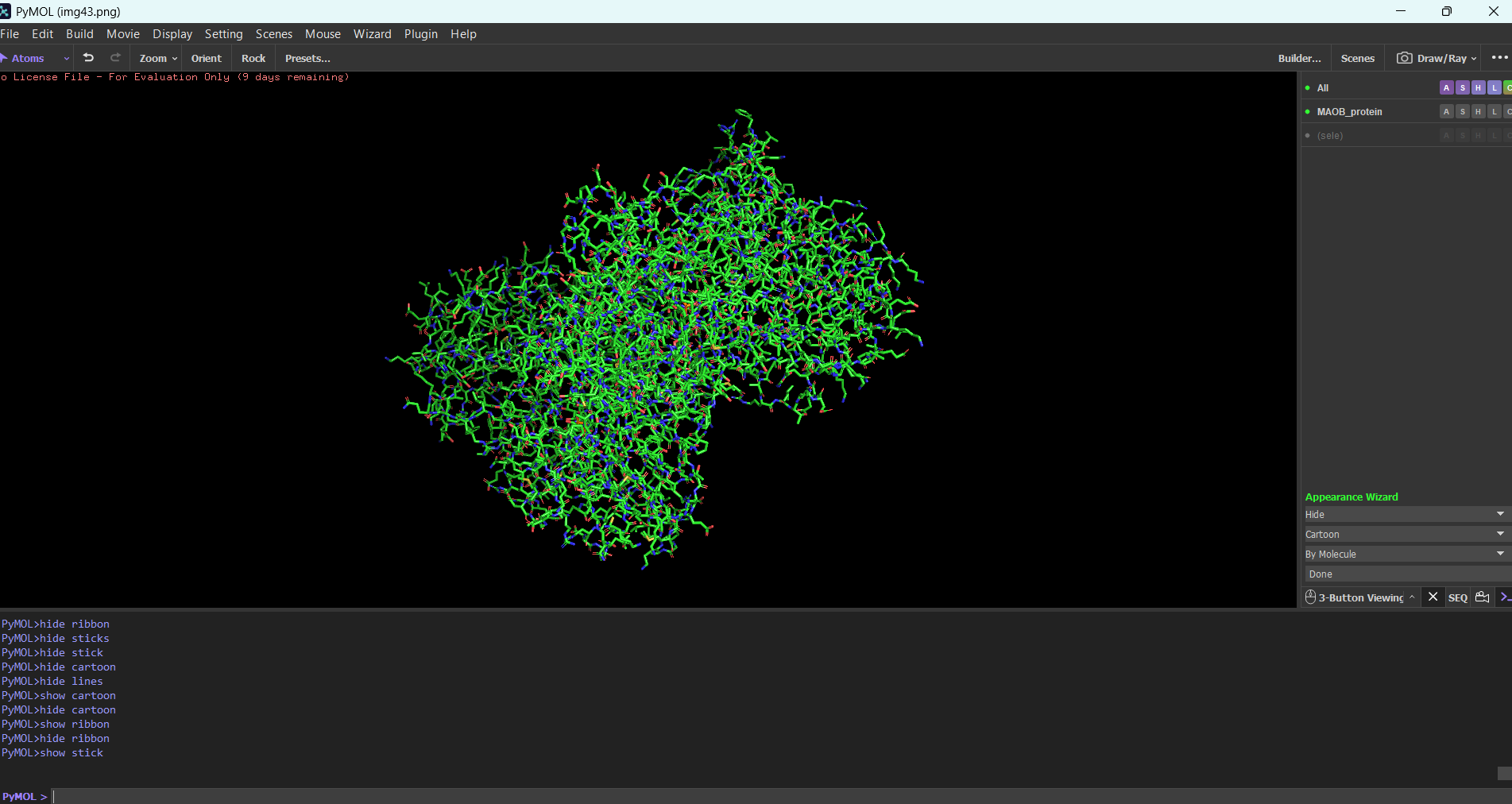
Cartoon:
Note: The first time I tried to visualize the protein, pyMOL showed an overlapped version of multiple modes combined. For example, to see only the ribbon, I needed to type in the integrated command line: “hide everything” then you type “show ribbon”. Normally you should be able to do this using the GUI, but in my case the GUI didn’t work for some reason.
Protein colored by secondary structure:
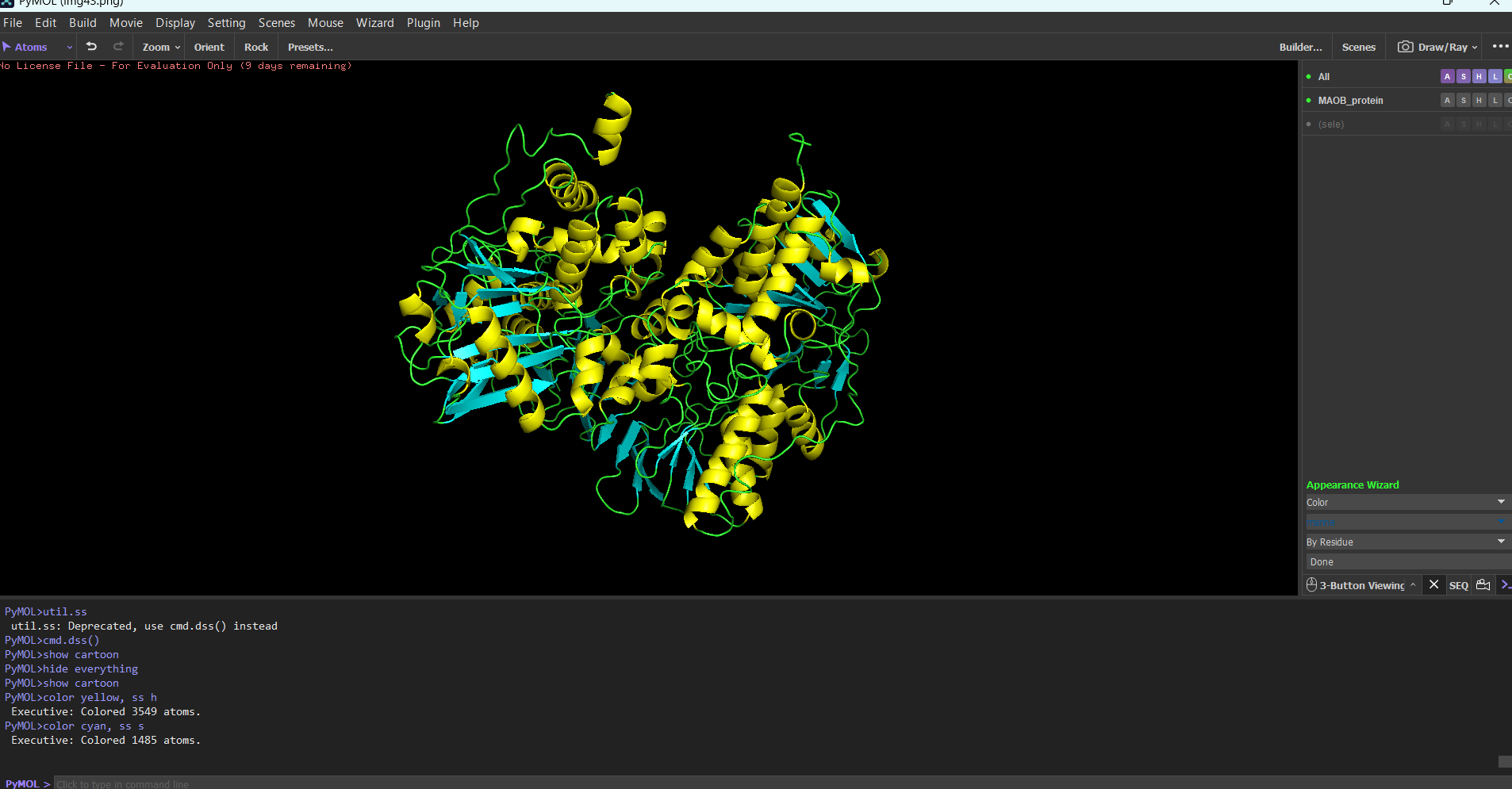 It has roughly the same number of helices and beta sheets (maybe a few more beta sheets, but it differs from organism to organism most likely)
It has roughly the same number of helices and beta sheets (maybe a few more beta sheets, but it differs from organism to organism most likely)
Note: I used “color yellow, ss h” and “cyan, ss s” to color the secondary structures.
Further research: From what I read, proteins rich in helices often have a function that involves dynamic processes (enzymes, DNA binding mechanisms), and proteins rich in beta-sheets often provide mechanical support (structure). This statement is not always true (collagen).
Protein residue distribution:
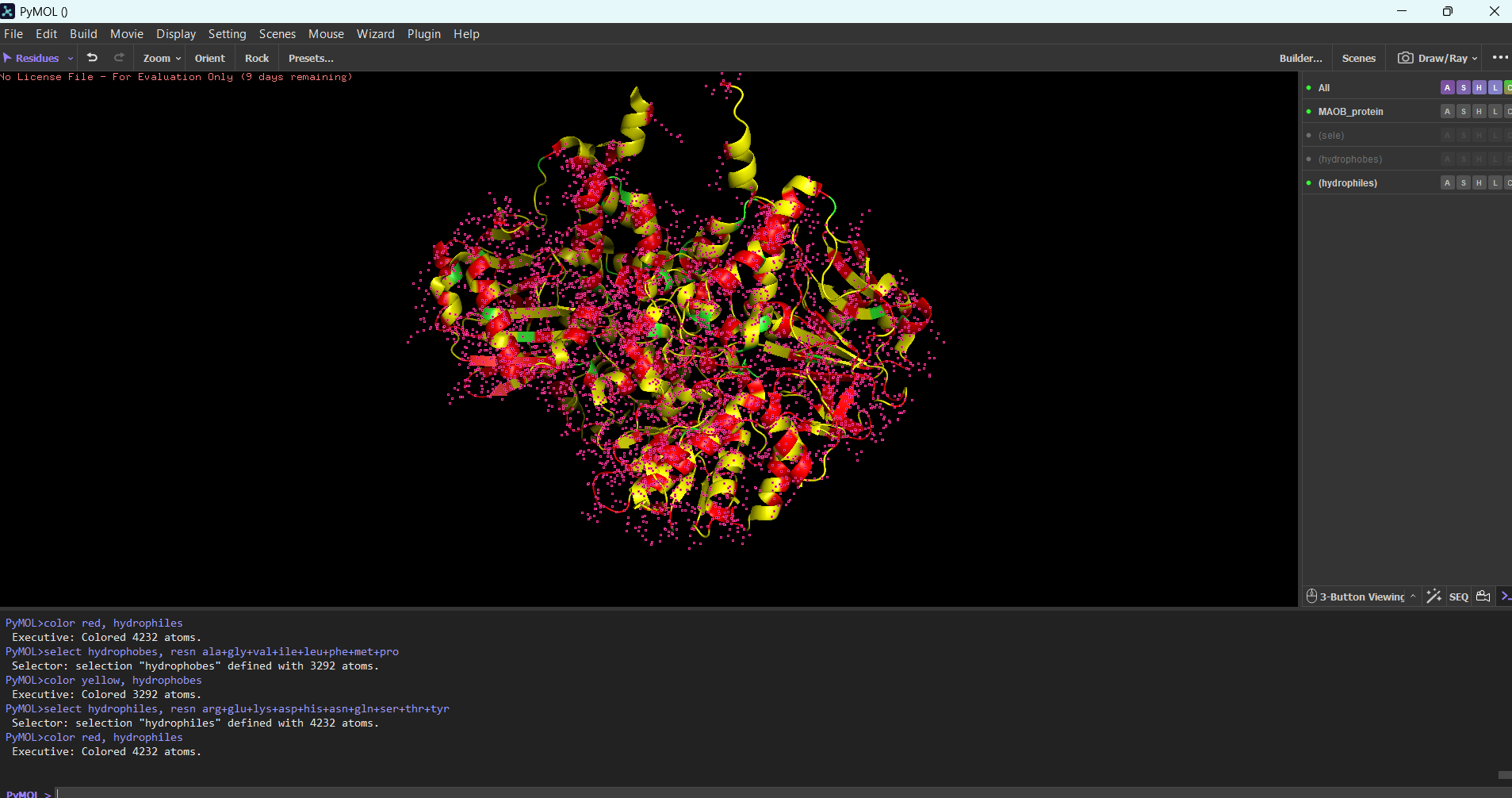
Hydrophobic and hydrophilic residues are distributed uniformly in this protein.
Surface mode:
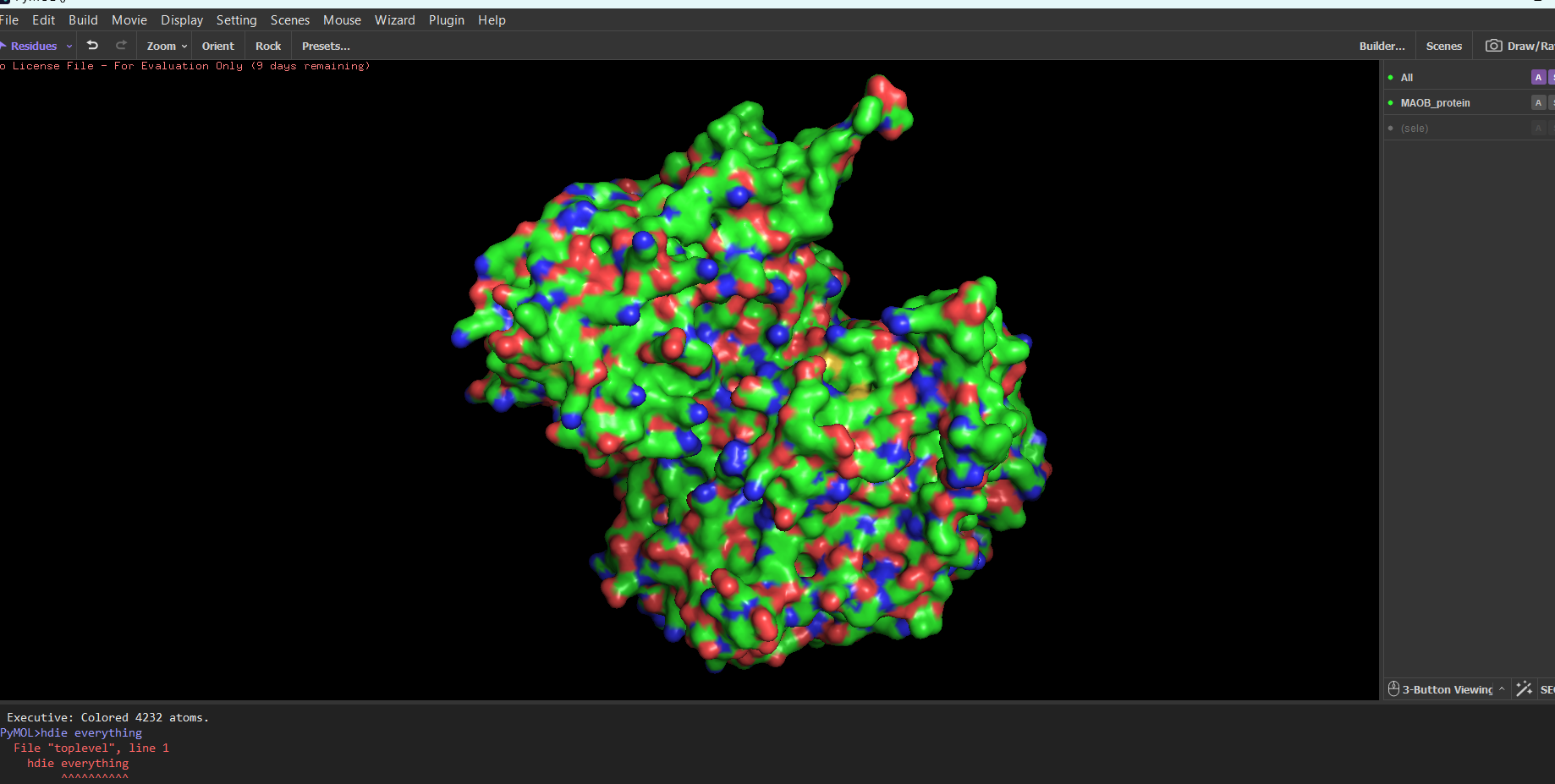
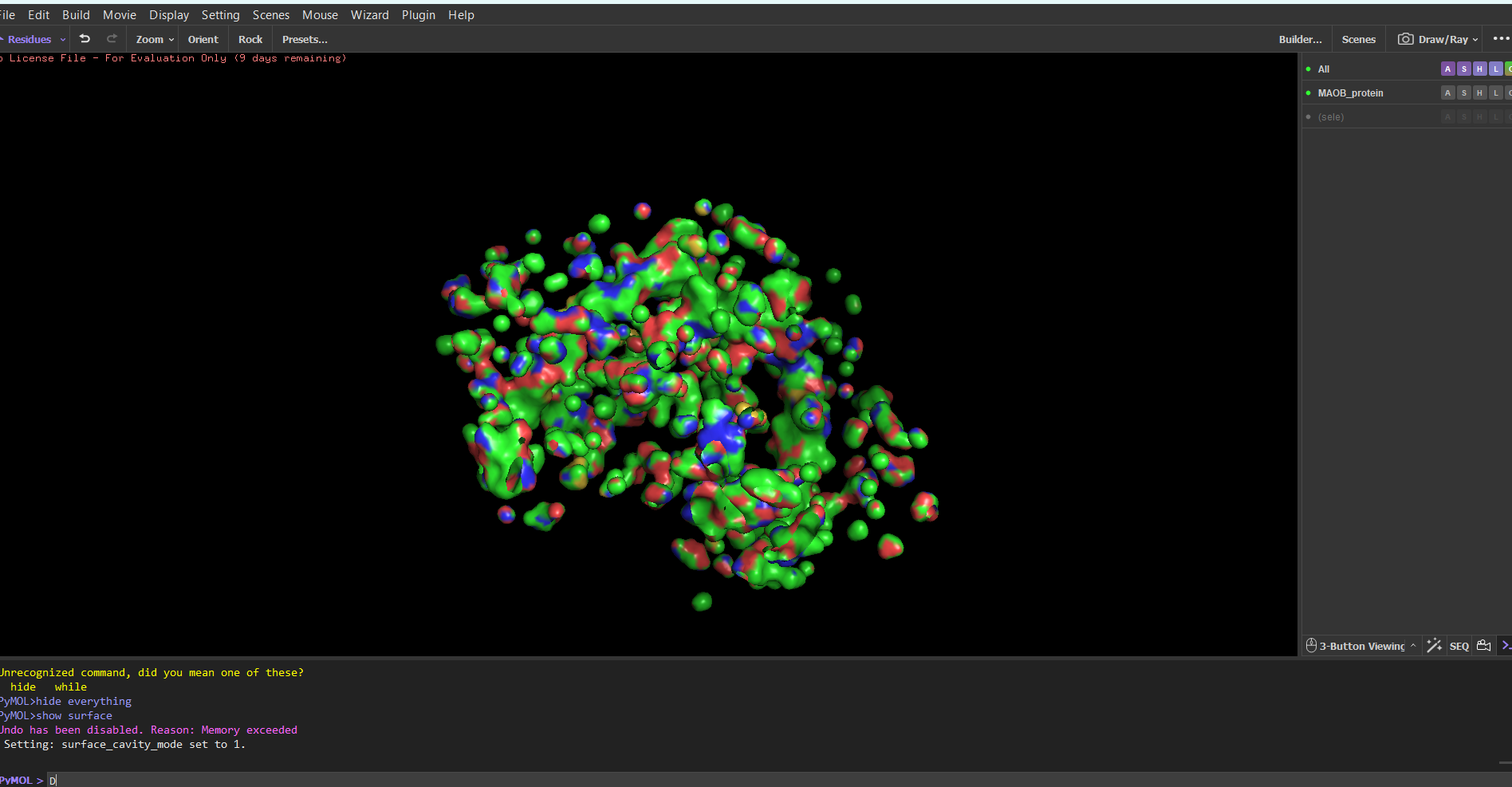
Note: I tried showing only the cavities, because I thought it would be easier to find the binding sites. I still don’t know which ones are the biding sites.
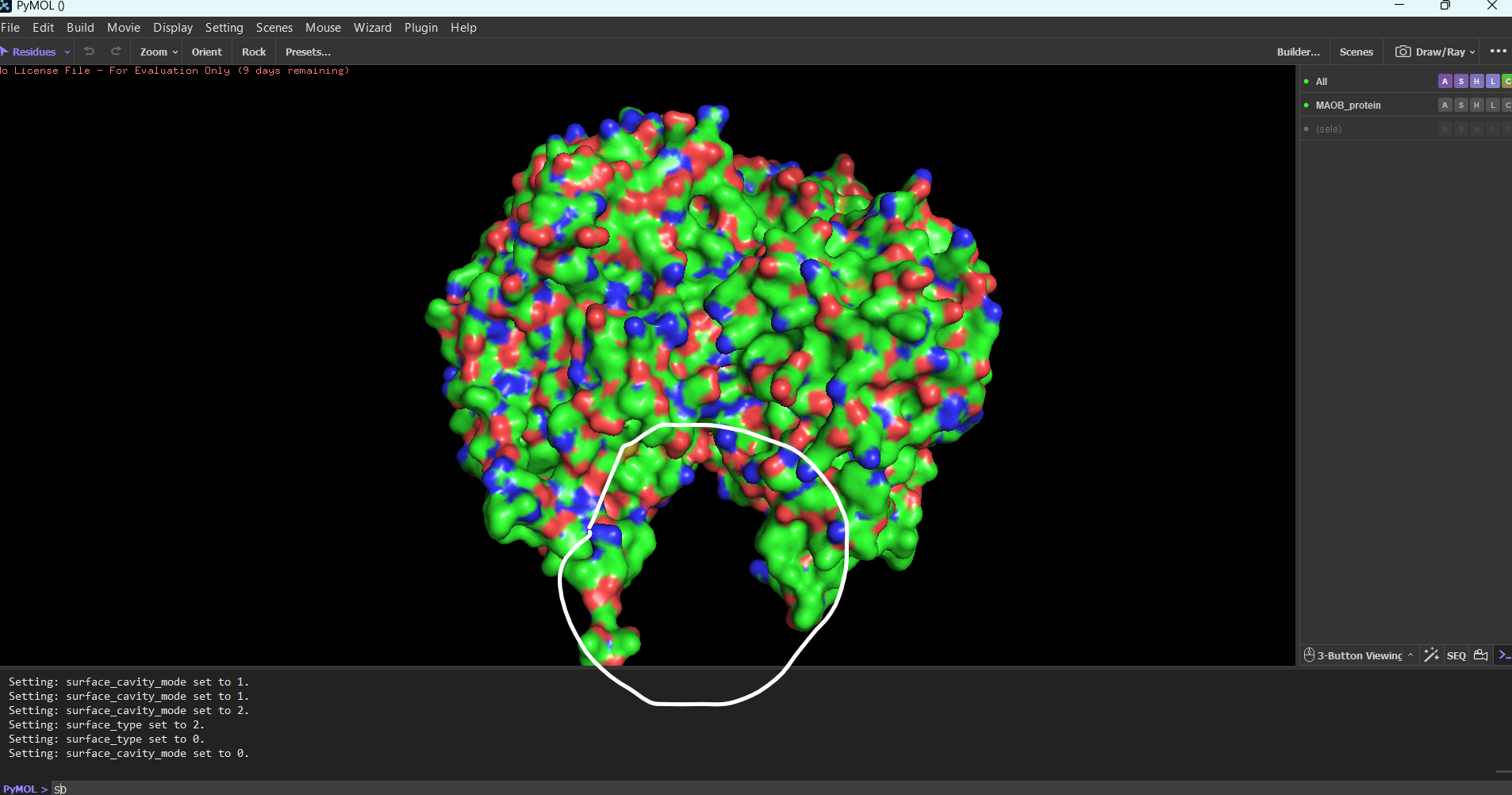 Substrate binding site found
Substrate binding site found
Part C:
C1:
Glu Glu Leu Ile Thr Gly Val Leu Gly Ile Ser Ile Asp Leu Gly Met Val Thr Gly Ser Asp Leu Ala Lys Ala Val Lys Leu Ala Thr Gly Leu Gly Glu Ala Val Val Glu Gly Ala Lys Ala Val Gly Ser Val Leu Ala Leu Ser Thr Ala Leu Val Leu Ala Leu Leu Gly Leu Ala Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu Ala Leu Gly Leu Leu Gly Leu
C2:
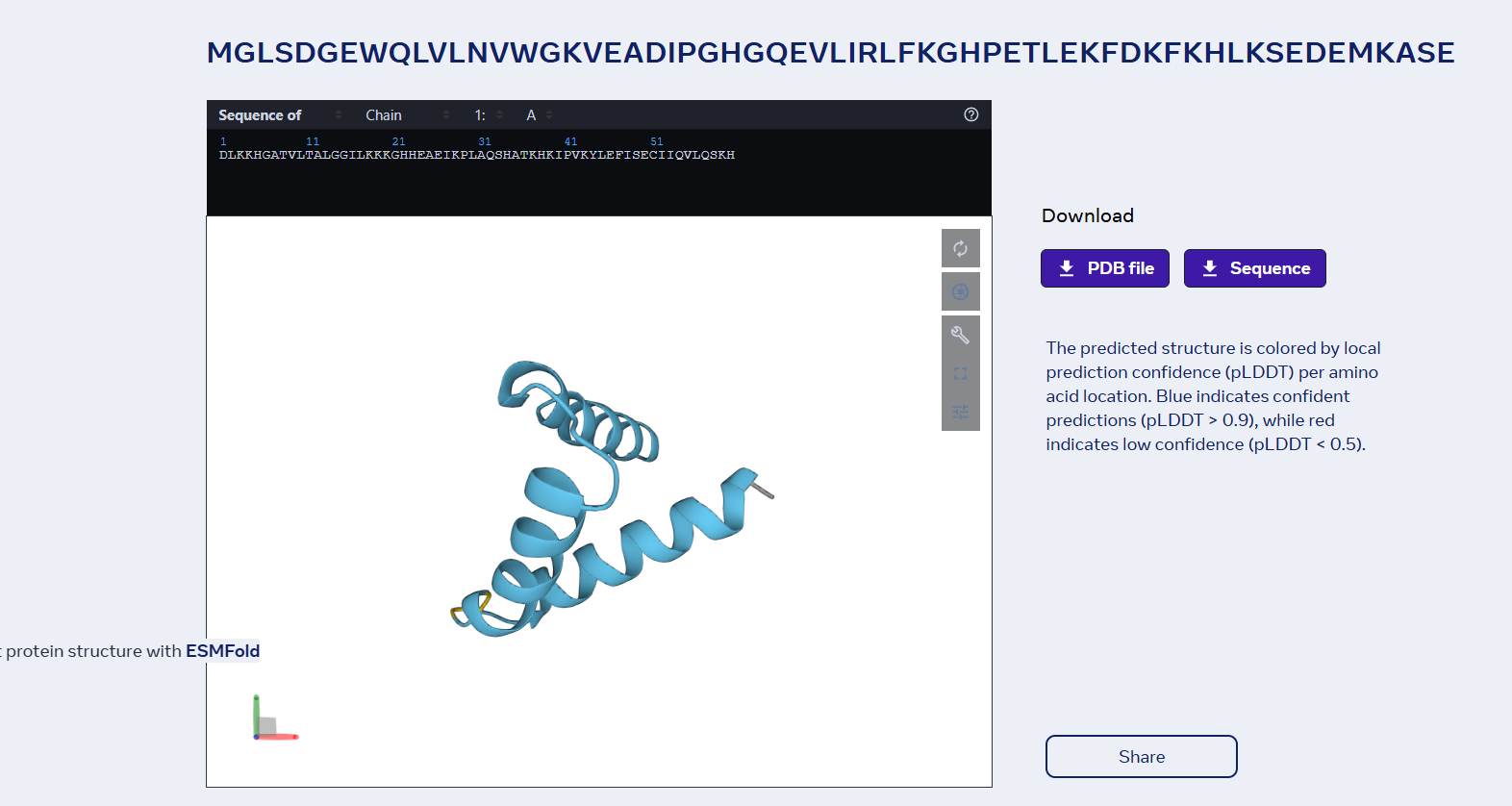
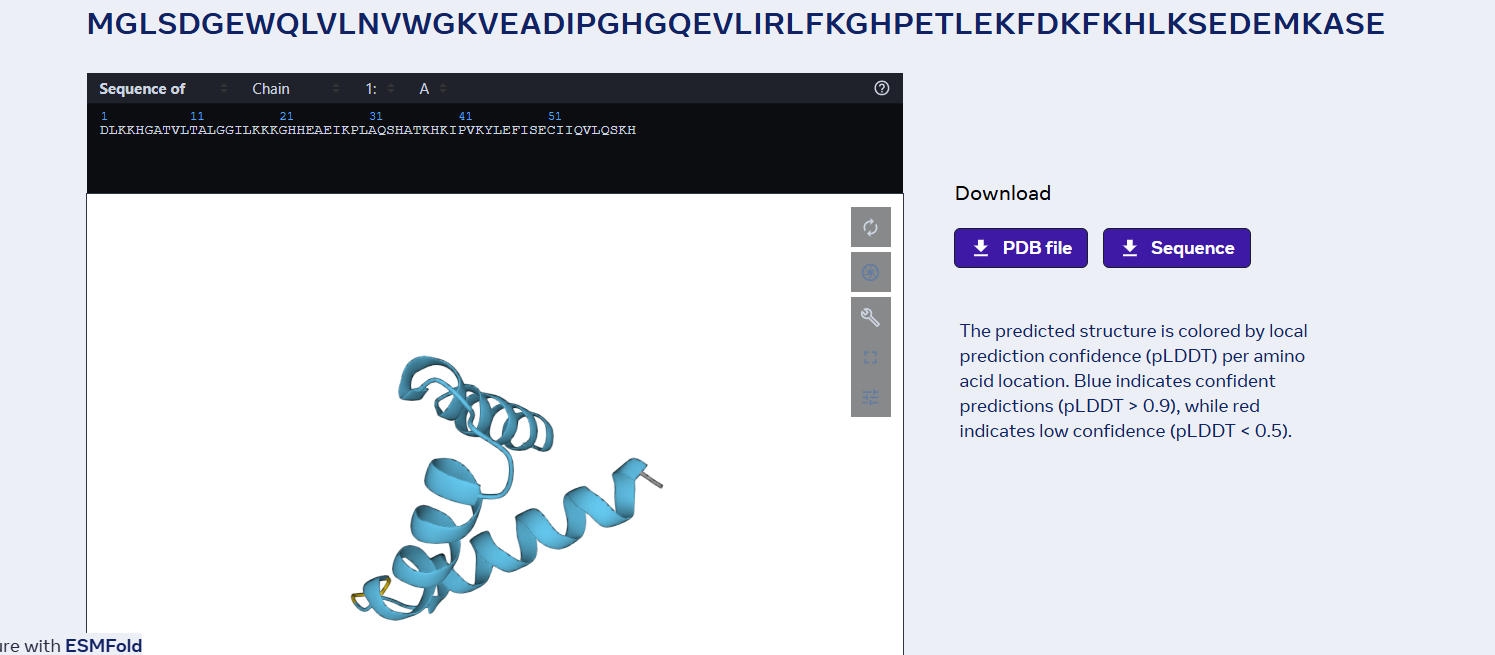
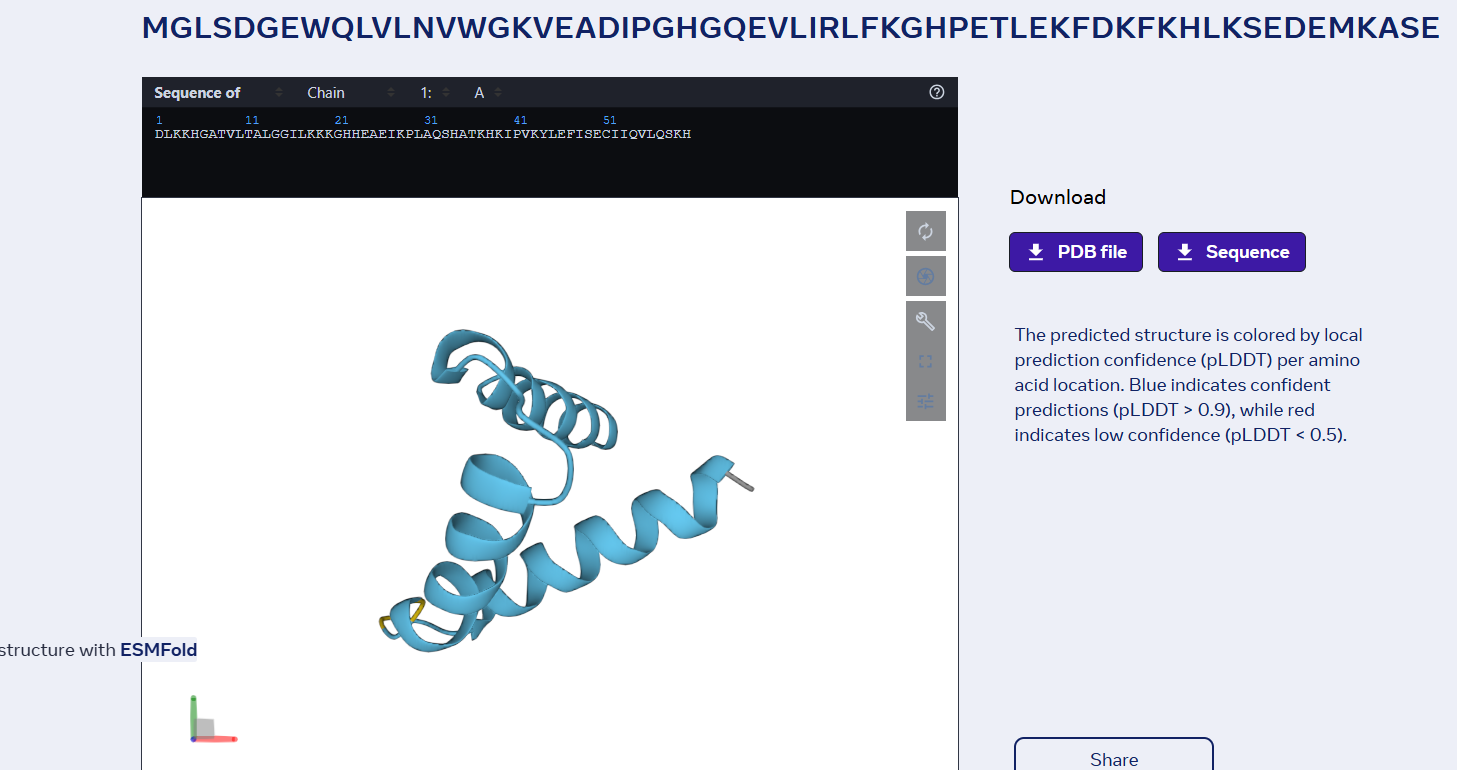
sp|P02144|MYG_HUMAN Myoglobin OS=Homo sapiens OX=9606 GN=MB PE=1 SV=2 MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASE DLKKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKH PGDFGADAQGAMNKALELFRKDMASNYKELGFQG
Folded with ESMFold:
Folded with Boltz-1:
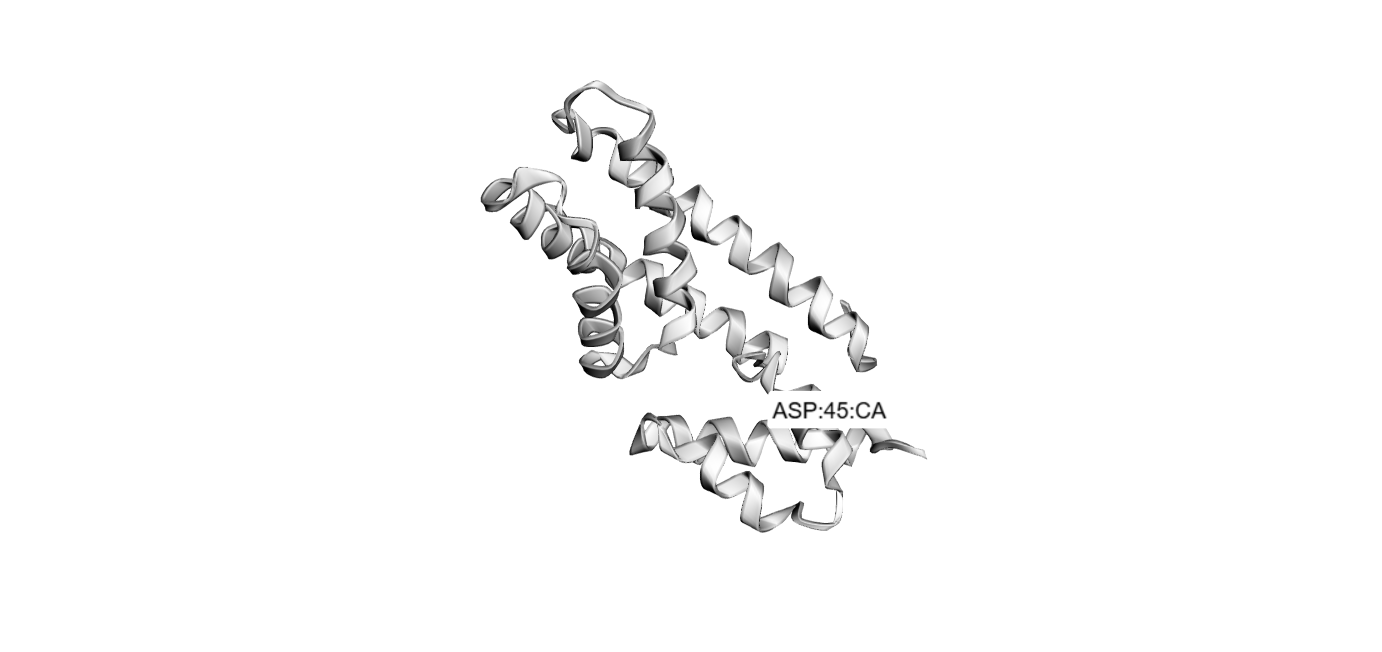
Folded with SwissDock:
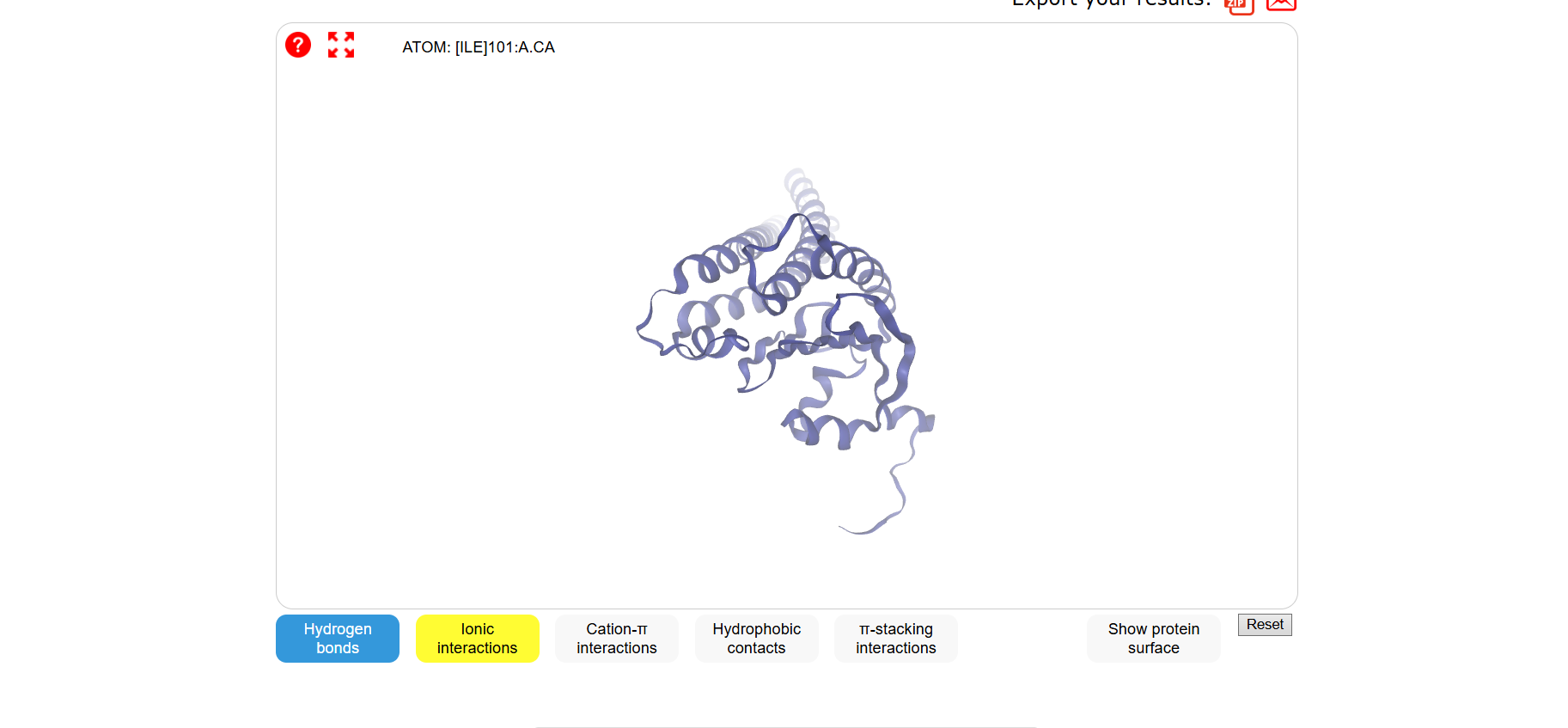
3 one point mutations:
last 6 proteins mutation:
Part D:
Project Proposal: Engineering a Minimal MS2-L Lysis Engine
Primary Goal: Our group aims to increase the functional stability of the MS2 lysis (L) protein. We will achieve this by eliminating the N-terminal domain (residues 1–36). This truncation removes the “regulatory brake” that normally makes lysis dependent on the host chaperone DnaJ, resulting in a more potent, autonomous lysis protein, thus lysis will be achieved a lot faster, beacuse MS2-L will be functional from the moment of translation which gives less time for the proteases to degrade it before it attached to the cellular membrane.
Tools & Approaches We propose a computational pipeline to validate and optimize this truncated variant:
AlphaFold3 / AlphaFold-Multimer: We will model the truncated L protein in a cramped lipid bilayer environment to predict how the remaining transmembrane helix (TMH) inserts itself. We will also use it to confirm the loss of binding affinity to E. coli DnaJ.
Protein Language Models (ESM-2 / ESM-1v): We will use these models to perform in silico mutagenesis on the remaining C-terminal sequence. Our goal is to identify “stabilizing” mutations that strengthen the alpha-helical propensity of the membrane-spanning region.
Molecular Dynamics (MD) Simulations (OpenMM or Gromacs): Since lysis involves membrane distortion, we will simulate the truncated protein within a model bacterial membrane to observe its ability to disrupt lipid packing.
- Potential Pitfalls Membrane Toxicity in in silico models: Most protein design tools are trained on soluble proteins. Modeling a protein whose entire job is to destroy the membrane (lysis) may lead to unstable simulations or “unphysical” results.
Expression Levels: In a real-world lab setting, a highly toxic protein might kill the host cells so quickly that we cannot produce high enough titers of the phage for study.
- Pipeline Schematic Design: Truncate N-terminus (1-36).
Optimize: Run ESM-1v to find high-probability stabilizing mutations.
Predict: Fold top candidates in AlphaFold3 to ensure TMH orientation.
Simulate: Insert into a virtual membrane to verify disruptive “toxicity.”