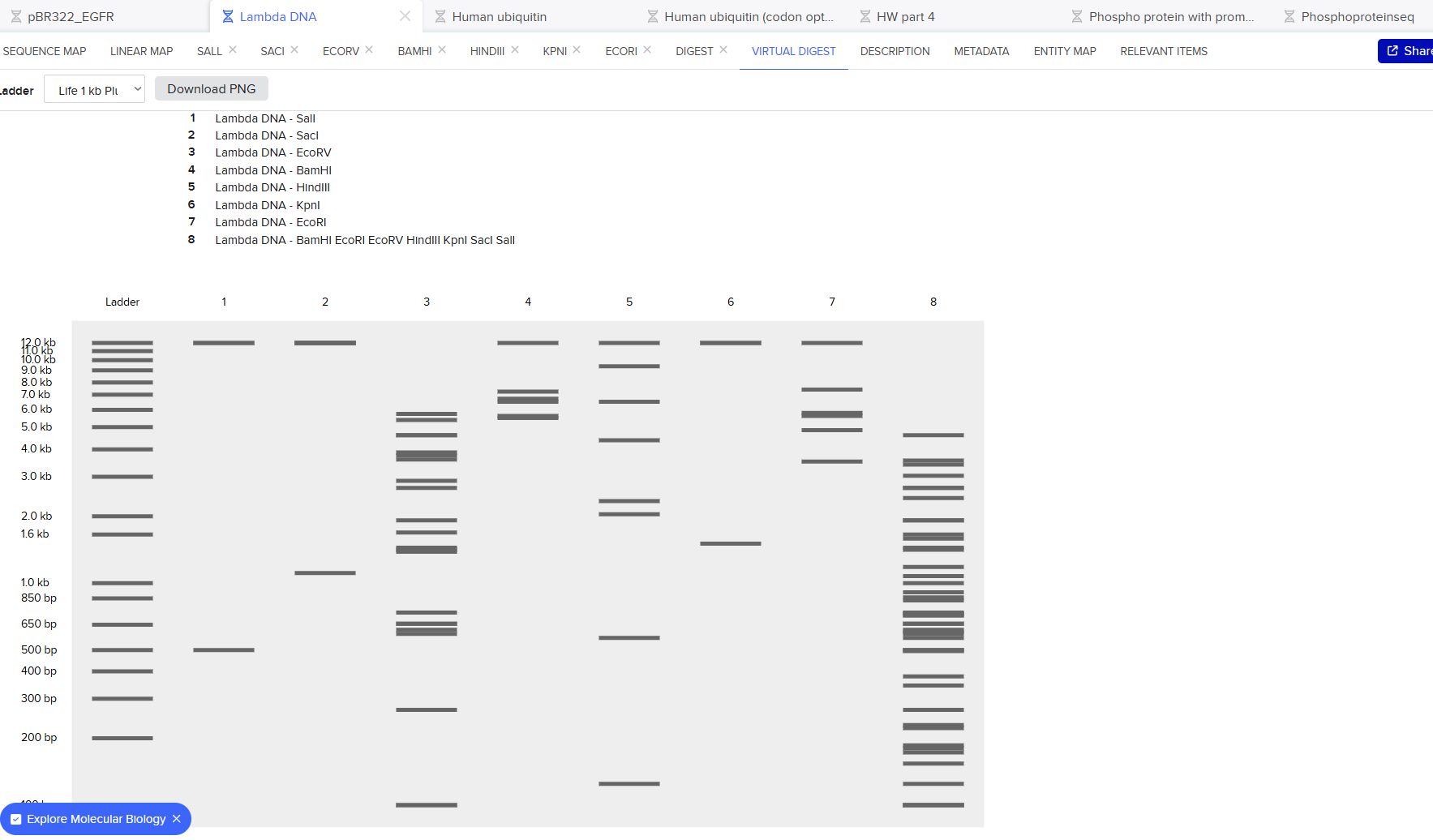
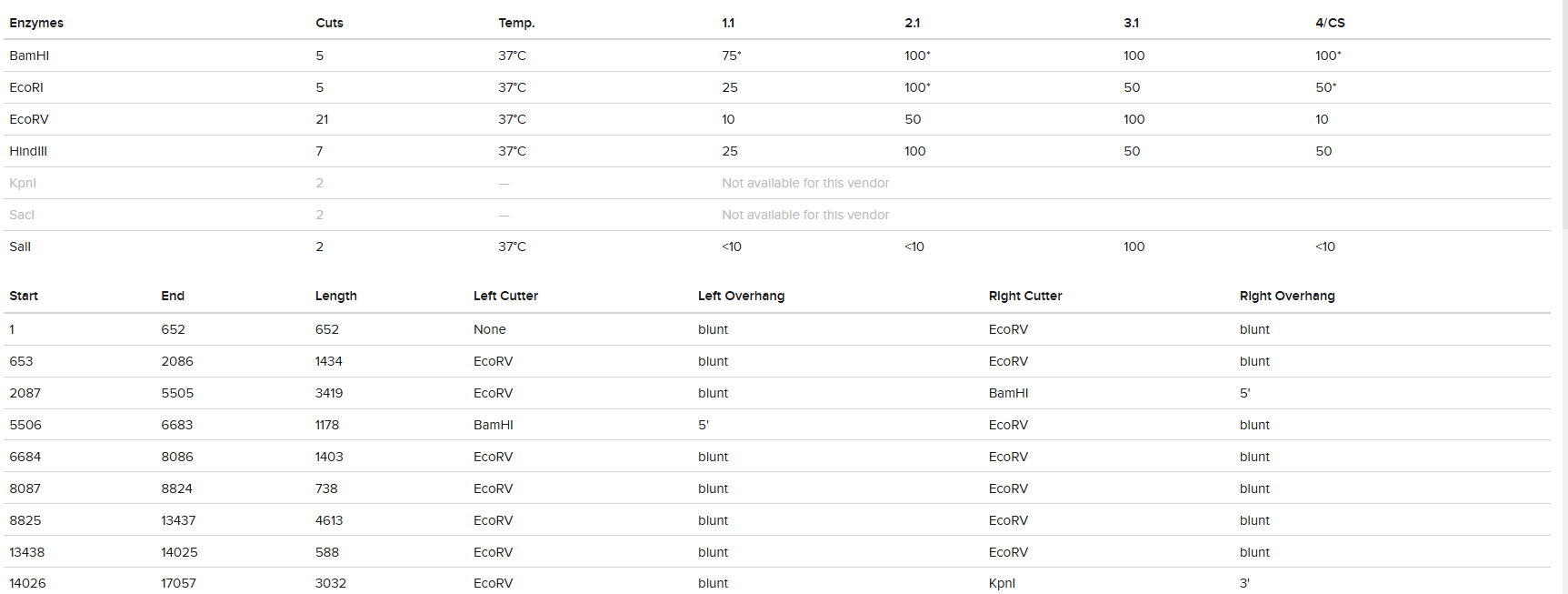
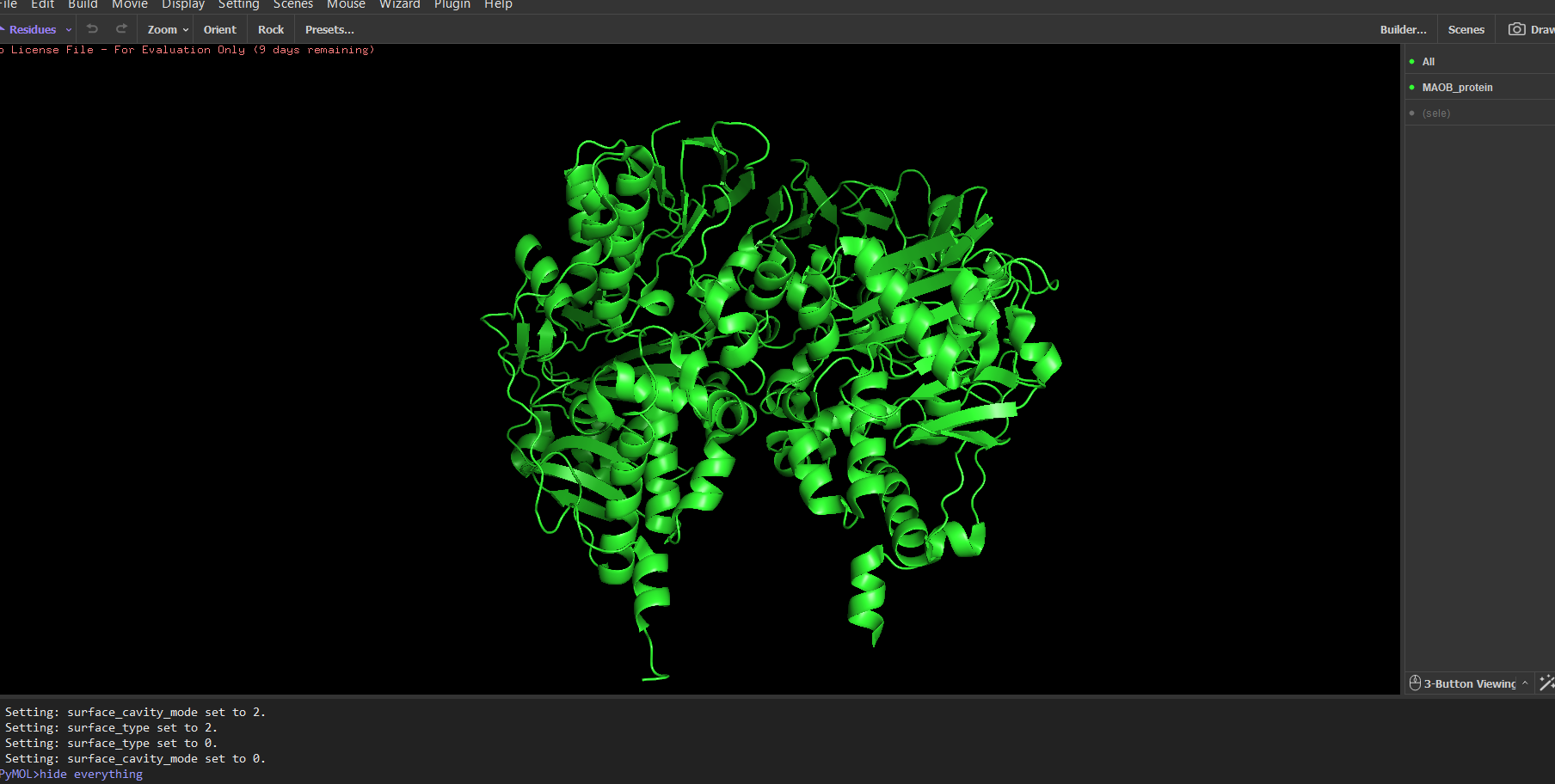
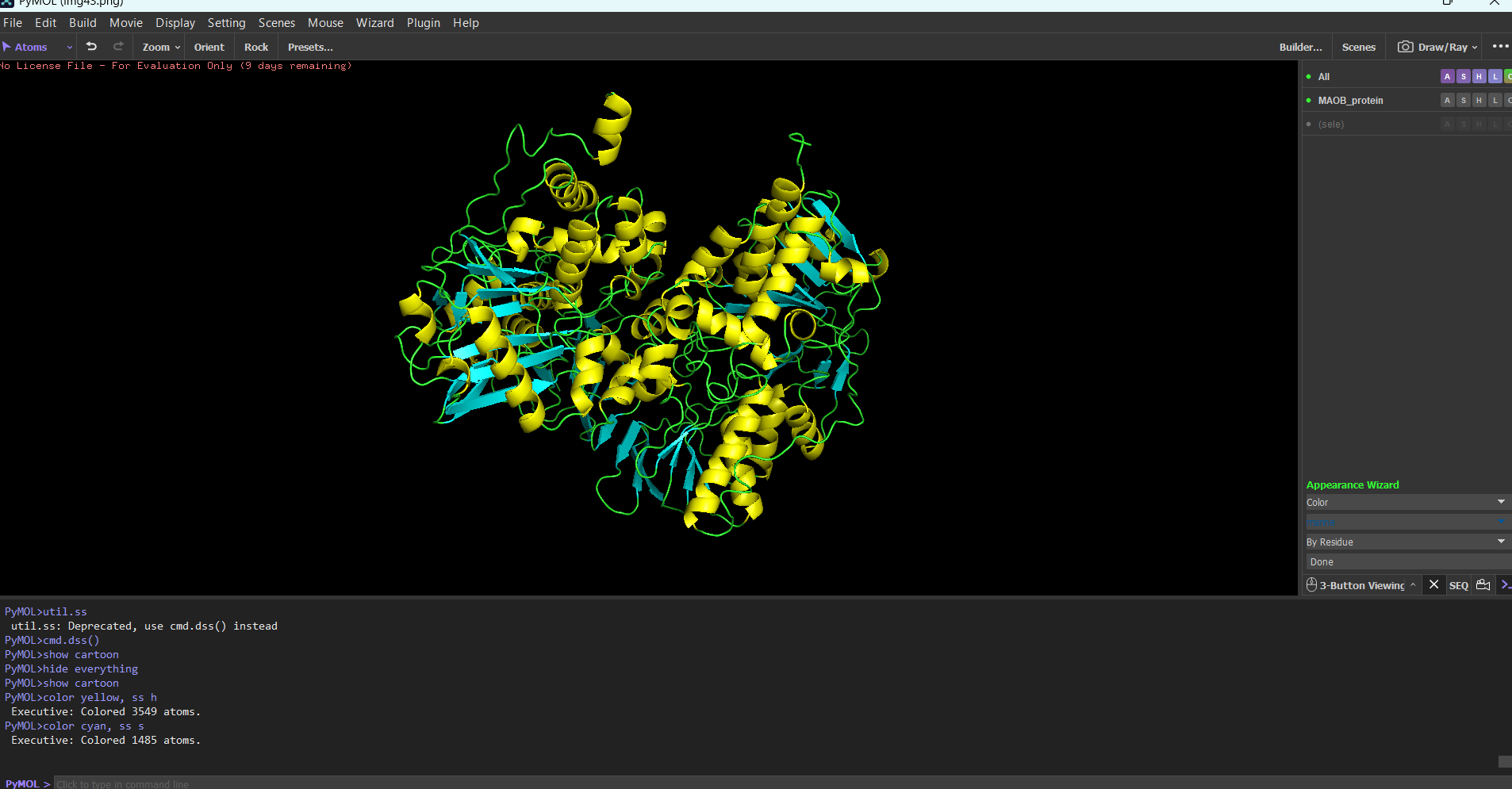
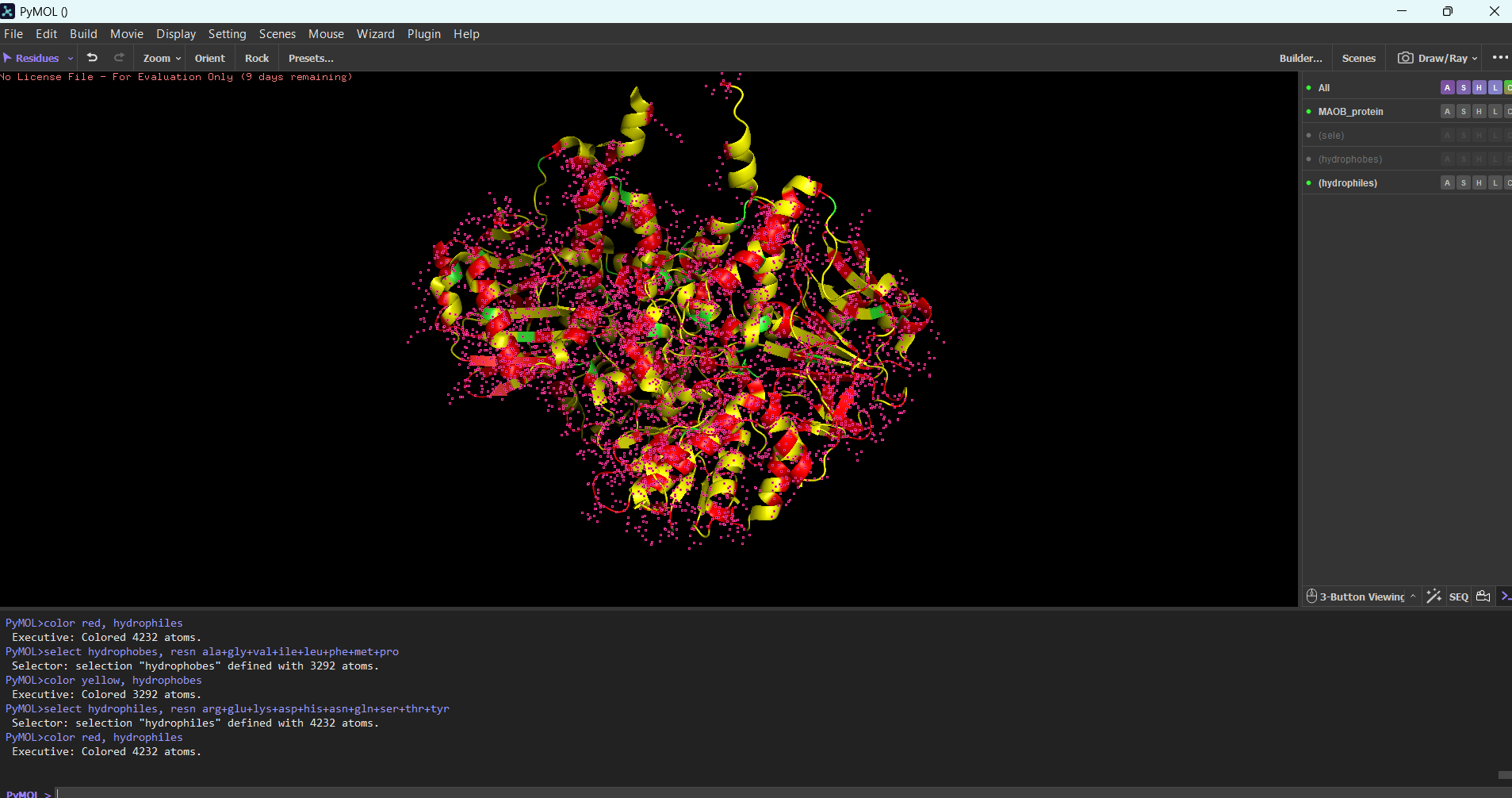
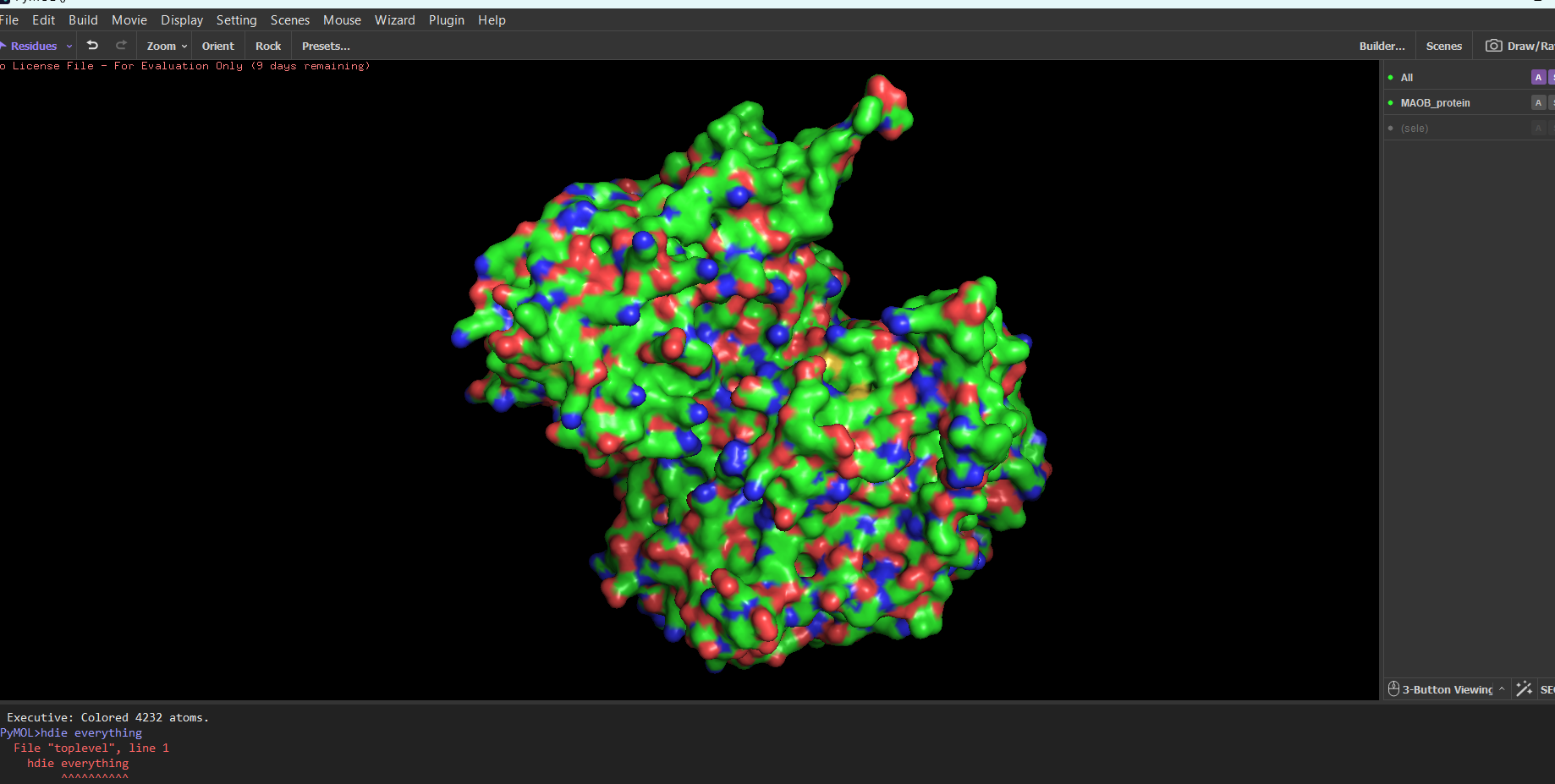
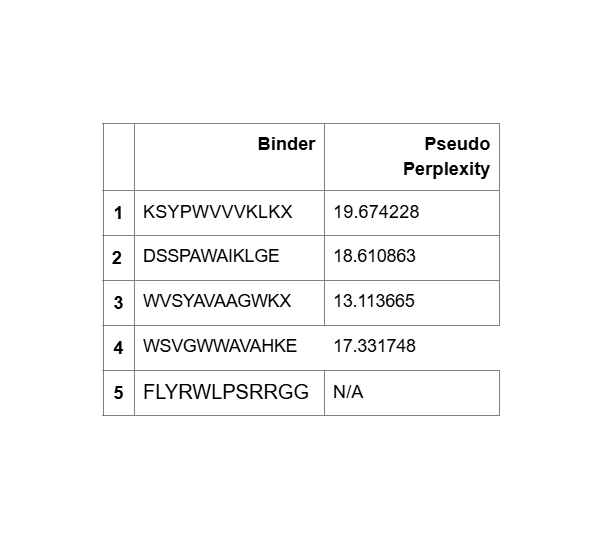
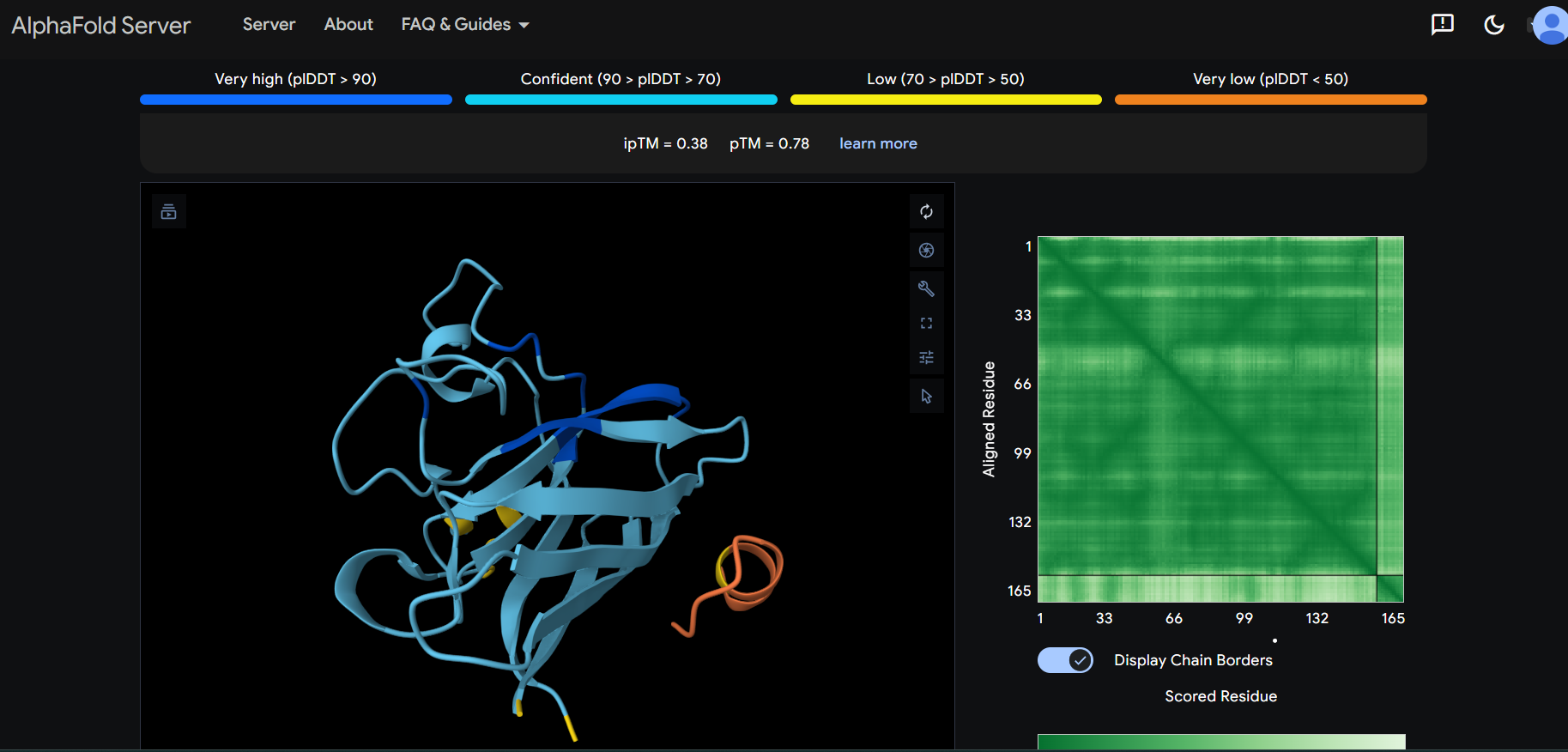
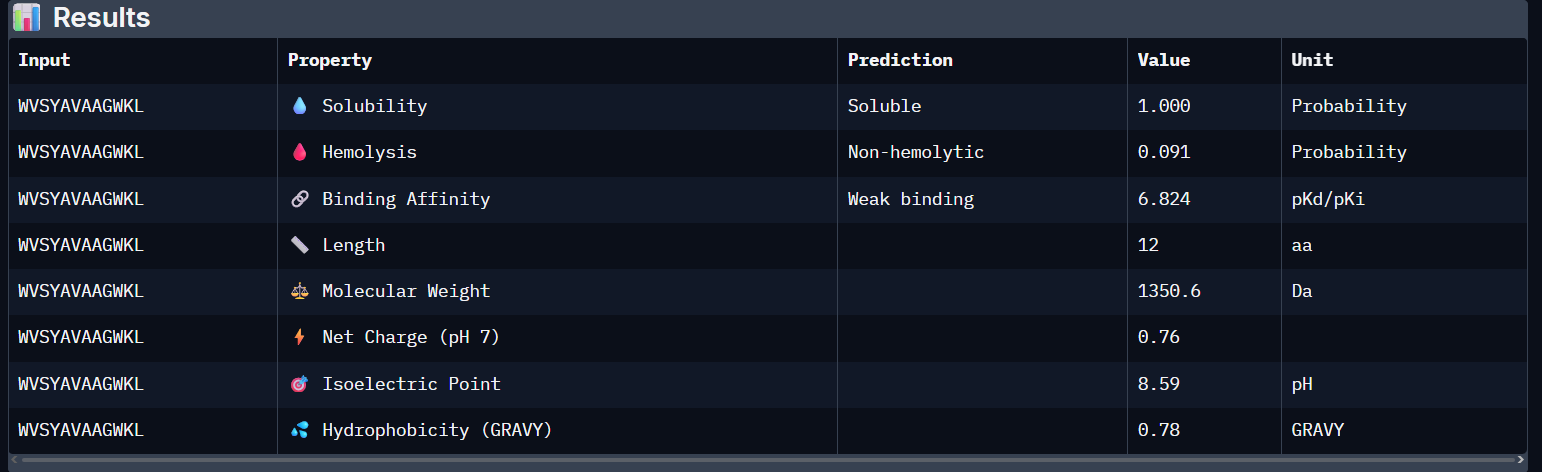
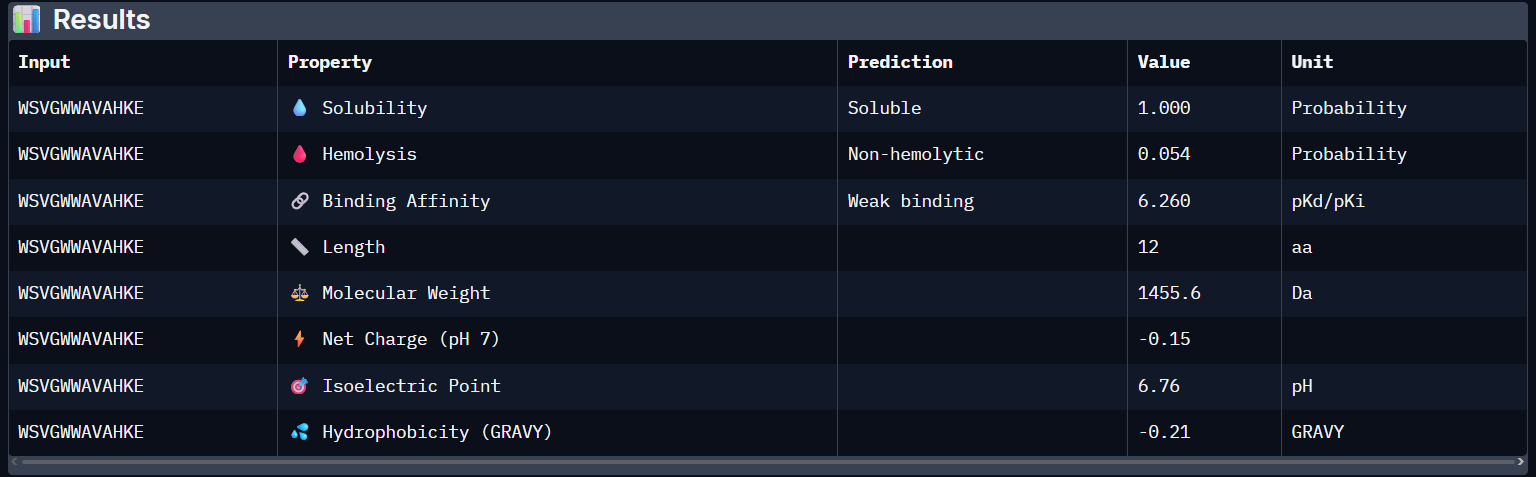
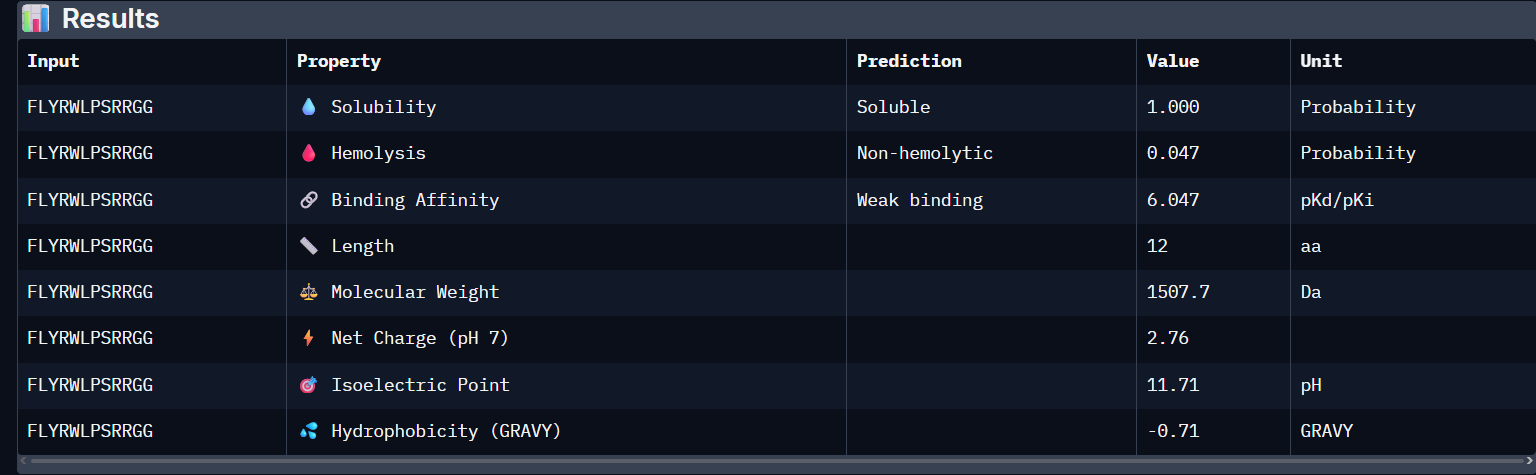
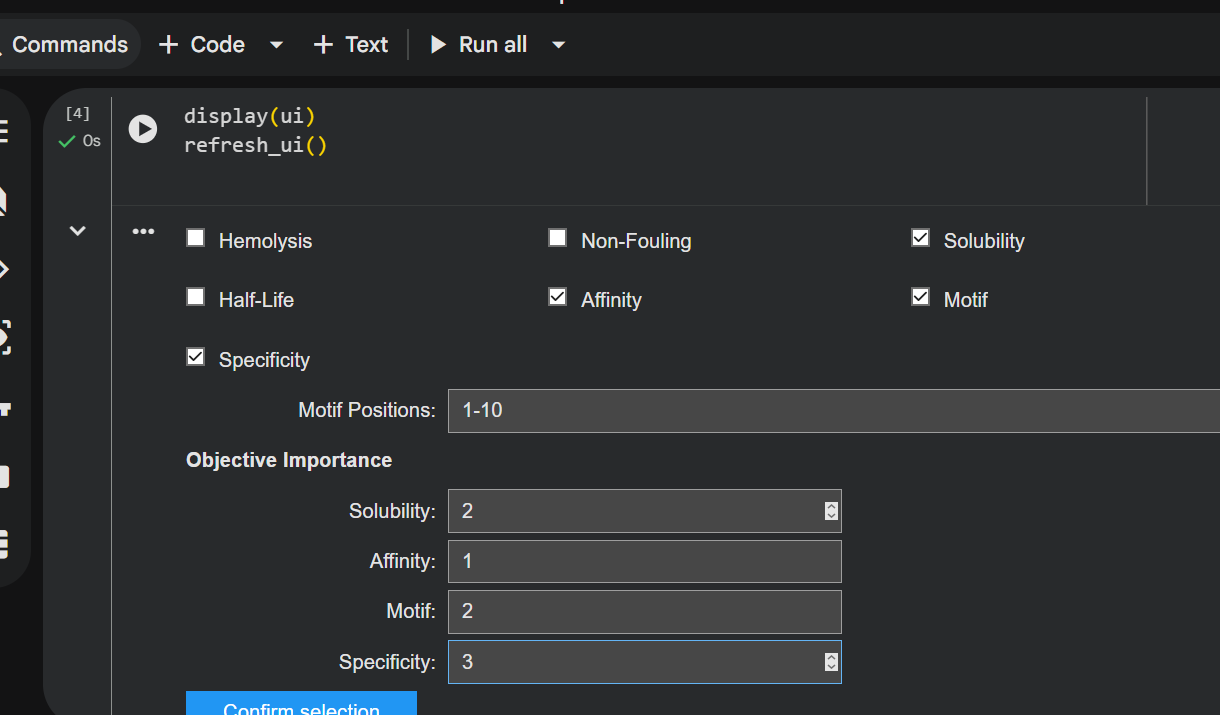
“Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
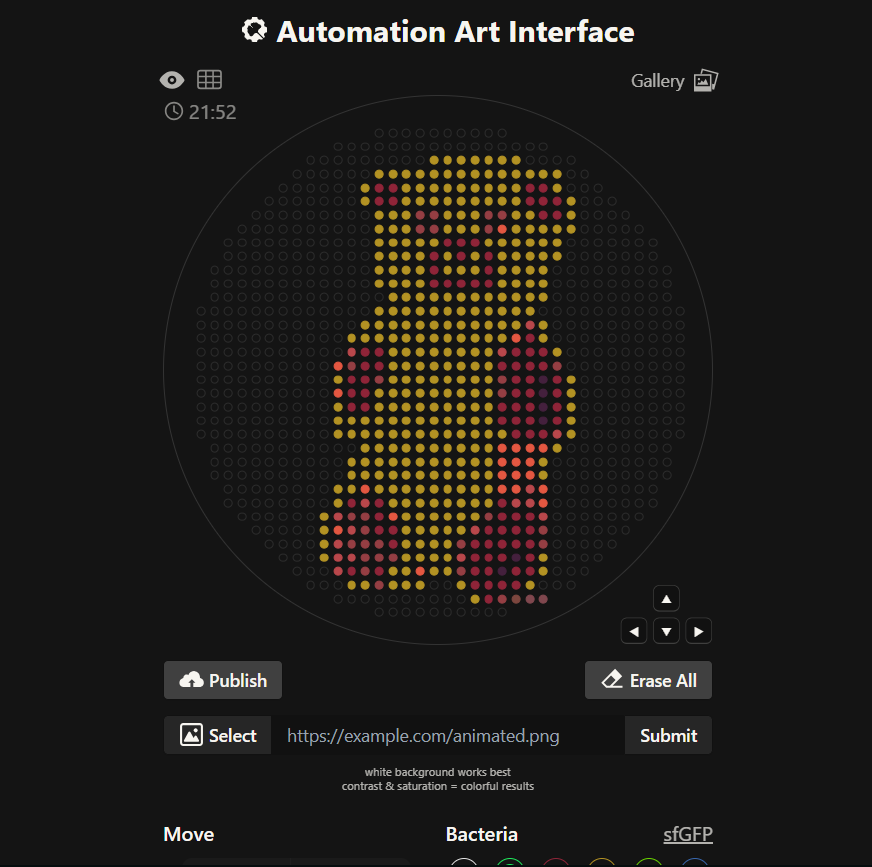
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.”
from opentrons import types
metadata = {
'author': 'Andrei Vasilan',
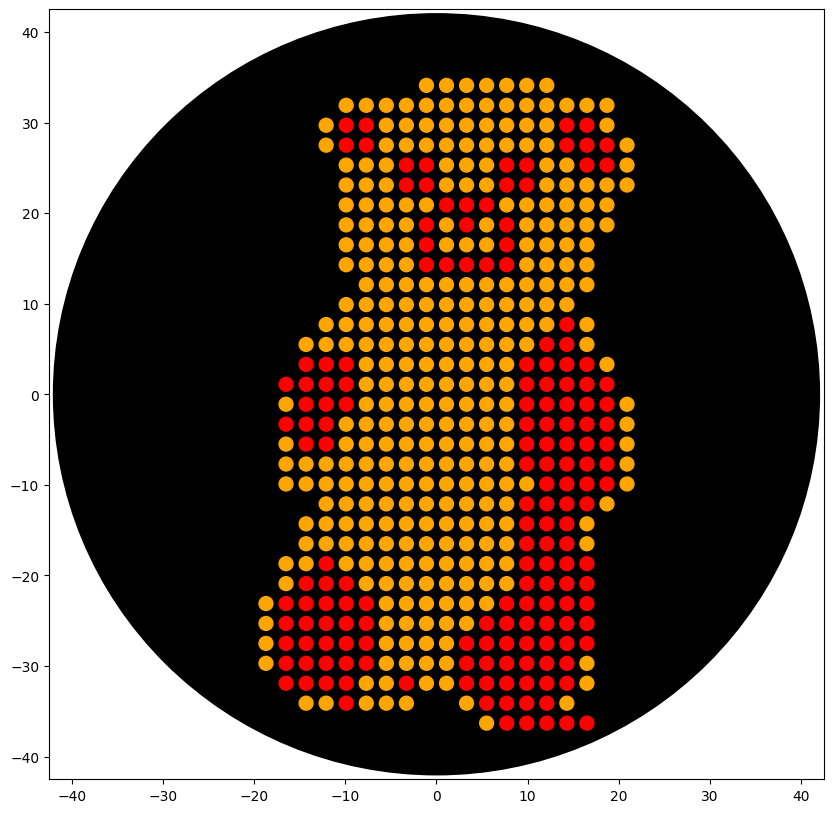
'protocolName': 'The shape of Barni',
'description': 'Patterns a full Barni shape onto an agar plate using multiple protein coordinate sets.',
'source': 'HTGAA 2026 Opentrons Lab',
'apiLevel': '2.20'
}
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
well_colors = {
'A1' : 'Red',
'B1' : 'Green',
'C1' : 'Orange'
}
def run(protocol):
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
temperature_module = protocol.load_module('temperature module gen2', COLORS_DECK_SLOT)
temperature_plate = temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul', 'Cold Plate')
color_plate = temperature_plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate')
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
def location_of_color(color_string):
for well, color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
def dispense_and_detach(pipette, volume, location):
above_location = location.move(types.Point(z=location.point.z + 5))
pipette.move_to(above_location)
pipette.dispense(volume, location)
pipette.move_to(above_location)
design_data = {
'Orange': [
(-1.1, 34.1),(1.1, 34.1),(3.3, 34.1),(5.5, 34.1),(7.7, 34.1),(9.9, 34.1),(12.1, 34.1),(-9.9, 31.9),(-7.7, 31.9),(-5.5, 31.9),(-3.3, 31.9),(-1.1, 31.9),(1.1, 31.9),(3.3, 31.9),(5.5, 31.9),(7.7, 31.9),(9.9, 31.9),(12.1, 31.9),(14.3, 31.9),(16.5, 31.9),(18.7, 31.9),(-12.1, 29.7),(-5.5, 29.7),(-3.3, 29.7),(-1.1, 29.7),(1.1, 29.7),(3.3, 29.7),(5.5, 29.7),(7.7, 29.7),(9.9, 29.7),(12.1, 29.7),(18.7, 29.7),(-12.1, 27.5),(-5.5, 27.5),(-3.3, 27.5),(-1.1, 27.5),(1.1, 27.5),(3.3, 27.5),(5.5, 27.5),(7.7, 27.5),(9.9, 27.5),(12.1, 27.5),(20.9, 27.5),(-9.9, 25.3),(-7.7, 25.3),(-5.5, 25.3),(1.1, 25.3),(3.3, 25.3),(5.5, 25.3),(12.1, 25.3),(14.3, 25.3),(20.9, 25.3),(-9.9, 23.1),(-7.7, 23.1),(-5.5, 23.1),(1.1, 23.1),(3.3, 23.1),(5.5, 23.1),(12.1, 23.1),(14.3, 23.1),(16.5, 23.1),(18.7, 23.1),(20.9, 23.1),(-9.9, 20.9),(-7.7, 20.9),(-5.5, 20.9),(-3.3, 20.9),(-1.1, 20.9),(7.7, 20.9),(9.9, 20.9),(12.1, 20.9),(14.3, 20.9),(16.5, 20.9),(18.7, 20.9),(-9.9, 18.7),(-7.7, 18.7),(-5.5, 18.7),(-3.3, 18.7),(1.1, 18.7),(5.5, 18.7),(9.9, 18.7),(12.1, 18.7),(14.3, 18.7),(16.5, 18.7),(18.7, 18.7),(-9.9, 16.5),(-7.7, 16.5),(-5.5, 16.5),(-3.3, 16.5),(1.1, 16.5),(3.3, 16.5),(5.5, 16.5),(9.9, 16.5),(12.1, 16.5),(14.3, 16.5),(16.5, 16.5),(-9.9, 14.3),(-7.7, 14.3),(-5.5, 14.3),(-3.3, 14.3),(9.9, 14.3),(12.1, 14.3),(14.3, 14.3),(16.5, 14.3),(-7.7, 12.1),(-5.5, 12.1),(-3.3, 12.1),(-1.1, 12.1),(1.1, 12.1),(3.3, 12.1),(5.5, 12.1),(7.7, 12.1),(9.9, 12.1),(12.1, 12.1),(14.3, 12.1),(16.5, 12.1),(-9.9, 9.9),(-7.7, 9.9),(-5.5, 9.9),(-3.3, 9.9),(-1.1, 9.9),(1.1, 9.9),(3.3, 9.9),(5.5, 9.9),(7.7, 9.9),(9.9, 9.9),(12.1, 9.9),(14.3, 9.9),(-12.1, 7.7),(-9.9, 7.7),(-7.7, 7.7),(-5.5, 7.7),(-3.3, 7.7),(-1.1, 7.7),(1.1, 7.7),(3.3, 7.7),(5.5, 7.7),(7.7, 7.7),(9.9, 7.7),(12.1, 7.7),(16.5, 7.7),(-14.3, 5.5),(-12.1, 5.5),(-9.9, 5.5),(-7.7, 5.5),(-5.5, 5.5),(-3.3, 5.5),(-1.1, 5.5),(1.1, 5.5),(3.3, 5.5),(5.5, 5.5),(7.7, 5.5),(9.9, 5.5),(16.5, 5.5),(-7.7, 3.3),(-5.5, 3.3),(-3.3, 3.3),(-1.1, 3.3),(1.1, 3.3),(3.3, 3.3),(5.5, 3.3),(7.7, 3.3),(18.7, 3.3),(-7.7, 1.1),(-5.5, 1.1),(-3.3, 1.1),(-1.1, 1.1),(1.1, 1.1),(3.3, 1.1),(5.5, 1.1),(7.7, 1.1),(-16.5, -1.1),(-7.7, -1.1),(-5.5, -1.1),(-3.3, -1.1),(-1.1, -1.1),(1.1, -1.1),(3.3, -1.1),(5.5, -1.1),(7.7, -1.1),(20.9, -1.1),(-9.9, -3.3),(-7.7, -3.3),(-5.5, -3.3),(-3.3, -3.3),(-1.1, -3.3),(1.1, -3.3),(3.3, -3.3),(5.5, -3.3),(7.7, -3.3),(20.9, -3.3),(-16.5, -5.5),(-9.9, -5.5),(-7.7, -5.5),(-5.5, -5.5),(-3.3, -5.5),(-1.1, -5.5),(1.1, -5.5),(3.3, -5.5),(5.5, -5.5),(7.7, -5.5),(20.9, -5.5),(-16.5, -7.7),(-14.3, -7.7),(-12.1, -7.7),(-9.9, -7.7),(-7.7, -7.7),(-5.5, -7.7),(-3.3, -7.7),(-1.1, -7.7),(1.1, -7.7),(3.3, -7.7),(5.5, -7.7),(7.7, -7.7),(20.9, -7.7),(-16.5, -9.9),(-14.3, -9.9),(-12.1, -9.9),(-9.9, -9.9),(-7.7, -9.9),(-5.5, -9.9),(-3.3, -9.9),(-1.1, -9.9),(1.1, -9.9),(3.3, -9.9),(5.5, -9.9),(7.7, -9.9),(9.9, -9.9),(20.9, -9.9),(-12.1, -12.1),(-9.9, -12.1),(-7.7, -12.1),(-5.5, -12.1),(-3.3, -12.1),(-1.1, -12.1),(1.1, -12.1),(3.3, -12.1),(5.5, -12.1),(7.7, -12.1),(18.7, -12.1),(-14.3, -14.3),(-12.1, -14.3),(-9.9, -14.3),(-7.7, -14.3),(-5.5, -14.3),(-3.3, -14.3),(-1.1, -14.3),(1.1, -14.3),(3.3, -14.3),(5.5, -14.3),(7.7, -14.3),(16.5, -14.3),(-14.3, -16.5),(-12.1, -16.5),(-9.9, -16.5),(-7.7, -16.5),(-5.5, -16.5),(-3.3, -16.5),(-1.1, -16.5),(1.1, -16.5),(3.3, -16.5),(5.5, -16.5),(7.7, -16.5),(16.5, -16.5),(-16.5, -18.7),(-14.3, -18.7),(-9.9, -18.7),(-7.7, -18.7),(-5.5, -18.7),(-3.3, -18.7),(-1.1, -18.7),(1.1, -18.7),(3.3, -18.7),(5.5, -18.7),(7.7, -18.7),(-16.5, -20.9),(-7.7, -20.9),(-5.5, -20.9),(-3.3, -20.9),(-1.1, -20.9),(1.1, -20.9),(3.3, -20.9),(5.5, -20.9),(7.7, -20.9),(-18.7, -23.1),(-5.5, -23.1),(-3.3, -23.1),(-1.1, -23.1),(1.1, -23.1),(3.3, -23.1),(5.5, -23.1),(-18.7, -25.3),(-5.5, -25.3),(-3.3, -25.3),(-1.1, -25.3),(1.1, -25.3),(3.3, -25.3),(-18.7, -27.5),(-5.5, -27.5),(-3.3, -27.5),(-1.1, -27.5),(1.1, -27.5),(-18.7, -29.7),(-5.5, -29.7),(-3.3, -29.7),(-1.1, -29.7),(1.1, -29.7),(16.5, -29.7),(-7.7, -31.9),(-5.5, -31.9),(-1.1, -31.9),(1.1, -31.9),(16.5, -31.9),(-14.3, -34.1),(-12.1, -34.1),(-7.7, -34.1),(-5.5, -34.1),(-3.3, -34.1),(3.3, -34.1),(14.3, -34.1),(5.5, -36.3)
],
'Red': [
# mRFP1
(-9.9, 29.7),(-7.7, 29.7),(14.3, 29.7),(16.5, 29.7),(-9.9, 27.5),(-7.7, 27.5),(14.3, 27.5),(16.5, 27.5),(18.7, 27.5),(16.5, 25.3),(18.7, 25.3),(1.1, 20.9),(3.3, 20.9),(5.5, 20.9),(-1.1, 18.7),(3.3, 18.7),(7.7, 18.7),(-1.1, 16.5),(7.7, 16.5),(-1.1, 14.3),(1.1, 14.3),(3.3, 14.3),(5.5, 14.3),(7.7, 14.3),(14.3, 5.5),(-12.1, 3.3),(-9.9, 3.3),(12.1, 3.3),(14.3, 3.3),(16.5, 3.3),(-12.1, 1.1),(-9.9, 1.1),(9.9, 1.1),(12.1, 1.1),(14.3, 1.1),(16.5, 1.1),(-14.3, -1.1),(-12.1, -1.1),(12.1, -1.1),(14.3, -1.1),(18.7, -1.1),(-14.3, -3.3),(-12.1, -3.3),(12.1, -3.3),(14.3, -3.3),(18.7, -3.3),(-14.3, -5.5),(-12.1, -5.5),(9.9, -5.5),(12.1, -5.5),(14.3, -5.5),(18.7, -5.5),(9.9, -7.7),(12.1, -7.7),(14.3, -7.7),(18.7, -7.7),(12.1, -9.9),(14.3, -9.9),(16.5, -9.9),(-14.3, -20.9),(-9.9, -20.9),(9.9, -20.9),(-9.9, -23.1),(9.9, -23.1),(12.1, -23.1),(14.3, -23.1),(-9.9, -25.3),(-7.7, -25.3),(7.7, -25.3),(9.9, -25.3),(12.1, -25.3),(14.3, -25.3),(-7.7, -27.5),(5.5, -27.5),(7.7, -27.5),(9.9, -27.5),(12.1, -27.5),(14.3, -27.5),(5.5, -29.7),(7.7, -29.7),(9.9, -29.7),(14.3, -29.7),(-14.3, -31.9),(-9.9, -31.9),(5.5, -31.9),(7.7, -31.9),(12.1, -31.9),(14.3, -31.9),(5.5, -34.1),(7.7, -34.1),(9.9, -34.1),(12.1, -34.1),(7.7, -36.3),
# mRuby2
(-3.3, 25.3),(-1.1, 25.3),(7.7, 25.3),(9.9, 25.3),(-3.3, 23.1),(-1.1, 23.1),(7.7, 23.1),(-14.3, 1.1),(18.7, 1.1),(-9.9, -1.1),(9.9, -1.1),(9.9, -3.3),(-14.3, -23.1),(-12.1, -23.1),(7.7, -23.1),(-12.1, -25.3),(16.5, -25.3),(-14.3, -27.5),(-12.1, -27.5),(-9.9, -27.5),(3.3, -27.5),(16.5, -27.5),(-12.1, -29.7),(-9.9, -29.7),(-7.7, -29.7),(-12.1, -31.9),(3.3, -31.9),(-9.9, -34.1),(9.9, -36.3),
# mLychee_TF
(9.9, 23.1),(12.1, 5.5),(-16.5, 1.1),(-16.5, -3.3),(9.9, -12.1),(12.1, -12.1),(14.3, -12.1),(16.5, -12.1),(9.9, -14.3),(12.1, -14.3),(14.3, -14.3),(9.9, -16.5),(12.1, -16.5),(14.3, -16.5),(-12.1, -18.7),(9.9, -18.7),(12.1, -18.7),(16.5, -18.7),(16.5, -20.9),(-7.7, -23.1),(-16.5, -25.3),(-3.3, -31.9),
# mScarlet_I
(14.3, 7.7),(-14.3, 3.3),(9.9, 3.3),(18.7, -9.9),(14.3, -18.7),(-12.1, -20.9),(12.1, -20.9),(14.3, -20.9),(-16.5, -23.1),(16.5, -23.1),(-14.3, -25.3),(5.5, -25.3),(-16.5, -27.5),(-16.5, -29.7),(-14.3, -29.7),(3.3, -29.7),(-16.5, -31.9),
# mKate2
(16.5, -1.1),(16.5, -3.3),(16.5, -5.5),(16.5, -7.7),(12.1, -29.7),(9.9, -31.9),
# dsRed
(12.1, -36.3),
# tagRFP
(14.3, -36.3),(16.5, -36.3)
]
}
for color_name, points in design_data.items():
if not points:
continue
pipette_20ul.pick_up_tip()
for i, (x, y) in enumerate(points):
# Aspirate in 20uL batches
if i % 20 == 0:
volume_to_aspirate = min(20, len(points) - i)
pipette_20ul.aspirate(volume_to_aspirate, location_of_color(color_name))
target_location = center_location.move(types.Point(x=x, y=y))
dispense_and_detach(pipette_20ul, 1, target_location)
pipette_20ul.drop_tip()
Paper name: “BOTany Methods: Accessible Automation for Plant Synthetic Biology” by Mingqi Xie and Qian de la Tour (bioRxiv, August 22, 2025)
Short description: democratizing access to laboratory automation in plant synthetic biology by developing a suite of modular protocols called BOTany Methods, which leverage the affordable Opentrons OT-2 liquid-handling robot to streamline common molecular biology workflows. The authors highlight how traditional biofoundries are costly and inaccessible, limiting high-throughput experimentation, reproducibility, and the Design-Build-Test-Learn (DBTL) cycle in plant science. By using the OT-2, they enable automation for tasks that are typically manual and error-prone, making them suitable for educational settings and nre researchers.
For my final project, I plan to automate the development and testing of an aptamer-based electrochemical biosensor integrated into a portable electrical device for automatic detection of heavy metals in water samples. The biosensor will use DNA aptamers as the biorecognition element, immobilized on gold screen-printed electrodes, to bind target metals and produce measurable changes in electrical impedance or current. This will be integrated into a compact, Arduino-based device with a microfluidic channel for sample flow, enabling automatic sensing: water is drawn in via a pump, interacts with the biosensor, and triggers real-time readout via a potentiostat module, with data logged to a cloud app for alerts if thresholds are exceeded. The goal is to create a low-cost, field-deployable system for environmental monitoring, optimizing aptamer sequences and immobilization for high sensitivity (sub-ppb levels).
I’ll use the Opentrons OT-2 for local automation to handle high-throughput screening of aptamer variants and electrode functionalization, while leveraging Ginkgo Nebula’s cloud lab for large-scale DNA synthesis and initial validation. Specifically:
Aptamer design and synthesis: Design aptamer libraries targeting heavy metals using tools like SELEX simulation software, then submit to Ginkgo Nebula for automated synthesis and amplification. Nebula’s high-throughput platform can produce 96-384 variants in plates, shipped for local use.
Local automation with Opentrons: The OT-2 will prepare functionalization solutions (e.g., thiol-modified aptamers mixed with linkers), deposit them precisely onto electrode arrays in a 96-well format for immobilization, and perform washing/incubation steps. It will also simulate testing by dispensing metal-spiked water samples and cofactors, allowing batch optimization of binding conditions.
If I were to choose these subject for my final project I would research a way for how we could use engineered viruses (VLPs) to inject proteins (not nucleic acid) into the cristallin cells. The proteins injected can range from proteases to any protein that can help the crystallin become clear again. Or if the virus method fails I would most likely try using perforin or similar substances to introduces the proteins in the cell and out of the cell.