Homework 5 Protein Design Part II
Protein Design Part II Homework:
Part 1
- protein sequence (with A4V mutation): ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGATSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLAAGVIGIAQ
Part 2:
- Firstly, I want to highlight a few hallmarks of the human protein SD01:
- The N-terminus is located near the C-terminus
- The residues 3 to 9, thus including the A4V mutation, form a beta-strand.
- Human SD01 is rich in beta-strands
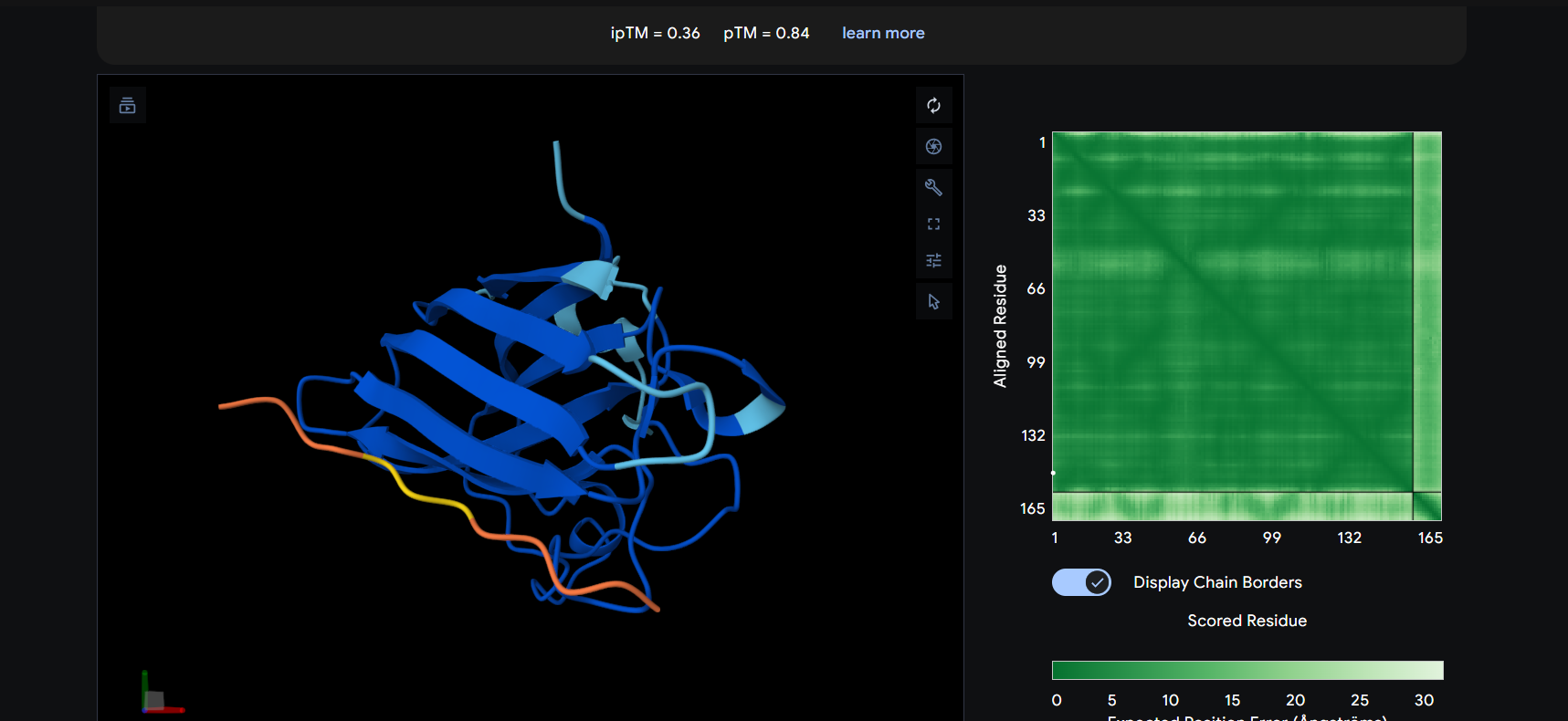
The peptide appears to bind on the surface of the protein in the opposite part of the N-terminus.
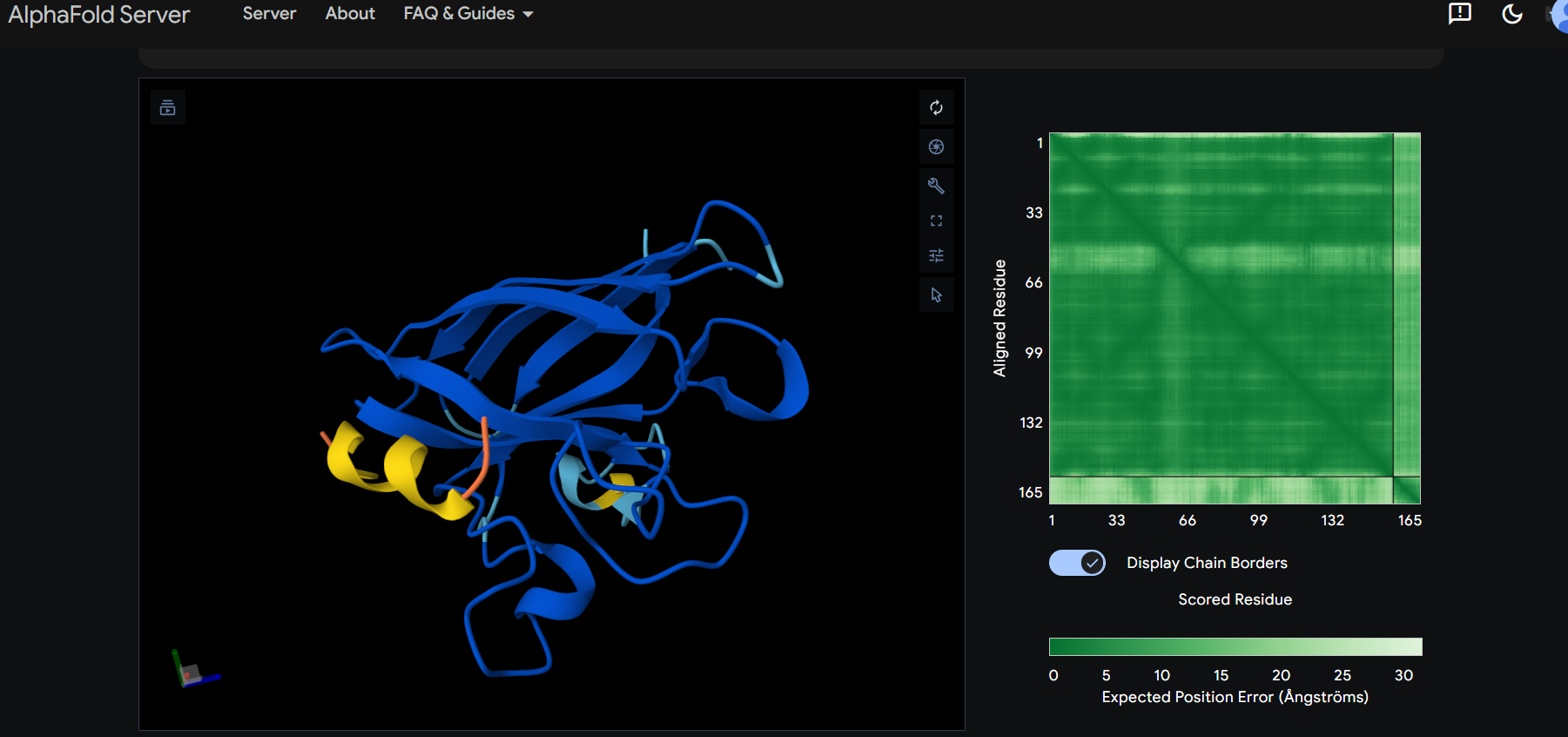
The peptide appears to bind on the surface of the protein in the opposite part of the N-terminus.
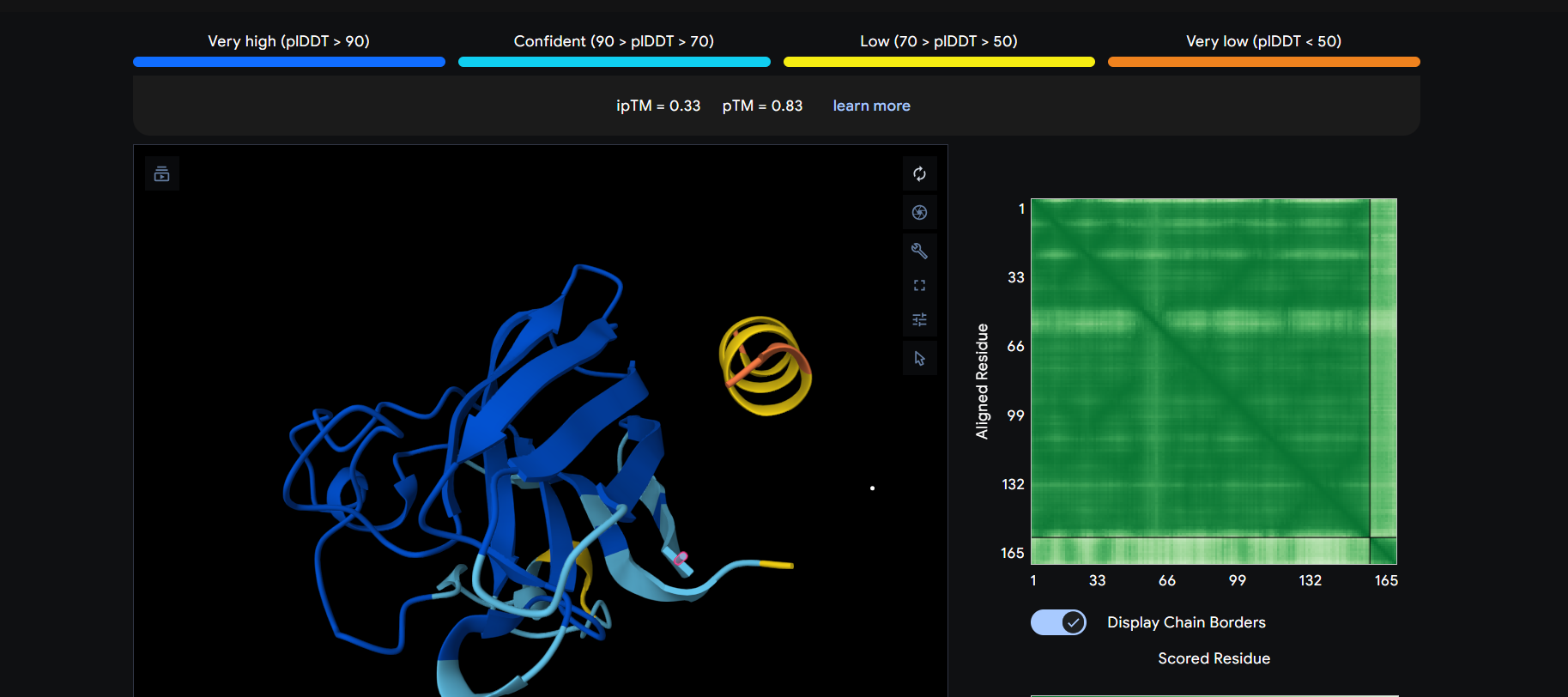
The peptide appears to bind on the surface of the protein near the N-terminus.
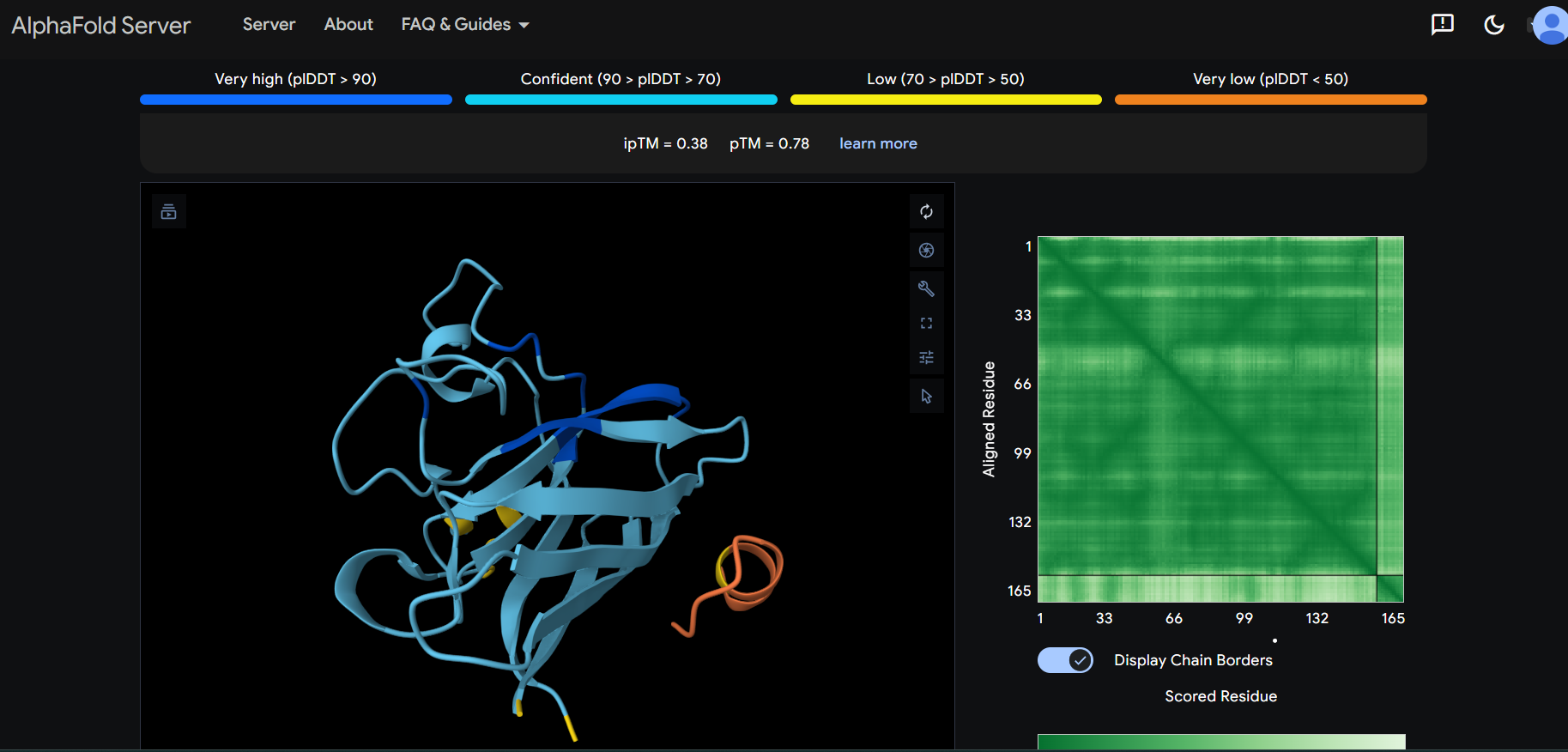
The peptide appears to bind on the surface of the protein aproximatevely near the N-terminus.
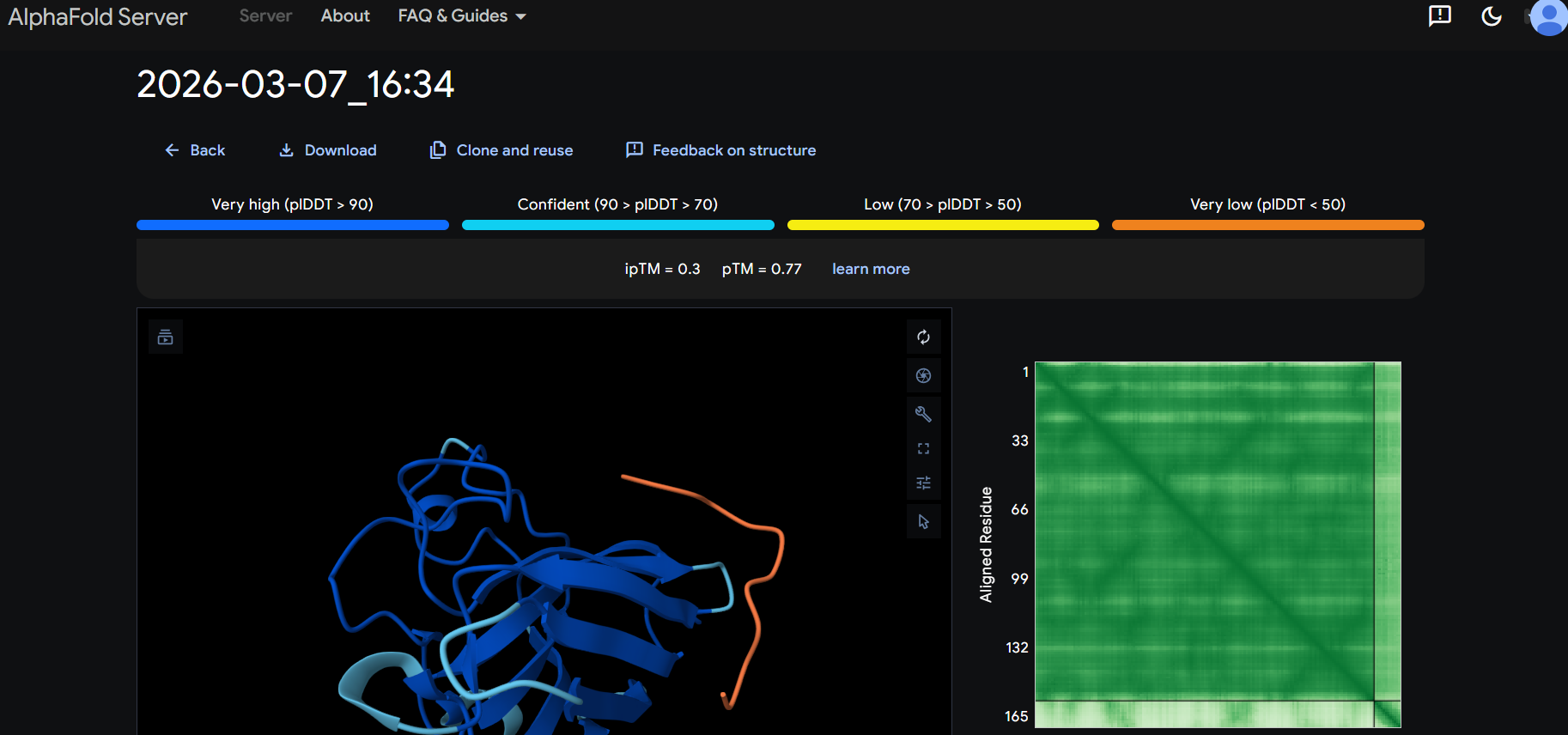
The peptide appears to bind on the surface of the protein aproximatevely near the N-terminus, near the beta-sheet formed by SD01.
The ipTM score for all the predicted SDo1-peptide interactions is below what it would be considered reliable.
The prediction are incongruent with the knwon binding sites.
Part 3:
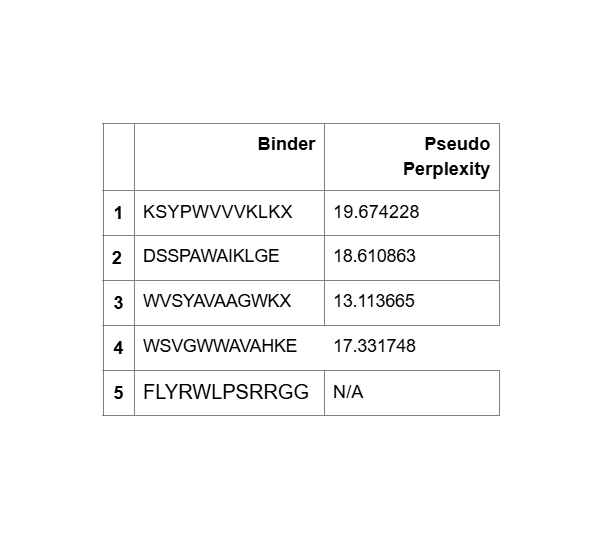
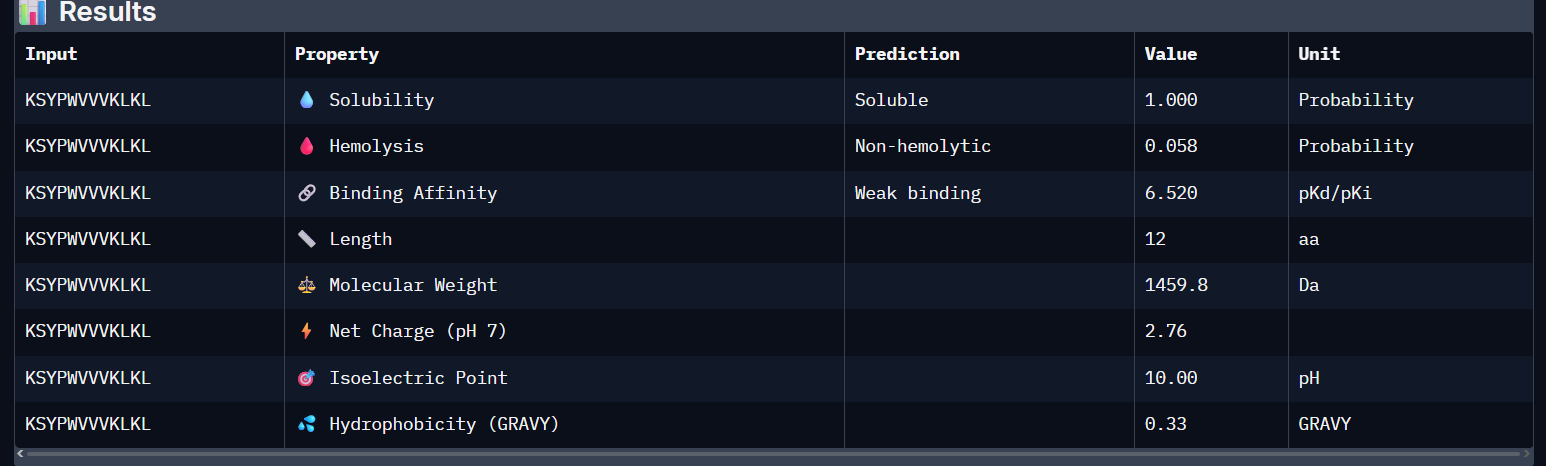
KSYPWVVVKLKL:
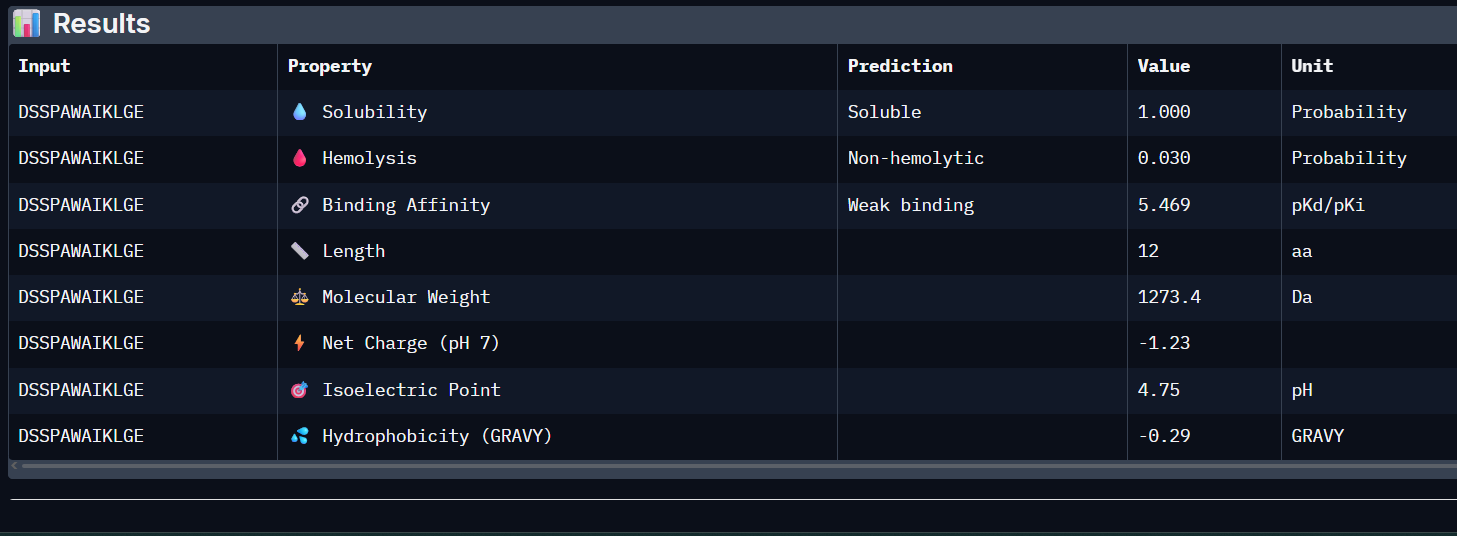
 DSSPAWAIKLGE:
DSSPAWAIKLGE:
 WVSYAVAAGWKL:
WVSYAVAAGWKL:
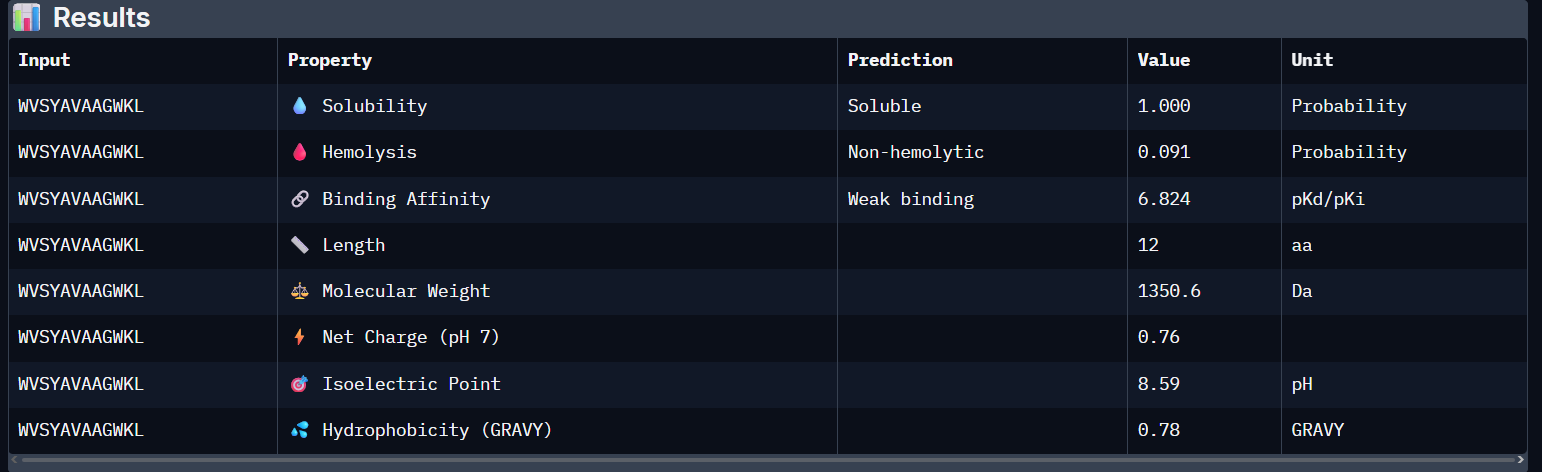 WSVGWWAVAHKE:
WSVGWWAVAHKE:
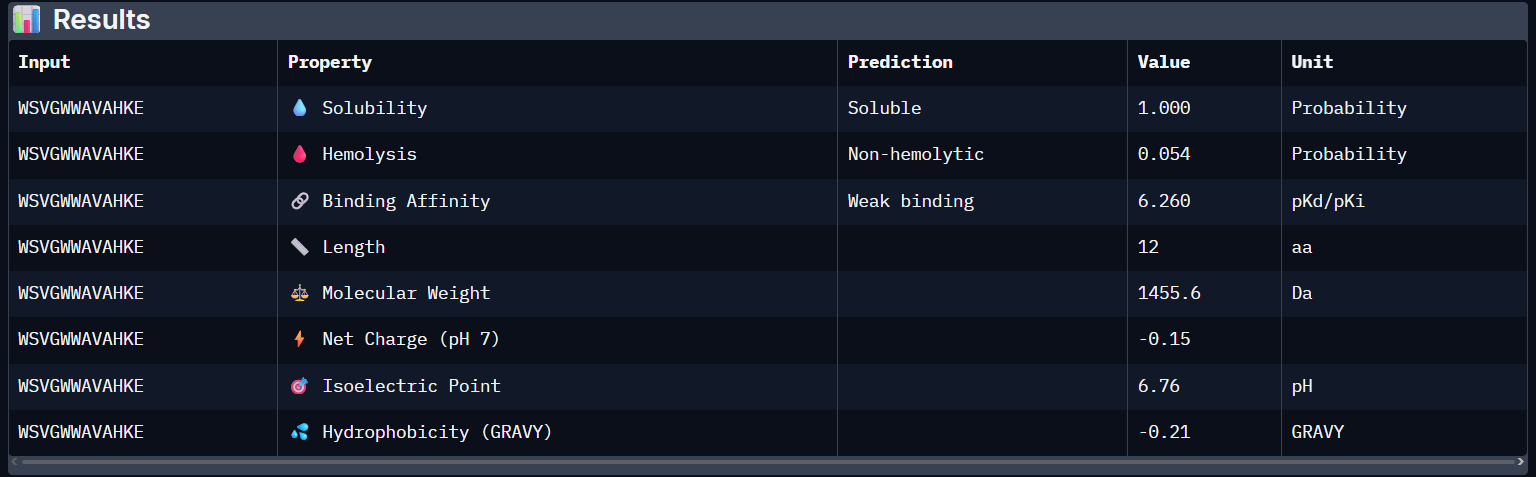 FLYRWLPSRRGG:
FLYRWLPSRRGG:
- Unfortunately, all the peptides showed above are soluble, weak binding affinity (which corresponds with the low ipTM score) and are non-hemolytic, thus there is no much to say about them (( .
“Choose one peptide you would advance and justify your decision briefly”:
- WVSYAVAAGWKL has the highest affinity ~6.8 and appears to be the best choice;
Part 4:
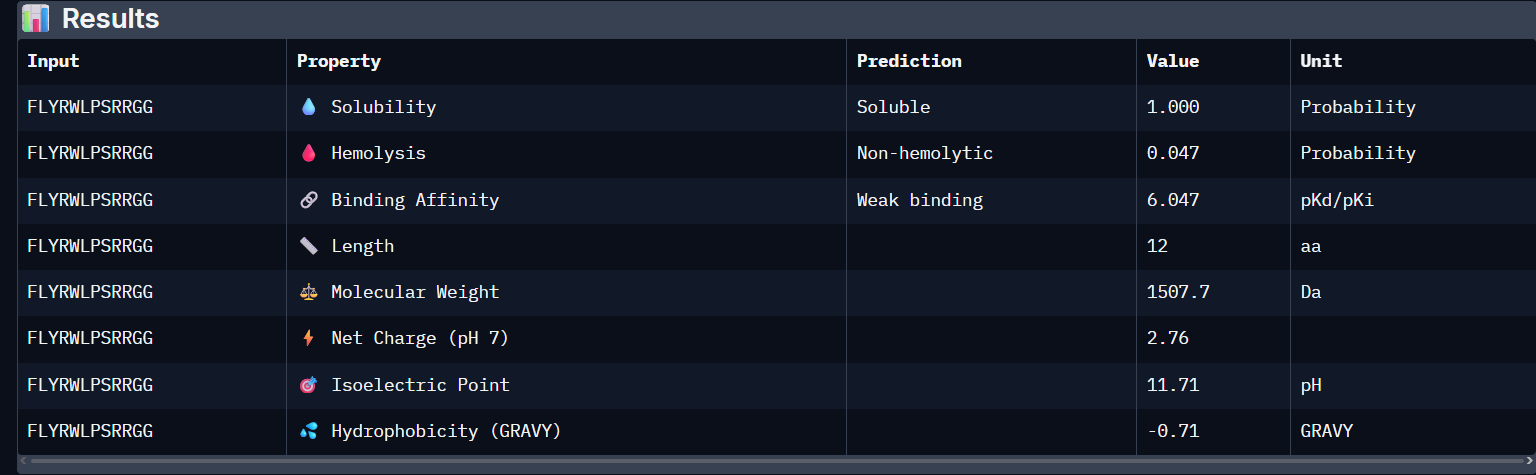
Generated peptide: CQGVFWTMCSHD
- nr. of amino acids: 12
- the peptide has the same number of AAs as the previous ones
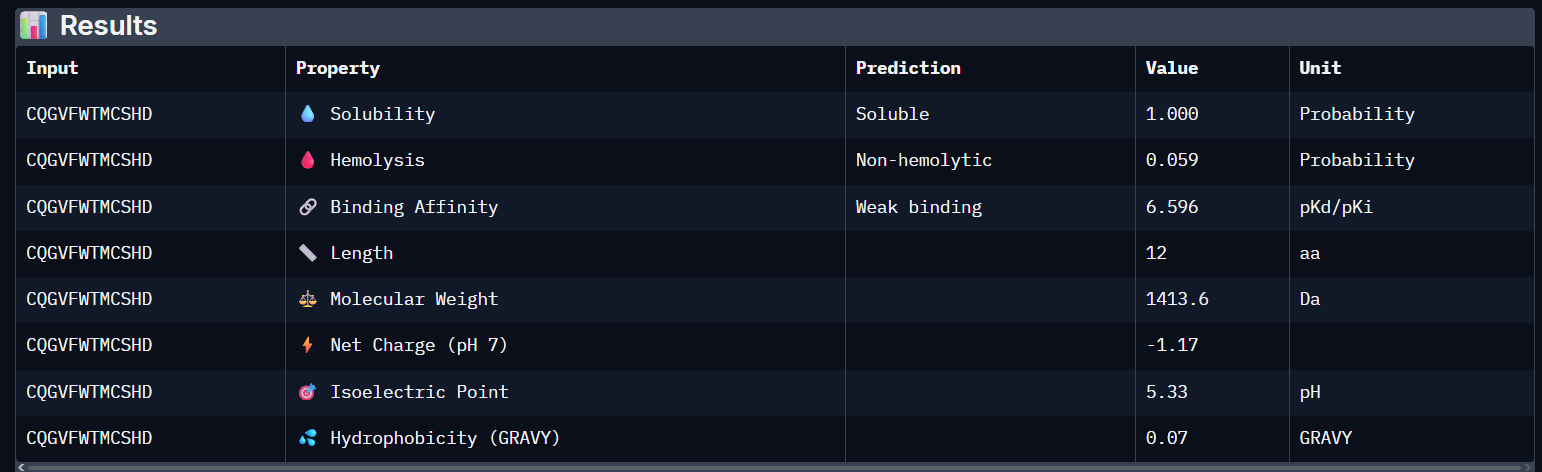
- it still has a weak binding affinity..
Part C:
I read the documentation resources about the MS2-L its similarity with the other amurines. List with a few important things for this project:
- MS2 is a bacteriophage that infects E.coli and has a genome containing only 4 proteins: the lysis protein (MS2-L/L-protein), the capsid protein, the maturation protein (initiates infection) and replicase;
- MS2-L is a protein found in the MS2 bacteriophage that produces the lysis of the infected bacteria using amuralytic ways, meaning that it doesn’t directly break down the peptoglycan wall (as the majority of bacteriophage lysis proteins do), but it most likely inserts into the cytoplasmatic membrane of E.coli;
- MS2-L is often compared with ΦX174 (also an amurine). The difference is that ΦX174 seems to alter the path of PG wall synthesis (inhibits the MraY enzyme), eventually leading to cell lysis.
- MS2-L is chaperone dependent. Without the DnaJ chaperone found in E.coli, the protein remain self-inhibited. The removal of the first domain of the lysis protein made the protein become chaperone-independent without altering the lysis function and increasing the speed of induced lysis with 20 minutes compared to the normal protein.
- the soluble domain of MS2-L –> first 40 AAs, the Transmembrane domain –> the last 35 AAs;
Then I ran the colab linked at option one of the protein engineering part and analyzed the results:
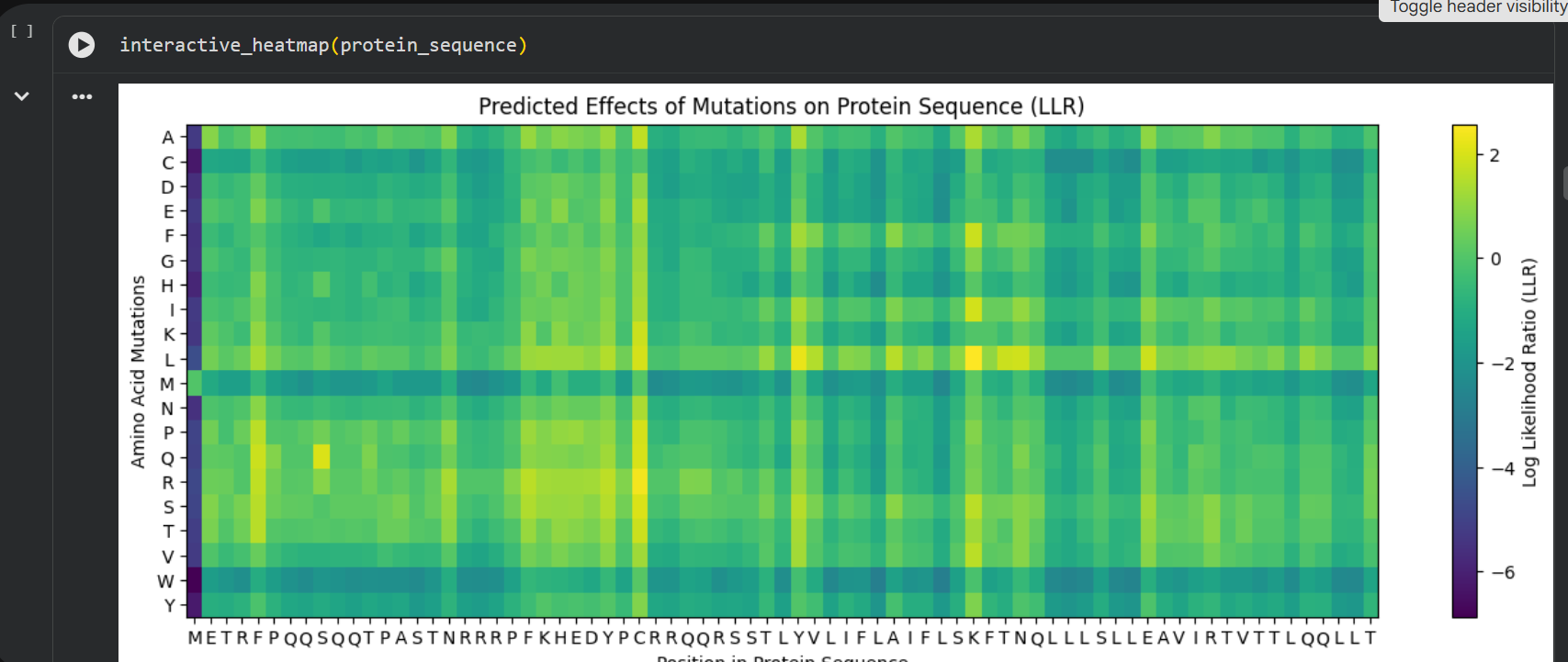
log likelihood ratio: < 0 - the original sequence is favoured; > 0 - the engineered protein is favoured The obvious observations that can be made by looking at the graph are:
the first resiude of the protein (M) is irreplaceable;
Mutations of a residue with: W - Tryptophan, M - Methionine, C - Cysteine , are not favoured
almost any mutation of the 29th residue (C) is favoured (Also generated using the colab code:) Position Wild_Type_AA Mutation_AA LLR_Score 989 50 K L 2.561464 574 29 C R 2.395427 769 39 Y L 2.241778 575 29 C S 2.043150 173 9 S Q 2.014323 573 29 C Q 1.997049 572 29 C P 1.971028 569 29 C L 1.960646 987 50 K I 1.928798 1049 53 N L 1.864930 1209 61 E L 1.818096 1029 52 T L 1.813965 984 50 K F 1.802066 576 29 C T 1.797247 568 29 C K 1.795877 93 5 F Q 1.795244 94 5 F R 1.659716 560 29 C A 1.648655 534 27 Y R 1.628060 434 22 F R 1.602028 92 5 F P 1.596889 997 50 K V 1.594573 995 50 K S 1.574555 96 5 F T 1.559023 95 5 F S 1.556416 889 45 A L 1.539248 775 39 Y S 1.517457 535 27 Y S 1.497052 789 40 V L 1.477630 529 27 Y L 1.474638 435 22 F S 1.423357 563 29 C E 1.383282 760 39 Y A 1.364997 571 29 C N 1.362601 980 50 K A 1.357792 567 29 C I 1.344121 89 5 F L 1.332615 334 17 N R 1.323652 767 39 Y I 1.320101 776 39 Y T 1.302803 514 26 D R 1.268762 566 29 C H 1.246106 764 39 Y F 1.245850 777 39 Y V 1.244389 454 23 K R 1.236555 494 25 E R 1.229349 474 24 H R 1.227778 996 50 K T 1.222128 533 27 Y Q 1.218850 536 27 Y T 1.215567
Choosing the most favoured mutations:
- I compared the most favoured mutations given by the colab with the results from the experimental data provided in the Excel and I chose a combination of the most favoured mutations to generate the final engineered MS2-L lysis protein:
- Mutations: S9Q, C29R, K50L Sequence: METRFPQQQQQTPASTNRRRPFKHEDYPRRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
- Mutations: S9Q, C29S, K50I Sequence: METRFPQQQQQTPASTNRRRPFKHEDYPSRRQQRSSTLYVLIFLAIFLSIFTNQLLLSLLEAVIRTVTTLQQLLT
- Mutations 3: S9Q, C29P, K50F Sequence: METRFPQQQQQTPASTNRRRPFKHEDYPPRRQQRSSTLYVLIFLAIFLSFFTNQLLLSLLEAVIRTVTTLQQLLT
- Mutation P13L Sequence: METRFPQQSQQTLASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Mutation S15A Sequence: METRFPQQSQQTPAATNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- nr. of amino acids: 12