Part 0: Basics of Gel Electrophoresis I watched the lectures by George Church, Joe Jacobson, and Emily Leproust, as well as the recitation by Ice Kiattisewee on DNA Gel, restriction enzymes, Benchling, and Twist.
Key takeaways from lectures:
Gel electrophoresis works by taking advantage of the negative charge of the DNA molecule due to the phosphate group. DNA fragments being placed in the agarose gel (which is heated and then cooled) move according to their size under the effect of the electric field applied, separating based on their size. The pore size of the agarose gel is determined by the concentration of agarose - (with higher concentrations –> decreased pore size –> smaller fragments/finer fragments being separated easily. The ladder containing the weight of the DNA in kDa helps in estimating the size of the DNA based on its position in the gel. Ethidium bromide is used for staining resulting in the DNA to glow under the UV light.
Part 1: Opentrons Agar Art - Biohazard Symbol Design Concept I conceptualised the biohazard symbol, which I thought of creating using parametric algorithms. I used the assistance of Claude (Anthropic) to generate the python code which I ran on Google Colab, in the copy provided by HTGAA. The results are shown below.
Week 4: Homework- Protein Design Part-1 Weekly Assignment Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
Figure 1: The medicine ward in a typical government Indian hospital. The one shown is the medicine ward at my medical college.
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
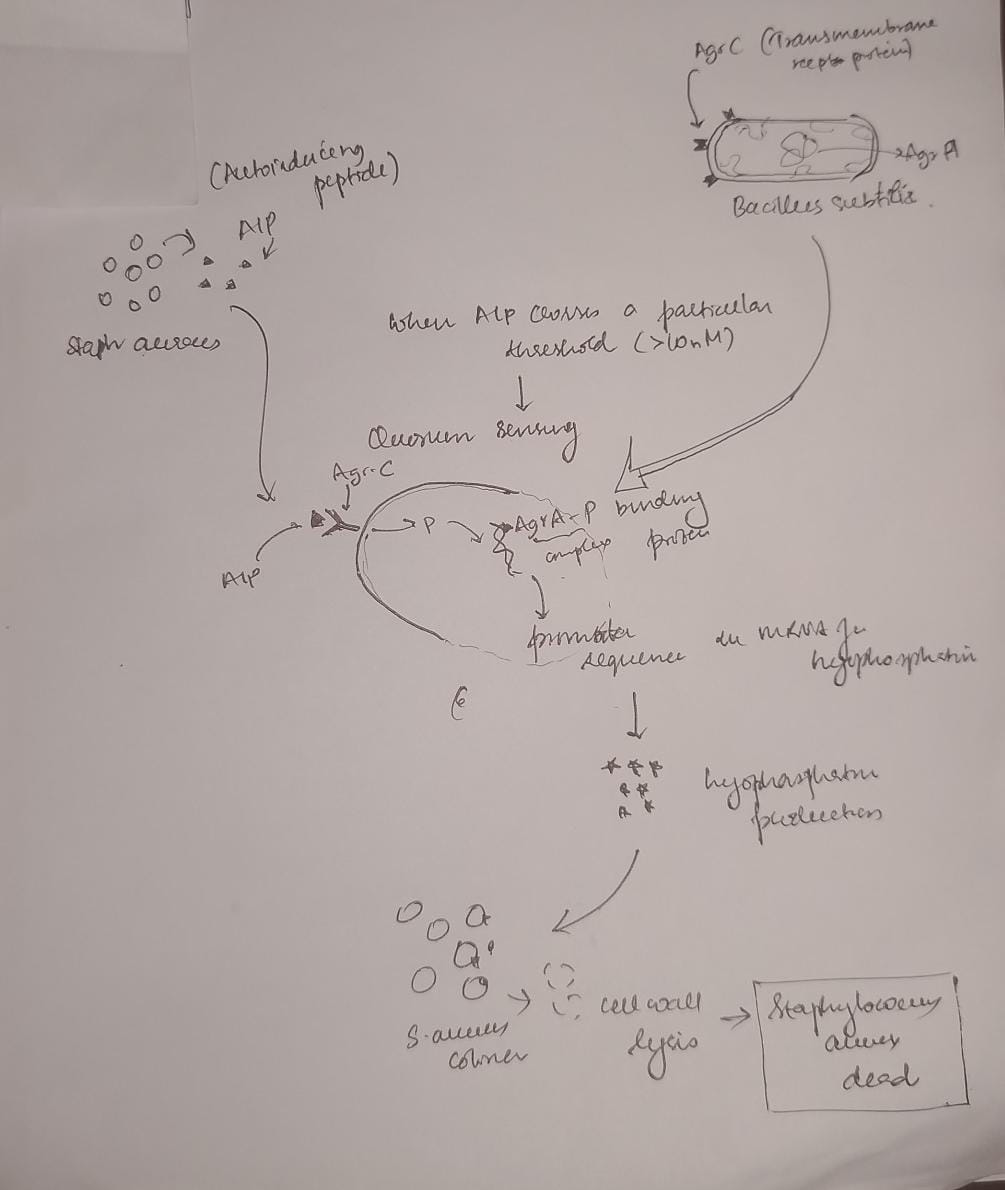
Although I started off with imparting neuron-like properties in mycelial networks I decided to pivot to solving nosocomial infections with synthetic biology as the immediate benefits outweigh that of the former. In the wards , especially in under-resourced settings where I am from, maintenance of sterility and aseptic conditions in the post surgery wards and medicine wards is an under addressed issue. Under staffed , under resourced settings like mine, do not prioritise it leading to hospital acquired infections in the patients, often resulting in severe complications than which they came to the hospital for. With living colonisers that produce bactericidal and bacteriostatic chemicals (including fungicidal and antiviral) that generally cause secondary infections and HAIs(hospital acquired infections),synthetic biology has the scope to solve this problem saving over 100,000 lives in India alone, where people die of these HAIs. Low maintenance systems like these could change the way a lot of people are recovering during treatment in the wards. I would like to explore and build something tangible with Bacillus subtilis as the chassis organism , with quorum sensing to produce antimicrobial peptides against the priority pathogens causing hospital acquired infections
Figure 2: A diagram showing an example of the seek and kill mechanism in the engineered living surfaces. Here Bacillus subtilis acts as the chasis organism carrying the gene for Lysostaphin which is activated only on concentration of AIP produced by S. aureus crossing a certain threshold. This comprises of the sensor (Agr-C transmembrane protein in the genetically engineered B. subtilis), the logic gate(quorum) and the effector module (Lysostaphin produced by mRNA for the same activated by Agr-P protein)
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Goal 1: Biosafety and Containment
Prevent uncontrolled spread of engineered colonizers beyond designated hospital surfaces.
Prevent horizontal gene transfer of engineered antimicrobial genes to environmental organisms.
Prevent environmental contamination by these engineered organisms.
Goal 2: Patient Safety
Ensure the colonizer produces no toxins or allergens harmful to immunocompromised patients.
Ensure colonizers cannot cause opportunistic infections in vulnerable populations.
Goal 3: Equitable Access
Keep unit cost below existing chemical disinfectant protocols.
Ensure technology is deployable without specialized infrastructure (cold chain, trained personnel).
Open-source genetic constructs and protocols to prevent IP monopolization.
Goal 4: Environmental Safety
Prevent the colonizer from establishing in non-target environments (soil, water systems, community surfaces outside hospitals).
Ensure engineered antimicrobial peptide genes do not contribute to resistance development in wild microbial populations.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”
Governance Actions
Action 1 - Regulatory: Establishing a Regulatory Authority for Engineered Living Surfaces in Healthcare
Purpose: Currently, India’s Genetic Engineering Appraisal Committee (GEAC) regulates environmental release of GMOs under the Environment Protection Act 1986, but no framework specifically addresses engineered microorganisms deployed on surfaces within healthcare infrastructure. Hospital surfaces occupy a grey zone-they are neither open environmental release nor contained laboratory use-and no existing regulatory body in India is equipped to evaluate the safety, efficacy, or deployment protocols of living antimicrobial surface systems in clinical settings.
Design: I propose that ICMR (Indian Council of Medical Research) and CDSCO (Central Drugs Standard Control Organisation), along with GEAC, establish a joint Advisory Board specifically to evaluate proposals for engineered microorganisms intended for deployment within healthcare infrastructure. This board would scrutinize applications for biosafety, patient safety, and environmental containment, and approve relevant proposals. Funding for development and deployment could be channeled through existing government mechanisms such as BIRAC (Biotechnology Industry Research Assistance Council). International precedent exists: the EU’s Contained Use Directive for GMOs could serve as a template adaptable to India’s regulatory landscape.
Assumptions: This assumes ICMR, CDSCO, and GEAC have the institutional capacity, funding, and political will to implement a new advisory framework. It also assumes that a centralized national framework can accommodate the enormous diversity of hospital settings across India-from tertiary urban centers to rural primary health centers-without becoming either too rigid or too permissive.
Risks of Failure & “Success”:
Failure: The approval process takes 5–10 years, during which no legal deployment pathway exists. This pushes hospitals toward unregulated, informal use of engineered organisms with no safety oversight-paradoxically increasing risk. Meanwhile, over 1,000,000 people continue to die each year from HAIs in India alone.
Success: The regulatory pathway, once established, becomes a barrier to entry that only large, well-resourced biotech companies can navigate. These companies gain outsized influence over the advisory committee’s decisions, creating a commercial monopoly over engineered living surfaces and undermining equitable access.
Action 2 - Technical: Engineering Auxotrophic Dependencies for Biocontainment
Purpose: Currently, commercially available probiotic cleaning products use wild-type organisms with no engineered containment mechanisms. If these organisms spread beyond their intended surfaces, there are no built-in safeguards. I propose building auxotrophic dependencies into the B. subtilis chassis-engineering the organism to require a synthetic amino acid or nutrient not found in nature-so the colonizer cannot survive outside the hospital environment where that nutrient is actively supplied.
Design: Academic synthetic biologists would develop and characterize the auxotrophic strains, validated by Institutional Biosafety Committees (IBCs). Standardized, well-characterized constructs would be deposited in repositories like Addgene or the iGEM Registry of Standard Biological Parts. Hospitals deploying the system would need to maintain a supply of the synthetic nutrient, which could be incorporated into routine surface treatment protocols. Redundant auxotrophies (requiring multiple synthetic nutrients simultaneously) would reduce the probability of reversion to environmental viability.
Assumptions: This assumes we can effectively engineer genetic circuits with auxotrophic dependencies that reliably prevent survival outside the defined environment. It assumes reversion rates through mutation remain low enough to be safe-though published data on single auxotrophies in B. subtilis suggest reversion is non-trivial, making redundant safeguards essential. It also assumes that the hospital environment can be consistently maintained to support the colonizer’s engineered nutritional requirements.
Risks of Failure & “Success”:
Failure: Mutations in the engineered organisms bypass the installed auxotrophic dependencies, allowing survival outside the intended environment. A containment breach at scale could result in mass environmental contamination by antimicrobial-peptide-producing organisms, which becomes extremely difficult to remediate.
Success: The auxotrophic dependency creates a new commercial bottleneck. Companies that hold patents on the specific synthetic nutrient required by the organism could monopolize the supply chain, establishing a cycle of dependence-you get the open-source organism for free, but you cannot use it without buying the proprietary nutrient. This undermines democratized access despite open-source genetic designs.
Action 3 - Incentive: Open-Source Consortium for Engineered Living Surfaces
Purpose: Currently, engineered antimicrobial surface organisms are being developed in isolated academic labs (ETH Zurich, MIT Media Lab) and private companies, with no standardized sharing of constructs, safety data, or deployment protocols. Patents and IP held exclusively by these institutions greatly hinder the development and distribution of this technology in under-resourced settings where it is needed most. Akin to how the Linux operating system emerged from the collective efforts of thousands of developers and disrupted proprietary software, I propose an international open-access repository modeled on iGEM’s Registry of Standard Biological Parts-specifically for engineered surface colonizers-where anyone can contribute designs, genetic circuits, safety profiles, and deployment protocols.
Design: Requires seed funding from organizations such as the WHO, Wellcome Trust, or Gates Foundation. Participating labs deposit genetic constructs, characterized safety profiles, and validated deployment protocols into the repository. In exchange, they receive access to the full library and structured co-authorship frameworks that incentivize contribution. WHO prequalifies validated strains for deployment in low-resource countries, creating a fast-track regulatory pathway analogous to its Essential Medicines prequalification programme. This decentralized model would accelerate development and acceptance of engineered living surfaces across diverse healthcare settings globally.
Assumptions: This assumes academic labs and companies will share constructs and protocols openly, which conflicts with current publication-priority and profit-driven incentive structures in science and industry. It assumes WHO has the institutional capacity to manage a prequalification programme for engineered organisms, which it currently does not. It also assumes that sufficient contributors will participate to reach a critical mass of useful, well-characterized parts.
Risks of Failure & “Success”:
Failure: No one contributes, and the repository remains empty-a common fate of open-source initiatives without strong network effects or institutional mandates.
Success: Open access enables actors with malicious intent to access engineered organisms and their protocols. However, the constructs in question are surface colonizers, not pathogens, which limits their weaponization potential compared to other synthetic biology tools. A more realistic success risk is that unvetted, poorly characterized constructs get deployed in hospitals by groups without adequate biosafety expertise, causing harm that discredits the entire approach.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Scoring Matrix
*Scored 1–3 (1 = best/strongest contribution, 3 = weakest/least contribution, n/a = not applicable)
Does the option:
Action 1: Regulatory Authority
Action 2: Auxotrophic Dependencies
Action 3: Open-Source Consortium
1. Biosafety/Containment
a)Prevent uncontrolled spread
1
1
3
b)Prevent horizontal gene transfer
2
1
3
c)Prevent environmental contamination
2
1
3
2. Patient Safety
a)No toxins/allergens to immunocompromised
1
2
3
b)Cannot cause opportunistic infection
1
2
3
3. Equitable Access
a)Cost below chemical disinfectants
3
2
1
b)Deployable without specialized infrastructure
3
2
1
c)Open-source constructs
3
2
1
3. Environmental Safety
a)No establishment in non-target environments
2
1
3
b)No contribution to resistance development
2
1
3
4. Other Considerations
a)Minimizing costs and burdens to stakeholders
3
2
1
b)Feasibility
2
2
2
c)Not impede research
3
2
1
d)Promote constructive applications
2
2
1
Total
30
23
30
Action 2 (Technical) scores best overall (loweest score is the best) as it directly addresses the safety and containment goals.
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Audience: Indian Council of Medical Research (ICMR)
I recommend a combination of all three governance actions, implemented in a phased sequence, because no single action adequately addresses all four policy goals.
Phase 1 : Technical safeguards first. The auxotrophic dependencies (Action 2) should be engineered and validated before any deployment is considered. This is non-negotiable. Without built-in biocontainment, no regulatory framework or open-source initiative can compensate for an organism that escapes its intended environment. Redundant auxotrophies should be the minimum standard.
Phase 2 : Open-source consortium in parallel. The open-source repository (Action 3) should be established concurrently with technical development, ensuring that safety data, construct characterization, and deployment protocols are shared from the beginning rather than locked behind institutional IP. This directly addresses the equitable access goals and prevents the commercial monopoly risks identified in Actions 1 and 2.
Phase 3 : Regulatory framework informed by real data. The regulatory authority (Action 1) should be established after sufficient technical and safety data exists from the first two phases. This prevents the common failure mode of regulating a technology before it is understood, which leads to either overly restrictive or dangerously permissive frameworks.
Key trade-offs: Speed versus safety remains the central tension. Moving too fast with deployment risks a containment failure that could set back the entire field of engineered living surfaces by a decade—eroding public trust in synthetic biology. Moving too slowly means people continue to die from preventable infections in hospitals that lack the resources for conventional sterility maintenance. The phased approach attempts to balance this by building safety into the organism first, sharing knowledge openly second, and formalizing regulation third.
Uncertainties: It remains unclear whether auxotrophic containment can be made robust enough at scale, whether open-source incentives can overcome academic publishing pressures, and whether Indian regulatory bodies can develop new frameworks within a reasonable timeframe. These uncertainties should be revisited as the technology matures.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
This week’s exploration of governance frameworks surfaced several ethical concerns that were new to me.
The first is the tension between speed and safety in resource-constrained settings. In well-funded Western hospitals, the cost of waiting for perfect regulation is inconvenience. In under-resourced Indian hospitals where I am training, the cost of waiting is measured in lives. This creates an ethical pressure to deploy before full safety validation ,a pressure I now recognize must be resisted, because a single high-profile failure would destroy trust in this entire approach and harm far more people long-term.
The second is the dual-use nature of open-source biology. I initially assumed open-sourcing was unambiguously good democratizing access, preventing monopoly. But the governance analysis forced me to consider that open access also means open access for actors without adequate biosafety training or with malicious intent. Akin to the quacks without formal medical education who practice in remote and under resourced areas causing the patients more damage and sometimes death. The realistic risk is not bioweapons (surface colonizers are poor candidates), but rather poorly characterized organisms deployed without adequate containment by well-meaning but under-equipped groups.
The third and most personally challenging is the realization that the success of this technology could create new forms of dependency even while solving the original problem. If the auxotrophic nutrient becomes a patented commercial product, under-resourced hospitals could end up dependent on supply chains they cannot control or afford. This mirrors the broader pattern I have seen in Indian healthcare, where solutions designed for equity are captured by commercial interests. Governance must anticipate this from the beginning, not as an afterthought.
Week 2 Lecture Prep
Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error rate: The raw “bit-flip” rate of a standard DNA Polymerase is roughly $10^{-5}$ (1 in 100,000). With intrinsic proofreading (3’ → 5’ exonuclease activity), it drops to ≈ $10^{-7}$.
The comparison to the human genome : The human genome is ≈ $3.2 \times 10^9$ base pairs (3.2 Gigabytes of code).
The way biology deals with this discrepancy: A post processing algorithm called MMR is run to fix these errors. This brings the final error rate to ≈ $10^{-9}$ , which is around 3 errors per cell division which is low enough to preserve genetic code but high enough to trigger evolution- an optimal signal to noise ratio.
How many different ways are there to code for an average human protein? Why don’t they all work?
The Coding Space: Due to codon degeneracy (61 codons for 20 amino acids), there are roughly $3N$ combinations for a protein of length $N$. For an average 400-amino acid protein, this is $3{400}$ (approx $10^{190}$)
Why most “codes” fail (Optimization constraints):
Codon Bias: The cell has a limited “RAM” of specific tRNAs. Rare codons cause ribosomal stalling because the hardware isn’t available.
mRNA Structure: Bad sequences can fold into tight hairpins, jamming the ribosome like a paper shredder.
GC Content: If G/C is too high (>70%) or low, the DNA becomes structurally unstable or difficult to replicate
Homework Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite Chemistry: A solid-phase synthesis method that builds DNA strands nucleotide by nucleotide on a support matrix (like a silicon chip or column).
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Yield Decay: Synthesis is an exponential decay function. With 99.5% efficiency per step, a 200nt strand has a yield of $0.995^{200} \approx 36%$. Beyond this, the “noise” (truncated sequences and side reactions) drowns out the signal, making purification inefficient.
Why can’t you make a 2000bp gene via direct oligo synthesis?
The Yield Cliff: The math makes it impossible: $0.995^{2000} \approx 0.004%$. You cannot “print” a gene this long atom-by-atom because the yield is effectively zero.
The Solution: We switch from “printing” to “assembly.” We synthesize short tiles (e.g., 200bp) and stitch them together using methods like Gibson Assembly or Golden Gate Assembly.
Homework Question from George Church
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The Flaw: The Jurassic Park logic was security theater. Since all animals (including humans) are Lysine auxotrophs (we cannot produce it), a strictly Lysine-depeddent dinosaur would simply survive by eating chickens, soy, or humans—sources rich in Lysine.
True containment requires engineering dependence on a Non-Canonical Amino Acid (ncAA)—a synthetic building block not found in nature. This ensures “Zero Leakage” because the organism cannot find the nutrient in the wild.
Citation: I used Gemini and Claude to stress test my ideas and refine them for the sections on auxotrophy All governance analysis, clinical observations, and project framing are my own work..
week-02-hw-dna-read-write-and-edit
Part 0: Basics of Gel Electrophoresis
I watched the lectures by George Church, Joe Jacobson, and Emily Leproust, as well as the recitation by Ice Kiattisewee on DNA Gel, restriction enzymes, Benchling, and Twist.
Key takeaways from lectures:
Gel electrophoresis works by taking advantage of the negative charge of the DNA molecule due to the phosphate group. DNA fragments being placed in the agarose gel (which is heated and then cooled) move according to their size under the effect of the electric field applied, separating based on their size. The pore size of the agarose gel is determined by the concentration of agarose - (with higher concentrations –> decreased pore size –> smaller fragments/finer fragments being separated easily. The ladder containing the weight of the DNA in kDa helps in estimating the size of the DNA based on its position in the gel. Ethidium bromide is used for staining resulting in the DNA to glow under the UV light.
I found the concept of using gel electrophoresis to create art interesting . The artwork done by the previous students was visually stunning. I felt that the benchling software is a really elegant tool to edit , add DNA sequences and simulate digestion of the genomes using restriction enzymes. I believe that I have not fully explored the potential it holds.
Part 1: Benchling & In-Silico Gel Art
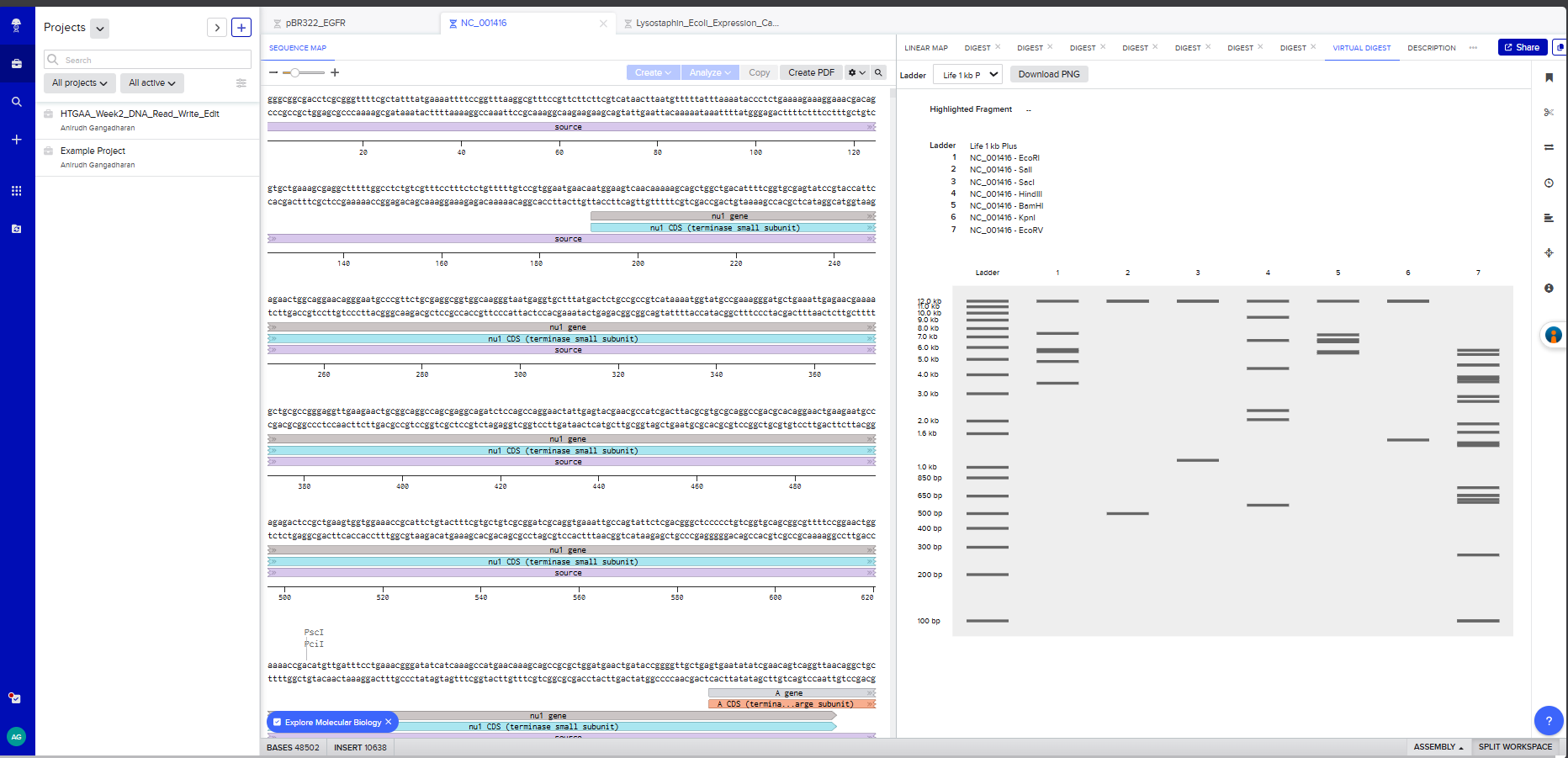
Restriction Enzyme Digestion of Lambda DNA (48,502 bp)
I created a Benchling account and imported the Lambda phage DNA sequence (48,502 bp). I then simulated restriction enzyme digestion with each of the seven enzymes specified:
Screenshot 1: The imported Lambda DNA sequence in Benchling showing the circular/linear map.
Screenshot 2: The EcoRI digest result showing the gel simulation + fragment table.
Screenshot 3: A multi-enzyme digest showing all 7 enzymes side by side on the simulated gel
Gel Art Design
On benchling , I virtually digested the DNA in 7 lanes with the following enzymes - EcoRI, SalI, SacI, HindIII, BamHI, KpnI, and EcoRV. I used Ronan’s website to iterate a few, but couldn’t effectively find a way to create the “HAI” pattern that I had envisioned.
Note: As a committed listener without wet-lab access, I completed this as an in-silico exercise.
Part 2: Gel Art - Wet Lab
As a committed listener at the London HTGAA node without direct wet-lab access for this week’s protocol, Part 2 is optional. The in-silico design in Part 1 serves as my documentation.
Lysostaphin is a zinc metalloenzyme derived from S. simulans acting against S. aureus. The gene for production of lysostaphin can be inserted via a plasmid vector into B. subtilis which is a commonly used chassis organism by biotechnologists. (https://www.mdpi.com/1424-8247/3/4/1139)
The other proteins I had in mind were PlyKp104 and PlyCD(1-174). PlyKp104 is an endolysin derived from Klebsiella pneumoniae phage which can kill Klebsiella pneumoniae and Pseudomonas aeuroginosa. (https://pubmed.ncbi.nlm.nih.gov/37098944). Similarly PlyCD (1-174) has endolytic activity on Clostridium difficile species. It’s derived from a prophage targeting the same. (https://pmc.ncbi.nlm.nih.gov/articles/PMC4649177.)
Why I chose this protein:
For my project that I had in mind - engineering living surfaces or biofilms to kill the common causative organisms causing nosocomial/HAIs (Hospital acquired infections) – the expression of these proteins I mentioned above (Lysostaphin, PlyKp104 and PlyCD) by Bacillus subtilis (chassis organism) are needed for the bactericidal action the bacteria causing HAIs.
S. aureus, including methicillin-resistant strains (MRSA), is among the most dangerous nosocomial pathogens. In the medicine wards of my medical college in India, I have seen patients admitted for routine conditions develop life-threatening staphylococcal secondary infections due to inadequate surface sterility. Lysostaphin is a potent weapon against precisely this pathogen.
The mature enzyme has two functional domains: an N-terminal catalytic domain (residues 1–148, M23 family zinc metalloendopeptidase) responsible for pentaglycine cleavage, and a C-terminal cell wall targeting domain (CWT, residues 149–246, SH3b family) that directs the enzyme to the staphylococcal surface by specifically recognizing the pentaglycine crosslinks.
3.2. Reverse Translation: Protein to DNA
Using the Central Dogma, I worked backwards from the protein sequence to determine the corresponding DNA sequence. I retrieved the native coding sequence for mature lysostaphin from the lss gene on plasmid pACK1 of S. simulans (GenBank Accession: M15686).
Native DNA sequence encoding mature lysostaphin (738 bp):
Note: The final three nucleotides (TAA) represent the stop codon.
3.3. Codon Optimization
Why codon optimization is necessary:
Different organisms have different levels of tRNA for a specefic codon. If the tRNA for a specific codon isn’t abudant enough, the translation process can get stalled.
Target organism for optimization: Escherichia coli (K12 strain)
I chose E. coli because it is the standard workhorse for recombinant protein production and the most well-characterized expression system. The initial cloning, expression validation, and protein characterization will be performed in E. coli BL21(DE3) before eventually transferring the validated construct into B. subtilis for the final project application. E. coli also has the highest density of codon usage tables and optimization tools available.
Codon-optimized lysostaphin DNA sequence for E. coli expression (738 bp):
Using the Twist Bioscience Codon Optimization Tool, avoiding Type IIs enzyme recognition sites (BsaI, BsmBI, BbsI):
Retrospective note: Since the deployment chassis for my final project is B. subtilis, direct codon optimization for B. subtilis would have been more strategically aligned. For the final project construct, a B. subtilis-optimized variant will be generated using the same Twist optimization pipeline.
3.4. From DNA Sequence to Protein: Production Methods
With the codon-optimized lysostaphin sequence in hand, there are two main routes to produce the functional protein:
Method 1: Cell-dependent expression in E. coli (Primary approach)
The codon-optimized DNA sequence is cloned into an expression vector (e.g., pET21 or pTwist Amp High Copy) downstream of a strong inducible promoter such as T7. The construct is transformed into E. coli BL21(DE3) competent cells. The process follows the Central Dogma:
Transcription: Upon induction with IPTG, T7 RNA polymerase binds the T7 promoter and synthesizes mRNA from the lysostaphin DNA template. The RNA polymerase reads the template strand 3’→5’ and produces mRNA 5’→3’, with each T in the DNA transcribed as U in the mRNA.
Translation: Ribosomes bind the mRNA at the ribosome binding site (Shine-Dalgarno sequence), recognize the AUG start codon, and begin translating the mRNA into protein. Each three-nucleotide codon in the mRNA specifies one amino acid via tRNA adaptor molecules. Translation proceeds until the ribosome encounters the UAA stop codon.
Folding and purification: The translated polypeptide folds into its native conformation. With a His-tag appended at the C-terminus, the protein can be purified using nickel affinity chromatography (IMAC), followed by size-exclusion chromatography to obtain pure, active lysostaphin.
Cell-free transcription-translation systems (e.g., PURExpress or E. coli S30 extract) can produce lysostaphin without living cells. The DNA template (linear or circular) is added directly to the cell-free reaction mix containing ribosomes, tRNAs, amino acids, RNA polymerase, and energy sources (ATP, GTP). Transcription and translation occur simultaneously in a single tube within 2–4 hours.
Cell-free is advantageous for rapid prototyping - testing whether the construct produces active protein before committing to cell-based production. It is particularly relevant to my project because cell-free expression could eventually enable on-demand antimicrobial peptide production in resource-limited settings without cold chain requirements.
3.5. How It Works in Nature
How a single gene codes for multiple proteins:
At the transcriptional level, a single gene can produce multiple protein variants through several mechanisms:
Alternative splicing (in eukaryotes): Different exons are included or excluded from the mature mRNA, generating distinct protein isoforms from one gene. The DSCAM gene in Drosophila can produce over 38,000 splice variants.
Alternative promoters: Different promoters upstream of the same gene can initiate transcription at different points, producing mRNAs with different 5’ exons and therefore different N-terminal protein sequences.
Polycistronic mRNAs (in prokaryotes): A single operon produces one mRNA encoding multiple proteins, each translated from its own RBS. The lysostaphin operon itself contains both lss and the immunity factor lif gene.
Central Dogma alignment for the first 30 nucleotides of lysostaphin:
DNA (coding): 5'- GCT GCG ACC CAT GAA CAT AGC GCG CAG TGG -3'
||| ||| ||| ||| ||| ||| ||| ||| ||| |||
DNA (template):3'- CGA CGC TGG GTA CTT GTA TCG CGC GTC ACC -5'
↓ Transcription (RNA Pol reads 3'→5')
mRNA: 5'- GCU GCG ACC CAU GAA CAU AGC GCG CAG UGG -3'
↓ Translation (Ribosome reads 5'→3')
Protein: Ala Ala Thr His Glu His Ser Ala Gln Trp
A A T H E H S A Q W
Note: All thymine (T) bases in DNA are transcribed as uracil (U) in mRNA. Each 3-nucleotide codon in the mRNA corresponds to exactly one amino acid in the protein.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Account Setup
I created accounts on both Benchling (benchling.com) and Twist Bioscience (twistbioscience.com).
4.2. Building the DNA Insert Sequence (Expression Cassette)
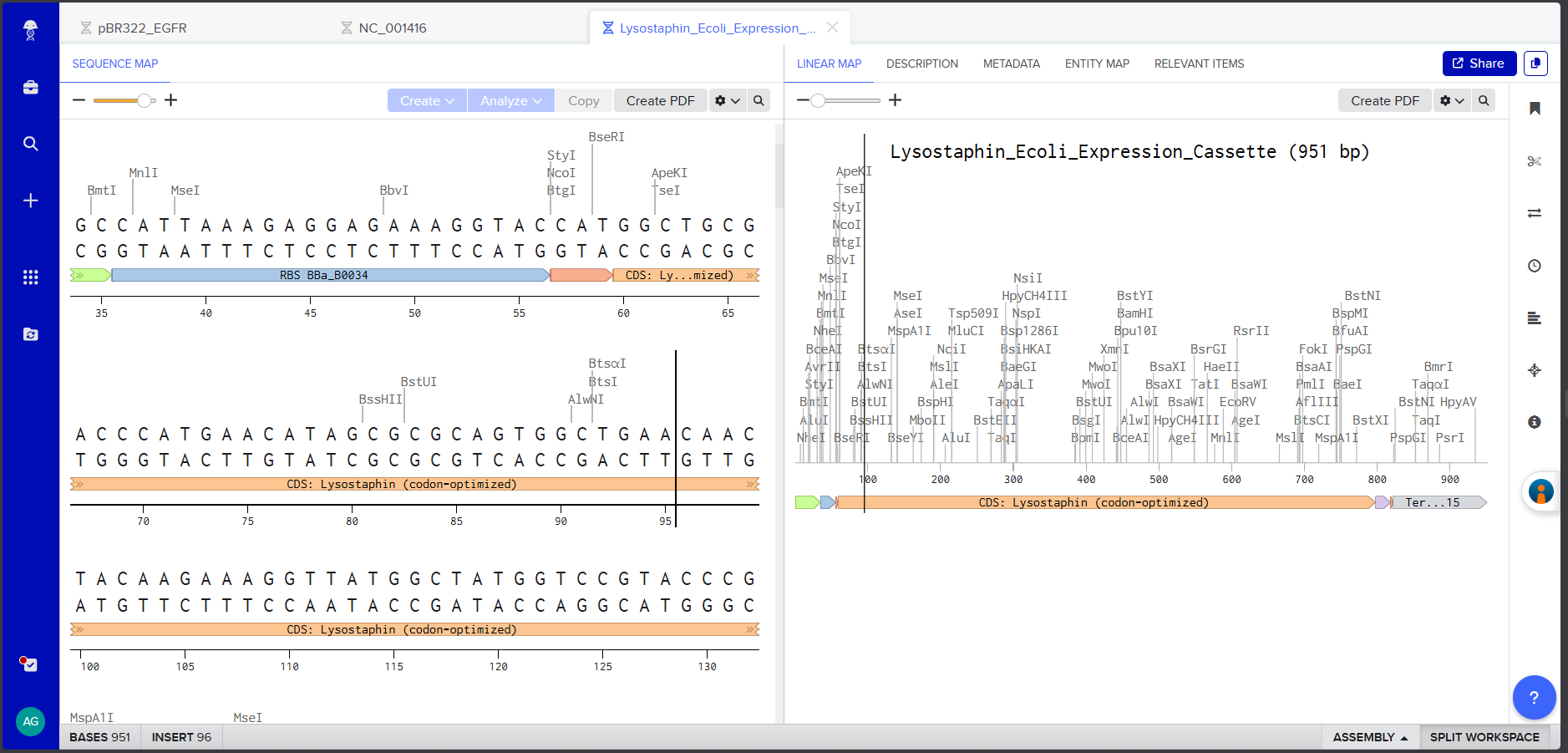
In Benchling, I created a new linear DNA sequence named “Lysostaphin_Expression_Cassette” and assembled the following expression cassette by concatenating the components in order:
Components:
Component
Sequence
Length
Promoter (BBa_J23106)
TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
35 bp
RBS (BBa_B0034 + spacers)
CATTAAAGAGGAGAAAGGTACC
22 bp
Start Codon
ATG
3 bp
Lysostaphin CDS (codon-optimized)
(738 bp sequence from 3.3, excluding start ATG and stop TAA)
Each component was annotated in Benchling by right-clicking and creating annotations with appropriate labels (Promoter, RBS, Start Codon, CDS: Lysostaphin, His-tag, Stop Codon, Terminator).
Screenshot 4: The annotated Linear Map showing all components labeled and color-coded.
On Twist Bioscience, I selected Genes → Clonal Genes and uploaded the FASTA file using the Nucleotide Sequence → Upload Sequence File option.
Screenshot 7: The uploaded sequence in Twist’s interface
I chose clonal genes over gene fragments because clonal genes arrive as circular plasmid DNA that can be directly transformed into E. coli without an additional assembly step - saving 1–2 weeks of experimental time, as noted in the course materials.
Screenshot 8: The vector selection showing pTwist Amp High Copy
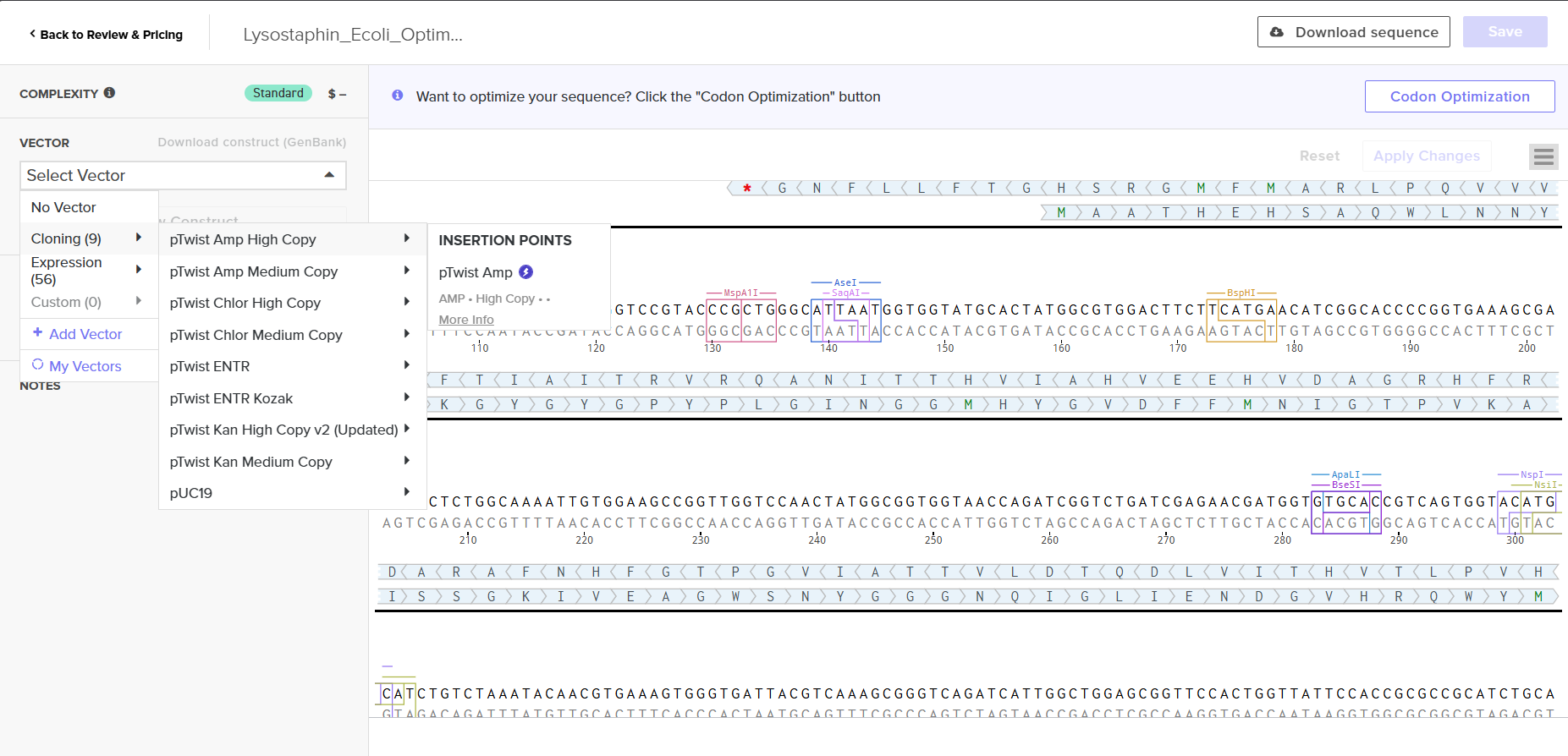
4.6. Vector Selection
I selected pTwist Amp High Copy as the backbone vector from Twist’s vector catalog. Rationale:
Ampicillin resistance - compatible with standard lab antibiotic stocks
High copy number - maximizes plasmid yield per miniprep, important for downstream cloning and sequence verification
ColE1 origin of replication - well-characterized, stable in E. coli
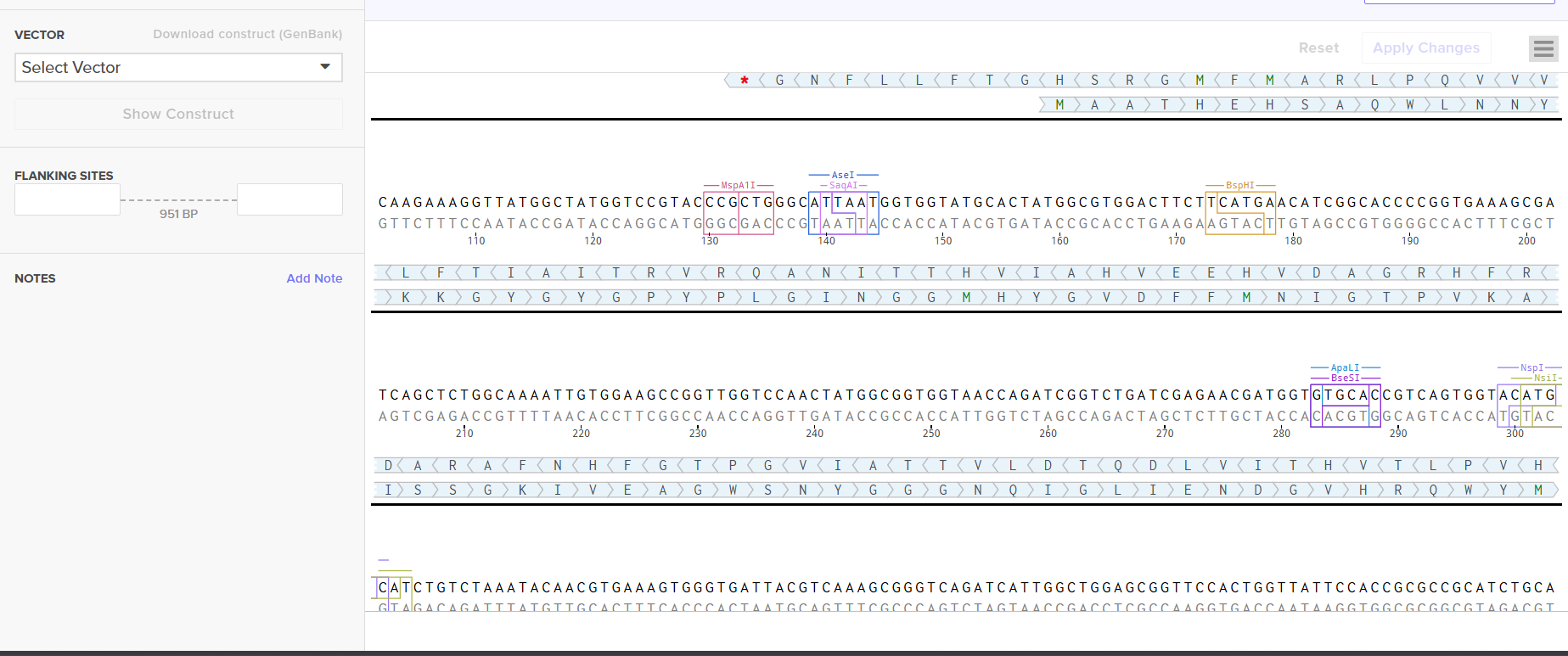
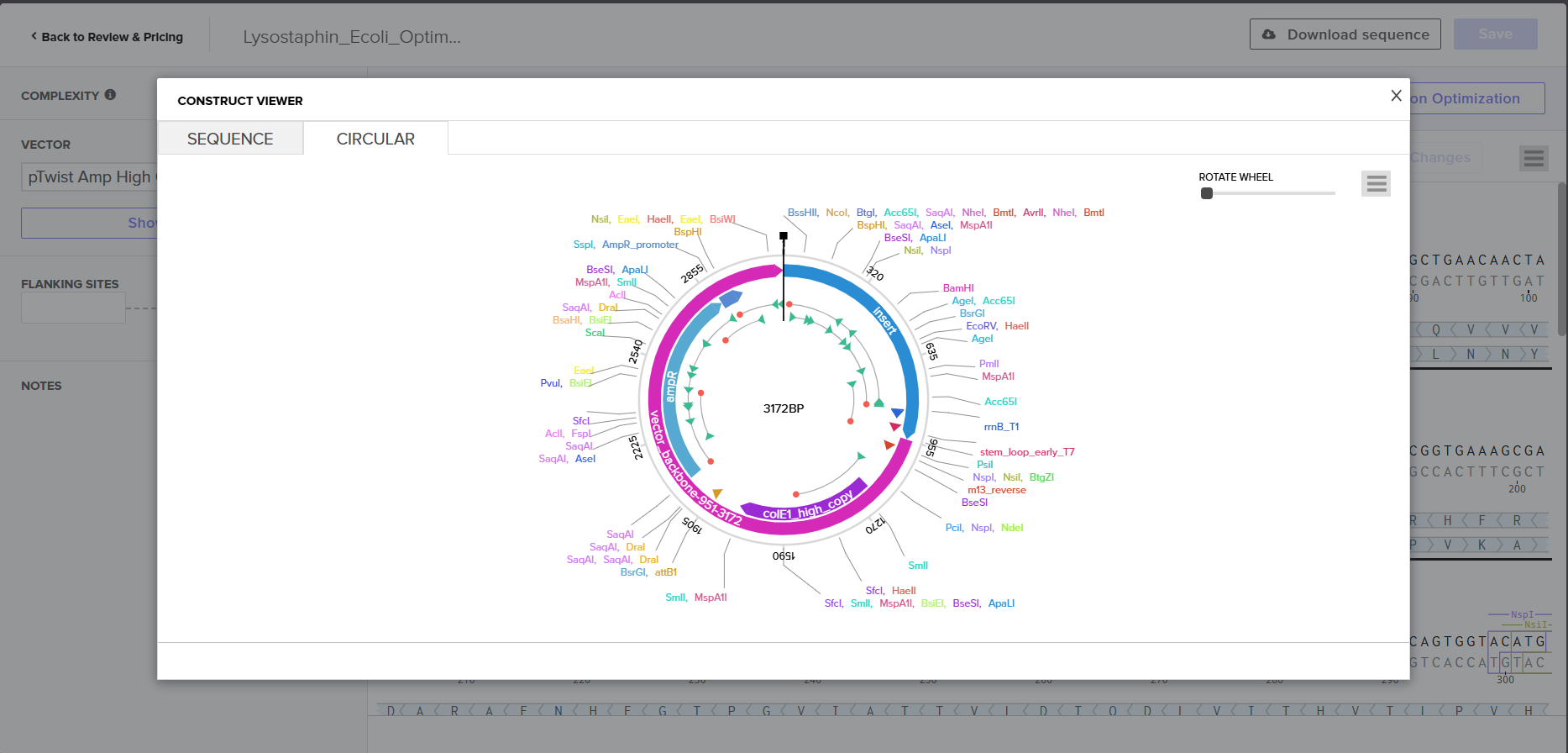
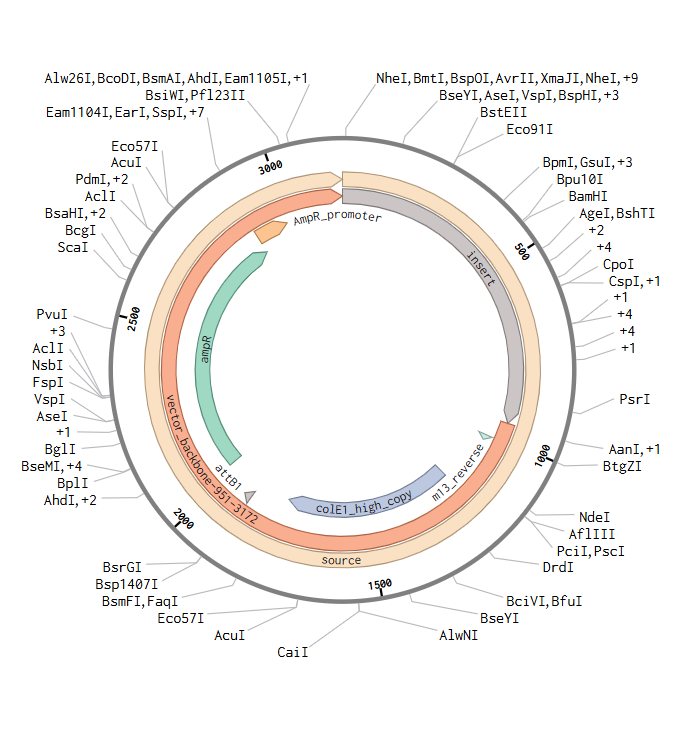
I downloaded the complete construct (GenBank format) from Twist and imported it into Benchling, confirming the circular plasmid map shows the lysostaphin expression cassette correctly inserted into the pTwist backbone.
Screenshot 9: Showing the plasmid uploaded to benchling.
For the final project, my construct will include:
Fully annotated Benchling insert fragment (lysostaphin expression cassette)
pTwist Amp High Copy as the Twist cloning vector
Flanking restriction sites for future subcloning into B. subtilis shuttle vectors
Part 5: DNA Read/Write/Edit
5.1. DNA Read (Sequencing)
(i) What DNA would I want to sequence and why?
I would want to sequence the resistomes of hospital surfaces in Indian government hospitals - specifically, performing metagenomic sequencing of microbial communities on high-touch surfaces (bedrails, IV poles, doorknobs, ventilator tubing) in the medicine and post-surgical wards where I have worked.
Why this matters: The WHO priority pathogen list includes organisms like MRSA, vancomycin-resistant Enterococcus (VRE), carbapenem-resistant Acinetobacter baumannii, and extended-spectrum beta-lactamase (ESBL)-producing Klebsiella pneumoniae - all of which I have encountered clinically in my wards. Sequencing hospital surface microbiomes would reveal: which resistance genes (AMR genes) are circulating on surfaces, the taxonomic composition of the surface biofilm community, whether these surface communities match the organisms causing actual patient infections (proving the transmission chain), and how the resistome changes over time and in response to cleaning protocols.
This data would directly inform the design of my engineered B. subtilis colonizer by identifying which antimicrobial peptides the system needs to produce. Without knowing precisely what organisms inhabit these surfaces, the engineering is blind.
(ii) Sequencing Technology: Oxford Nanopore MinION
Generation: Third-generation sequencing technology. Unlike first-generation (Sanger, chain termination) and second-generation (Illumina, sequencing by synthesis with short reads), nanopore sequencing reads single DNA molecules in real-time without requiring amplification or fluorescent labeling.
Input preparation - essential steps:
DNA extraction: Use a bead-beating + column-based kit (e.g., Qiagen DNeasy PowerSoil) to lyse both Gram-positive and Gram-negative organisms from surface swabs.
Size selection: Optional - use SPRI beads or gel extraction to enrich for fragments >1 kb if long reads are desired.
Library preparation: Using the Rapid Sequencing Kit (SQK-RAD004): a transposase fragments the DNA and simultaneously attaches sequencing adapters in a single 10-minute step. Alternatively, the Ligation Sequencing Kit (SQK-LSK114) provides higher throughput with more steps: end repair/dA-tailing → adapter ligation → SPRI bead cleanup.
No PCR amplification required - native DNA is sequenced directly, preserving epigenetic modifications (methylation).
How it decodes bases (base calling):
A single strand of DNA is ratcheted through a biological nanopore (CsgG protein pore) embedded in a synthetic membrane. As each nucleotide passes through the constriction of the pore, it causes a characteristic disruption of the ionic current flowing through the pore. A neural network-based base caller (e.g., Dorado or Guppy) decodes the raw electrical signal (squiggle) into nucleotide sequences. The current signal is influenced by ~5–6 nucleotides simultaneously (the k-mer in the pore), so the base caller uses sequence context to make predictions.
Output:
FASTQ files containing long reads (N50 typically 5–20 kb, with individual reads >100 kb possible)
Real-time streaming output - reads are available within minutes of starting the run
Per-read quality scores (Q-scores), with current chemistry (R10.4.1) achieving median Q20 (~99% accuracy)
Methylation calls from the same raw signal (no bisulfite conversion needed)
Why MinION specifically: It costs ~$1,000 for the device (or free through Oxford Nanopore’s starter program), runs on a laptop via USB, requires no large infrastructure, and can be used at point-of-care - making it deployable in Indian hospitals where I work. For metagenomic surveillance, long reads resolve species-level taxonomy and link AMR genes to their host organisms on single reads, which short-read sequencing cannot do.
5.2. DNA Write (Synthesis)
(i) What DNA would I want to synthesize and why?
I would want to synthesize a modular antimicrobial peptide expression cassette library for Bacillus subtilis - specifically, a set of genetic constructs each encoding a different antimicrobial effector under the control of a quorum-sensing-responsive promoter. The library would include:
Lysostaphin (lss) - targets S. aureus including MRSA (the protein designed in Parts 3–4 above)
Dispersin B (dspB) - a glycoside hydrolase from Aggregatibacter actinomycetemcomitans that degrades poly-N-acetylglucosamine (PNAG) biofilm matrix, disrupting biofilms formed by S. aureus and S. epidermidis
Art-175 - an engineered endolysin-antimicrobial peptide fusion (artilysin) effective against Gram-negative pathogens including Acinetobacter baumannii and Pseudomonas aeruginosa
Each construct would use the same standardized architecture: [Quorum-sensing promoter] → [RBS] → [Signal peptide for secretion] → [Antimicrobial CDS] → [Terminator]. This modularity allows swapping effectors in and out depending on the pathogen landscape of a specific hospital - a “pharmacological palette” for engineered living surfaces.
The specific genetic sequences are: lysostaphin as described in Part 3.3, and I would design and order the others through Twist.
Oligo synthesis on silicon chips: Short oligonucleotides (up to ~200 nt) are synthesized in parallel on miniaturized wells etched into silicon wafers using phosphoramidite chemistry. Each cycle: (a) detritylation (remove 5’-DMT protecting group), (b) coupling (add next phosphoramidite monomer), (c) capping (block failed couplings), (d) oxidation (stabilize the new phosphodiester bond).
Oligo pool harvesting and error removal: Completed oligos are cleaved from the chip, and error-containing sequences are removed using enzymatic mismatch cleavage or hybridization-based selection.
Hierarchical assembly: Overlapping oligos are assembled into longer gene fragments through PCR-based assembly (e.g., polymerase cycling assembly or Gibson Assembly). Multiple fragments are then joined into the complete gene.
Clonal isolation: Assembled genes are cloned into the selected vector, transformed into E. coli, and individual clones are Sanger-sequenced to confirm 100% sequence accuracy. Only sequence-verified clones are shipped.
Limitations:
Speed: Typical turnaround is 2–3 weeks for clonal genes, which limits rapid iteration cycles
Accuracy: While final clonal products are sequence-perfect, the per-base error rate during oligo synthesis (~1 in 200–500 bases) necessitates the error-correction and clonal selection steps, adding time
Scalability: Individual gene synthesis scales well, but whole-genome synthesis (>100 kb) remains prohibitively expensive and slow - the “gene writing gap” described by Hoose et al. (2023)
Sequence constraints: Certain sequences are difficult to synthesize: extreme GC content (<25% or >75%), long homopolymer runs, extensive secondary structure, and repetitive sequences can cause synthesis failures
Cost: While cost per base has dropped dramatically (~$0.07/bp for gene fragments), synthesizing entire pathways or genomes remains expensive
5.3. DNA Edit (Genome Editing)
(i) What DNA would I want to edit and why?
I would want to edit the genome of Bacillus subtilis 168 to create an optimized chassis for my engineered living surface colonizer. Specifically, three categories of edits:
Edit 1: Knockout of sporulation genes (spo0A, sigF). B. subtilis forms endospores that are extremely resistant to disinfection and could spread the engineered organism beyond hospital surfaces. Deleting key sporulation regulators prevents spore formation, ensuring the organism remains in vegetative form and is susceptible to standard decontamination - a critical biocontainment feature.
Edit 2: Introduction of synthetic auxotrophies. As I described in my Week 1 governance analysis, I would engineer dependence on a non-canonical amino acid (ncAA) not found in nature. Specifically, I would knock out genes in the biosynthetic pathway for an essential amino acid and replace them with an engineered aminoacyl-tRNA synthetase that charges tRNA with a synthetic analog. The organism can only survive when the synthetic nutrient is supplied - true containment beyond the “Jurassic Park lysine contingency.”
Edit 3: Integration of the quorum-sensing antimicrobial peptide circuits into the chromosome. Rather than maintaining the constructs on plasmids (which can be lost and impose a fitness burden), I would integrate the engineered circuits directly into the B. subtilis chromosome at neutral loci (e.g., amyE, lacA). Chromosomal integration provides stable, single-copy expression without antibiotic selection - essential for real-world deployment where you cannot maintain antibiotic pressure.
(ii) Editing Technology: CRISPR-Cas9 combined with MAGE (Multiplex Automated Genome Engineering)
For targeted knockouts and insertions, I would use CRISPR-Cas9 as the primary editing tool, supplemented by recombineering for multiplex edits.
How CRISPR-Cas9 edits DNA - essential steps:
Guide RNA design: Design a 20-nt spacer sequence complementary to the target site in the B. subtilis genome, adjacent to a 5’-NGG-3’ PAM (protospacer adjacent motif) recognized by SpCas9. I would use Benchling’s CRISPR guide designer to score guides for on-target efficiency and minimize off-target sites.
Construct assembly: Clone the sgRNA (spacer + scaffold) into an expression vector under a constitutive promoter. Co-deliver or separately provide: (a) Cas9 protein or expression cassette, (b) the sgRNA, and (c) a repair template - a DNA fragment with 500–1000 bp homology arms flanking the desired edit (deletion, insertion, or substitution).
Delivery into B. subtilis:B. subtilis is naturally competent - it can take up exogenous DNA from the environment. Transform competent cells with the CRISPR plasmid and repair template simultaneously.
Double-strand break and repair: The Cas9-sgRNA complex binds the target DNA, verifies PAM recognition, and creates a blunt double-strand break (DSB) 3 bp upstream of the PAM. The cell’s homology-directed repair (HDR) machinery uses the supplied repair template to fix the break, incorporating the desired edit.
Selection and verification: Plate transformants on selective media. Screen colonies by PCR across the edited locus and confirm by Sanger sequencing.
Preparation required:
Input materials: Cas9 expression plasmid (or purified Cas9 protein), sgRNA expression construct, repair template DNA (synthesized by Twist or generated by PCR), competent B. subtilis cells, selective antibiotics
Design steps: Target site selection → off-target analysis → guide scoring → repair template design with homology arms → cloning and sequence verification
Limitations:
Efficiency: In B. subtilis, CRISPR-Cas9 editing efficiency varies (50–95%) depending on the locus, guide quality, and repair template design. Unlike E. coli, B. subtilis has efficient HDR, but Cas9 toxicity can reduce viable transformants.
Off-target effects: SpCas9 can tolerate mismatches at positions distal from the PAM, potentially causing unintended edits. This can be mitigated by using high-fidelity Cas9 variants (e.g., eSpCas9) and whole-genome sequencing of final strains.
Multiplexing complexity: Editing multiple loci simultaneously (sporulation knockouts + auxotrophy + circuit integration) requires sequential rounds of editing with plasmid curing between rounds, which is time-consuming. MAGE-like approaches (Wannier et al., 2021) using ssDNA oligonucleotides can accelerate multiplex editing but are less established in B. subtilis than in E. coli.
PAM restriction: The requirement for an NGG PAM limits targetable sites to ~1 in every 8 bp on average. This can be overcome by using Cas9 orthologs with different PAM preferences (e.g., SaCas9 recognizes NNGRRT, CjCas9 recognizes NNNNRYAC) or engineered PAM-relaxed variants (e.g., SpCas9-NG, SpRY).
Large insertions: Integrating multi-kb constructs (full antimicrobial circuits) via HDR is less efficient than small edits. For large insertions, combining CRISPR-mediated counterselection with traditional B. subtilis integrative vectors at established loci (amyE, thrC) may be more reliable.
References
Sabala, I. et al. “Crystal structure of the antimicrobial peptidase lysostaphin from Staphylococcus simulans.” FEBS J. 281, 4112–4122 (2014).
Nazari, R. et al. “Cloning and expression of Staphylococcus simulans lysostaphin enzyme gene in Bacillus subtilis WB600.” Mol. Biotechnol. 63, 1043–1052 (2021).
Shendure, J. et al. “DNA sequencing at 40: past, present, and future.” Nature 550, 345–353 (2017).
Hoose, A. et al. “DNA synthesis technologies to close the gene writing gap.” Nat. Rev. Chem. 7, 144–161 (2023).
Wannier, T. et al. “Recombineering and MAGE.” Nat. Rev. Methods Primers 1, 7 (2021).
Wang, J.Y. & Doudna, J.A. “CRISPR technology: A decade of genome editing is only the beginning.” Science 379, eadd8643 (2023).
Bastos, M.C.F. et al. “Lysostaphin: A Staphylococcal Bacteriolysin with Potential Clinical Applications.” Pharmaceuticals 3, 1139–1161 (2010).
Recsei, P.A. et al. “Cloning, sequence, and expression of the lysostaphin gene from Staphylococcus simulans.” Proc. Natl. Acad. Sci. USA 84, 1127–1131 (1987).
Citation: I used Claude to help refine the codon-optimized sequence generation and to verify technical details of sequencing/editing technologies. All project framing, protein choice rationale, clinical observations, and design decisions connecting to my final project are my own work.
Week 3: Lab Automation & Opentrons Art
Part 1: Opentrons Agar Art - Biohazard Symbol
Design Concept
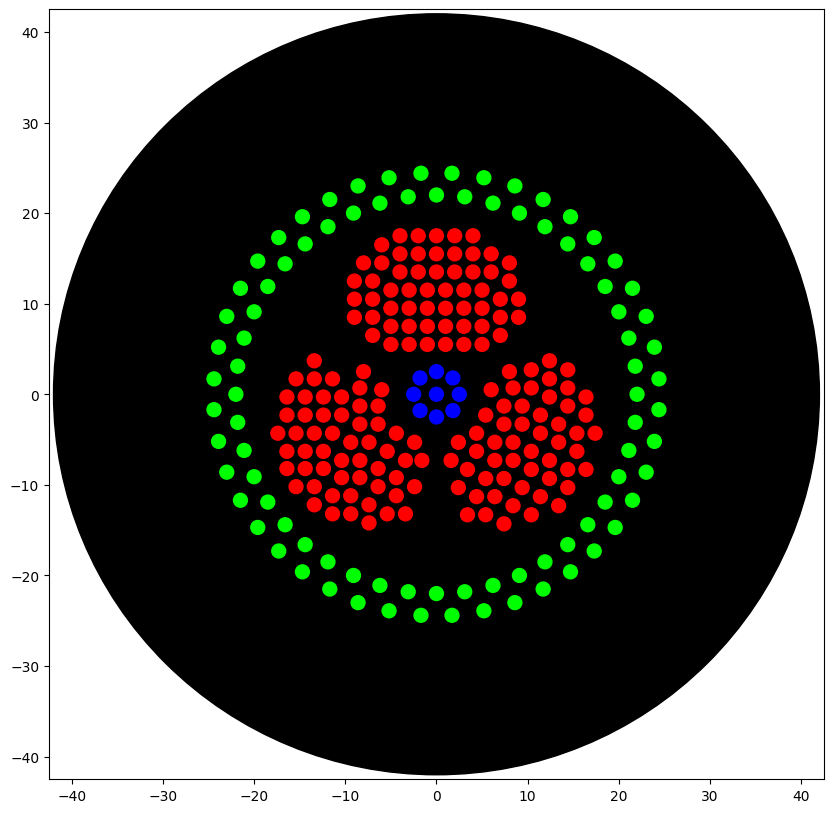
I conceptualised the biohazard symbol, which I thought of creating using parametric algorithms. I used the assistance of Claude (Anthropic) to generate the python code which I ran on Google Colab, in the copy provided by HTGAA. The results are shown below.
The artwork I conceptualised is a biohazard symbol rendered in fluorescent bacteria on black agar. I specifically chose this design because my final project focuses on engineering Bacillus subtilis to combat hospital-acquired infections (HAIs), which kill over 200,000 people annually in India alone.
Python Code
The complete Python script is embedded below and was also submitted via the course form. I used Claude (Anthropic) to help write the code, debug coordinate math and validate the Opentrons API calls, while I designed the concept, chose the geometry, and structured the protocol logic.
fromopentronsimporttypesimportmathmetadata={# see https://docs.opentrons.com/v2/tutorial.html#tutorial-metadata'author':'Anirudh Gangadharan','protocolName':'Biohazard Biosensor Symbol','description':'Parametric biohazard symbol representing engineered B. subtilis biosensor: purple crescents (threat detection), blue hub (sensing logic), pink safety ring (biocontainment)','source':'HTGAA 2026 Opentrons Lab','apiLevel':'2.20'}################################################################################# Robot deck setup constants - don't change these##############################################################################TIP_RACK_DECK_SLOT=9COLORS_DECK_SLOT=6AGAR_DECK_SLOT=5PIPETTE_STARTING_TIP_WELL='A1'well_colors={'A1':'Red','B1':'Blue','C1':'Green'}defrun(protocol):################################################################################# Load labware, modules and pipettes############################################################################### Tipstips_20ul=protocol.load_labware('opentrons_96_tiprack_20ul',TIP_RACK_DECK_SLOT,'Opentrons 20uL Tips')# Pipettespipette_20ul=protocol.load_instrument("p20_single_gen2","right",[tips_20ul])# Modulestemperature_module=protocol.load_module('temperature module gen2',COLORS_DECK_SLOT)# Temperature Module Platetemperature_plate=temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul','Cold Plate')# Choose where to take the colors fromcolor_plate=temperature_plate# Agar Plateagar_plate=protocol.load_labware('htgaa_agar_plate',AGAR_DECK_SLOT,'Agar Plate')## TA MUST CALIBRATE EACH PLATE!# Get the top-center of the plate, make sure the plate was calibrated before running thiscenter_location=agar_plate['A1'].top()pipette_20ul.starting_tip=tips_20ul.well(PIPETTE_STARTING_TIP_WELL)################################################################################# Patterning#################################################################################### Helper functions for this lab#### pass this e.g. 'Red' and get back a Location which can be passed to aspirate()deflocation_of_color(color_string):forwell,colorinwell_colors.items():ifcolor.lower()==color_string.lower():returncolor_plate[well]raiseValueError(f"No well found with color {color_string}")# For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_detach(pipette, 1, loc)defdispense_and_detach(pipette,volume,location):"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
5mm because a 4uL drop is 2mm diameter; and a 2deg tilt in the agar pour is >3mm difference across a plate.
"""assert(isinstance(volume,(int,float)))above_location=location.move(types.Point(z=location.point.z+5))# 5mm abovepipette.move_to(above_location)# Go to 5mm above the dispensing locationpipette.dispense(volume,location)# Go straight downwards and dispensepipette.move_to(above_location)# Go straight up to detach drop and stay high###### BIOHAZARD BIOSENSOR SYMBOL#### --- Geometry Parameters ---LOBE_DIST=8.5# distance from center to each lobe centerLOBE_R_OUTER=10.0# outer radius of each crescent lobeLOBE_R_INNER=5.5# inner cutout radiusGAP_ANGLE=25# degrees of gap between lobesDOT_SPACING=2.0# mm between dots (>= 2mm to avoid merging)PLATE_R=33.0# max radius from center (within 40mm safety limit)lobe_degs=[90,210,330]# three lobes at 120 degree intervals# --- Generate Red points: Three crescent lobes (threat/pathogen detection) ---red_pts=[]forbdinlobe_degs:br=math.radians(bd)cx,cy=LOBE_DIST*math.cos(br),LOBE_DIST*math.sin(br)forgxinrange(-15,16):forgyinrange(-15,16):x,y=gx*DOT_SPACING/2,gy*DOT_SPACING/2px,py=cx+x,cy+ydl=math.sqrt(x**2+y**2)do=math.sqrt(px**2+py**2)ifdl>LOBE_R_OUTERordo<=LOBE_R_INNERordo>PLATE_R:continueang=math.degrees(math.atan2(py,px))%360gap=Falseforgcin[150,270,30]:ad=abs(ang-gc)ifad>180:ad=360-adifad<GAP_ANGLE/2:gap=True;breakifgap:continueifnotany(math.sqrt((px-ex)**2+(py-ey)**2)<DOT_SPACING*0.85forex,eyinred_pts):red_pts.append((round(px,1),round(py,1)))# --- Generate Blue points: Central hub (sensing/logic core) ---blue_pts=[(0.0,0.0)]foriinrange(8):a=2*math.pi*i/8blue_pts.append((round(2.5*math.cos(a),1),round(2.5*math.sin(a),1)))# --- Generate Green points: Outer safety ring (biocontainment) ---green_pts=[]foriinrange(44):a=2*math.pi*i/44green_pts.append((round(22*math.cos(a),1),round(22*math.sin(a),1)))foriinrange(44):a=2*math.pi*(i+0.5)/44green_pts.append((round(24.5*math.cos(a),1),round(24.5*math.sin(a),1)))# --- Helper: pick up tip, aspirate color, dispense all points, drop tip ---defpick_up_and_draw(color_name,points):pipette_20ul.pick_up_tip()batch_size=19# aspirate up to 19uL at a time (19 dots at 1uL each)foriinrange(0,len(points),batch_size):batch=points[i:i+batch_size]pipette_20ul.aspirate(len(batch),location_of_color(color_name))for(px,py)inbatch:loc=center_location.move(types.Point(x=px,y=py))dispense_and_detach(pipette_20ul,1,loc)pipette_20ul.drop_tip()# --- Execute patterning ---pick_up_and_draw('Red',red_pts)# Crescents: threat detectionpick_up_and_draw('Blue',blue_pts)# Hub: sensing logic gatepick_up_and_draw('Green',green_pts)# Ring: biocontainment boundary# Done! All tips dropped.
After running the above code, I ran the code shown below to visualise the image and it is provided below.
=== VOLUME TOTALS BY COLOR ===
Blue: aspirated 9 dispensed 9
Red: aspirated 168 dispensed 168
Green: aspirated 88 dispensed 88
[all colors]: [aspirated 265] [dispensed 265]
=== TIP COUNT ===
Used 3 tip(s) (ideally exactly one per unique color)
AI Usage Disclosure
I used Claude (Anthropic) to write the code for the biohazard geometry.
Part 2: Post-Lab Questions
Published Paper Utilizing Laboratory Automation for Novel Biological Applications
Paper: Semiautomated Production of Cell-Free Biosensors
Citation: Brown, D.M., Phillips, D.A., Garcia, D.C., et al. (2025). Semiautomated Production of Cell-Free Biosensors. ACS Synthetic Biology, 14(3), 979–986. DOI: 10.1021/acssynbio.4c00703
Affiliations: Northwestern University (Department of Chemical & Biological Engineering, Center for Synthetic Biology); U.S. Army DEVCOM Chemical Biological Center.
Summary
Brown et al. (2025) present the first systematic study comparing manual versus robot-assembled cell-free biosensor reactions, using the Opentrons OT-2 liquid handling robot to automate the assembly of cell-free gene expression (CFE) reactions across full 384-well plates.
Cell-free biosensors represent a powerful class of synthetic biology diagnostics: DNA-encoded genetic circuits are executed in cell-free transcription-translation (TX-TL) systems, freeze-dried for ambient storage and field distribution, then rehydrated at the point of use to detect target analytes via colorimetric or fluorescent output. Previous work - notably Pardee et al. (2016) demonstrating Zika virus detection using toehold switches in cell-free systems - established the diagnostic potential of this platform. However, manufacturing has remained a manual, low-throughput bottleneck that limits translation from proof-of-concept to deployable diagnostics.
Automation Platform and Workflow
The OT-2 performed semiautomated assembly of CFE reactions, handling precise nanoliter-to-microliter transfers of cell extract, energy buffer, DNA template, and water into each well of a 384-well plate. The team benchmarked two cell-free reporter systems:
A constitutive LacZ colorimetric reporter (absorbance-based readout)
A GFP fluorescent reporter (fluorescence-based readout)
For each system, reactions were assembled both manually and via OT-2, enabling direct comparison of precision, reproducibility, and biosensor performance. The culminating experiment deployed a complete 384-well fluoride riboswitch biosensor array - a genetically encoded sensor element that activates gene expression in the presence of fluoride ions - with every reaction assembled by the OT-2.
Key Findings
Semiautomated OT-2 assembly produced biosensor reactions with comparable or improved consistency relative to manual assembly, reducing the well-to-well coefficient of variation that plagues hand-pipetted plates and directly degrades diagnostic reliability.
A full 384-well fluoride riboswitch biosensor array was successfully built and functionally validated in a single automated run, demonstrating the feasibility of medium-throughput biosensor production on affordable hardware.
The study provides the field’s first rigorous benchmarking data for automated versus manual CFE biosensor manufacturing, systematically quantifying where robotic and manual assembly diverge in performance characteristics.
Why Automation Was Essential
Three factors render automation indispensable for this application. First, cell-free reactions are exquisitely sensitive to pipetting precision - small volumetric errors in extract or DNA template concentration produce disproportionately large signal variability, which is unacceptable for a diagnostic biosensor that must deliver a reliable binary answer. The OT-2’s reproducible liquid handling directly addresses this failure mode. Second, scaling from proof-of-concept to deployment requires manufacturing hundreds to thousands of biosensor reactions per batch; manual assembly of 384-well plates is tedious, error-prone, and fundamentally unscalable. Third, the low cost of the OT-2 (~$5,000–$10,000) means that academic laboratories, field-deployed diagnostic operations, and resource-limited clinical settings can adopt semiautomated manufacturing without the capital expenditure of biofoundry-grade liquid handlers such as the Hamilton STAR or Beckman Biomek platforms.
Relevance to My Final Project
This paper is directly relevant to my proposed work on autonomous antimicrobial surfaces for hospital-acquired infection prevention. My project design incorporates both the OT-2 (for lysostaphin zone-of-inhibition assays and serial dilutions) and the Ginkgo Nebula cloud laboratory (for high-throughput toehold switch screening using the Echo → Bravo → PHERAstar workflow). Brown et al. demonstrate precisely the principle that underpins my automation strategy: affordable liquid handling robots can achieve the precision and throughput required to screen synthetic biology constructs at a scale that manual methods cannot support, particularly for cell-free biosensor applications where reaction assembly variability is a primary performance bottleneck. Their validation of OT-2-assembled cell-free biosensor arrays provides direct precedent for my planned Week 11 toehold switch screening experiments on Nebula’s cell-free platform.
Reference:
Brown, D.M., Phillips, D.A., Garcia, D.C., et al. (2025). Semiautomated Production of Cell-Free Biosensors. ACS Synthetic Biology, 14(3), 979–986. https://doi.org/10.1021/acssynbio.4c00703
Question 2 : Automation Plan for Final Project
My final project - an engineered Bacillus subtilis surface colonizer for hospital-acquired infection prevention - has two primary components that benefit from automation: antimicrobial peptide optimization and biosensor screening.
Automation Scale 1: OpenTrons OT-2 (Bench-Scale, London Node)
Goal: Serial dilution and zone-of-inhibition assays for lysostaphin antimicrobial activity.
Pseudocode:
PROTOCOL: Lysostaphin Activity Screening
LABWARE:
- Slot 1: 96-well deep-well plate (lysostaphin variants in columns 1-6)
- Slot 2: 12-well reservoir (LB broth, S. aureus overnight culture)
- Slot 4: Tiprack 200 uL
- Slot 7: Tiprack 20 uL
PROCEDURE:
# Step 1: Serial dilution of lysostaphin across rows
FOR each column c in [1..6]: # 6 construct variants
pick_up_tip(200uL)
aspirate(100uL, reservoir['LB'])
FOR each row r in [B..H]: # 7-point serial dilution
dispense(50uL, plate[r][c])
mix(3, 50uL) # Mix by pipetting up/down
drop_tip()
# Step 2: Add S. aureus to all test wells
FOR each well in plate[A1:H6]:
pick_up_tip(20uL)
aspirate(10uL, reservoir['S_aureus'])
dispense(10uL, well)
drop_tip()
# Step 3: Incubate 16h at 37°C (manual)
# Step 4: Read OD600 on plate reader (manual)
This protocol generates a dose-response matrix: 6 lysostaphin variants × 7 concentrations × triplicate, with automated serial dilution ensuring precise and reproducible concentration gradients.
Goal: Screen toehold switch biosensor library for mecA mRNA detection (connects to Week 11 biosensor module).
Nebula Workflow (RAC Plate Flow):
Step 1: ECHO - Acoustic transfer of 384 toehold switch DNA constructs
(NUPACK-optimized designs targeting mecA mRNA)
Source: 384-well plate with switch DNA variants
Destination: 384-well assay plate
Volume: 100 nL per well
Step 2: BRAVO - Stamp CFPS reagent master mix into all 384 wells
Source: Reservoir with PURExpress or custom CFPS lysate
Volume: 5 uL per well
Step 3: MULTIFLO - Dispense mecA trigger RNA at 4 concentrations
Quadrants of plate get 0, 10, 100, 1000 nM trigger
Volume: 2 uL per well
Step 4: PLATELOC - Heat-seal the plate
Step 5: INHECO - Incubate at 37°C for 2 hours
(Cell-free expression of toehold switch + GFP reporter)
Step 6: XPEEL - Remove seal
Step 7: PHERASTAR - Fluorescence readout (excitation 485nm, emission 520nm)
Kinetic read: every 10 min for 6 hours
Output: ON/OFF ratio matrix for all 384 designs
ANALYSIS:
- Rank switches by ON/OFF ratio (trigger vs. no-trigger)
- Select top 10 switches for integration into B. subtilis chassis
- Feed results back to NUPACK for next design round (DBTL cycle)
This cloud lab workflow screens 384 biosensor designs in a single automated run - something that would take months manually. The top-performing switches will be integrated into my B. subtilis chassis to create the sense-and-respond circuit for MRSA detection.
Why Two Scales Matter
The OpenTrons protocol handles the effector optimization (how well does lysostaphin kill S. aureus?), while the Nebula workflow handles the sensor optimization (how sensitively can we detect MRSA?). Together, they close the loop on both halves of the sense-and-respond system - all through automation.
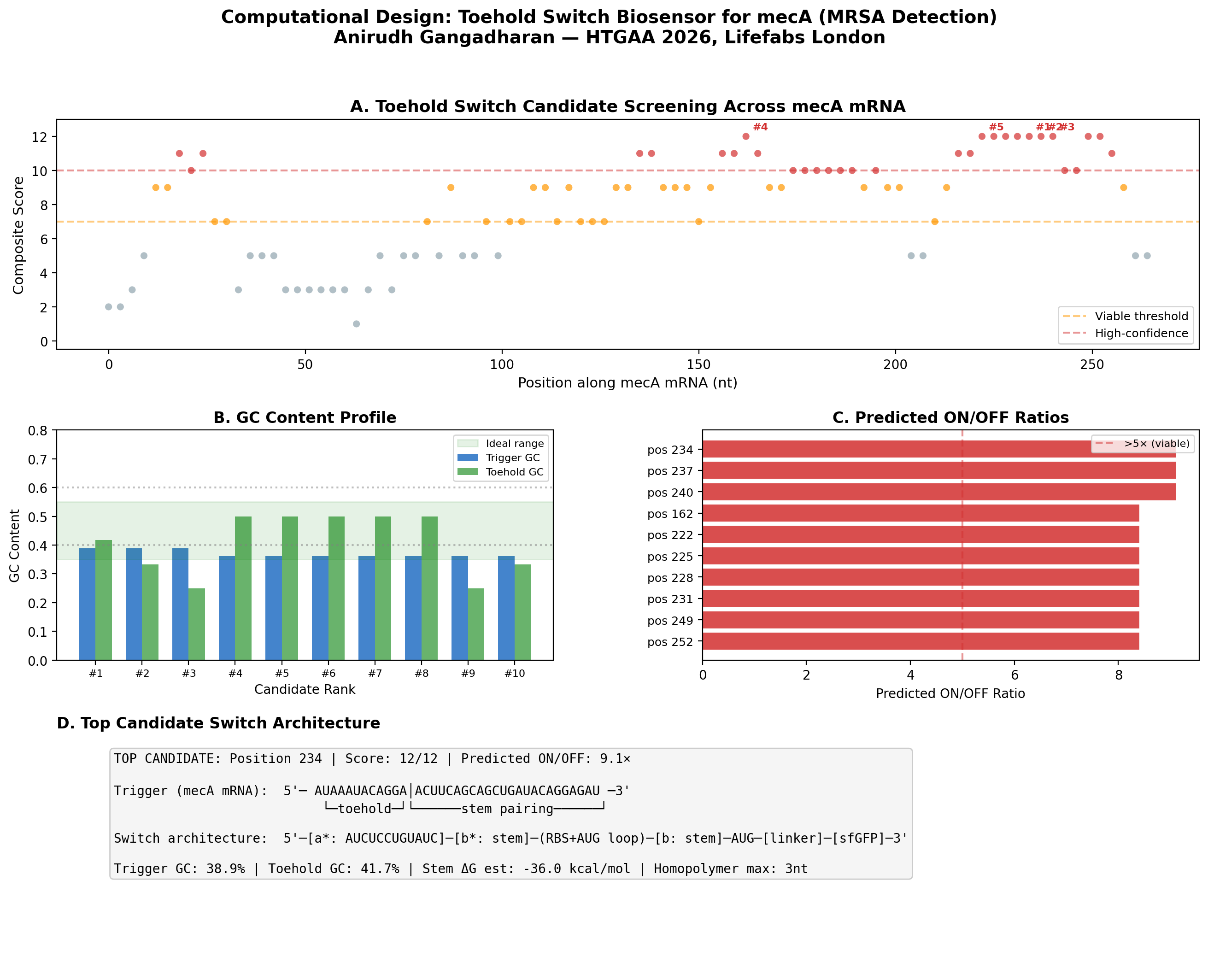
Preliminary Computational Results: Toehold Switch Design for mecA
To validate the feasibility of my biosensor approach, I wrote a Python pipeline to computationally design toehold switch candidates targeting the mecA mRNA of S. aureus N315 (GenBank: D86934). The mecA gene encodes PBP2a, the penicillin-binding protein that confers methicillin resistance - making it the definitive genetic marker for MRSA.
Method
I implemented the Series B toehold switch architecture from Green et al. (2014) Cell 159:925-939, screening 89 candidate positions across the first 303 nucleotides of the mecA coding sequence. Each candidate was evaluated on six criteria: trigger GC content (ideal 35-55%), toehold GC content (ideal 25-50%), absence of homopolymer runs >4 nt, absence of internal AUG codons, estimated stem thermodynamic stability, and predicted ON/OFF ratio.
Key Results
89 candidates screened, of which 30 scored ≥ 10/12 (high-confidence)
Best candidate: Position 234, targeting the region encoding the transpeptidase catalytic domain
The position 150-260 region of mecA contains a cluster of high-scoring candidates, suggesting favorable mRNA secondary structure accessibility in this region
Significance
This computational screen identifies candidate toehold switches ready for experimental validation on Ginkgo Nebula during the Week 11 biosensor module. The top 10 designs can be synthesized by Twist Bioscience and screened in cell-free reactions using the cloud lab workflow described in my automation plan above.
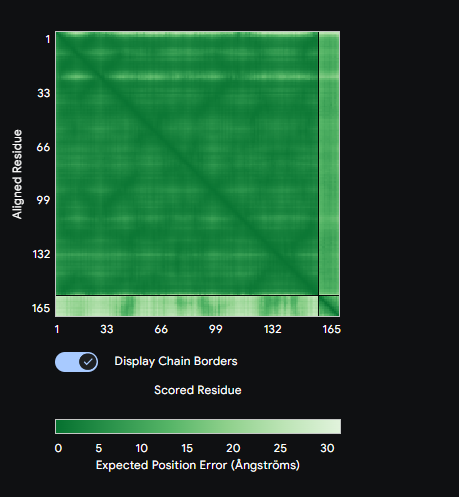
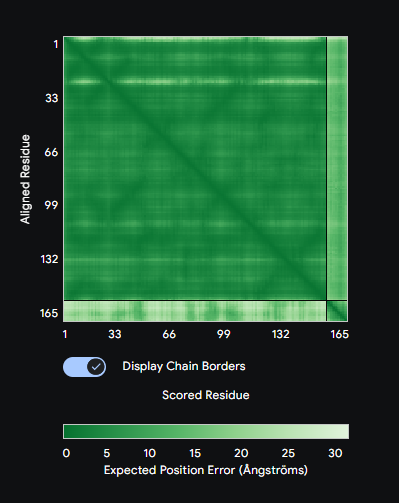
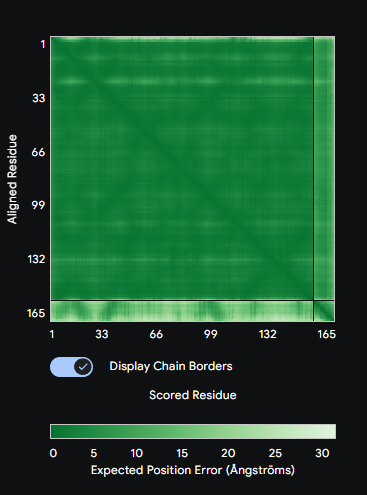
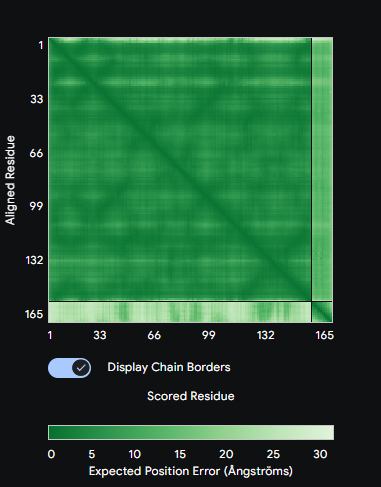
Figure: Computational screening of toehold switch biosensor candidates targeting mecA mRNA. (A) Composite score distribution across the mecA 5’ coding region. (B) GC content profiles of top 10 candidates. (C) Predicted ON/OFF ratios. (D) Architecture of the top-ranked candidate at position 234.
Next Steps
Validate top 5 candidates with NUPACK (nupack.org) for full minimum free energy analysis
BLAST trigger sequences against the complete S. aureus transcriptome to confirm specificity
Order synthesis of top 10 switches from Twist Bioscience
Screen in cell-free system on Ginkgo Nebula during Week 11 biosensor module
Integrate the best-performing switch into the B. subtilis sense-and-respond chassis
The complete Python code and candidate data (JSON) are available in my documentation.
Part 3: Three Final Project Ideas
Idea 1 : Cell-Free Lysostaphin Optimization via AI-Guided DBTL on Nebula
What: Optimize recombinant lysostaphin expression in a cell-free protein synthesis system using automated Design-Build-Test-Learn cycles on Ginkgo Nebula’s cloud laboratory.
How: Codon-optimize lysostaphin variants for cell-free expression. Screen hundreds of reaction conditions (DNA concentration, Mg²⁺, temperature, incubation time) in parallel using Nebula’s RAC workflow: Echo acoustic transfer → Bravo master mix stamping → Inheco incubation → PHERAstar fluorescence readout. Validate top candidates via OpenTrons OT-2 serial dilution and zone-of-inhibition assays against S. aureus.
Expected output: Optimized cell-free lysostaphin expression protocol with quantified MIC data against clinical S. aureus isolates.
Idea 2 : Toehold Switch MRSA Detection Coupled to Antimicrobial Secretion in B. subtilis
What: Design RNA-based toehold switch biosensors targeting mecA mRNA (the methicillin resistance determinant) and integrate them with a T7 RNAP amplification cascade driving lysostaphin secretion in B. subtilis - creating an autonomous sense-and-kill genetic circuit.
How: Computationally screen toehold switch candidates across the mecA coding sequence using thermodynamic scoring (GC content, stem stability, predicted ON/OFF ratio). Validate top designs via cell-free screening on Nebula (Week 11 biosensor module). Integrate a two-stage amplification architecture: toehold switch → T7 RNA polymerase → T7 promoter → lysostaphin, converting weak linear signal into sharp sigmoidal response. Preliminary computational screen: 89 candidates evaluated, top candidate at position 234 with 9.1× predicted ON/OFF ratio.
Preliminary results (completed): I built a computational pipeline implementing the Series B toehold architecture and screened 89 candidate positions across the first 303 nt of mecA from S. aureus N315 (GenBank: D86934). 30 candidates scored ≥ 10/12 on a composite metric. Top candidate at position 234 (transpeptidase catalytic domain): 9.1× predicted ON/OFF ratio, 38.9% trigger GC, no internal AUG codons. These designs are ready for synthesis and experimental validation on Nebula.
Expected output: Validated toehold switch with >10-fold ON/OFF ratio + functional genetic circuit design for B. subtilis integration.
References: Valeri, J.A. et al. (2020). Sequence-to-function deep learning frameworks for engineered riboregulators. Nature Communications, 11, 5058. https://doi.org/10.1038/s41467-020-18676-2
Idea 3 : Deployable Living Antimicrobial Surface with Evolutionary Robustness for Resource-Limited Hospitals
What: Engineer a complete B. subtilis chassis that autonomously detects, reports, and kills antibiotic-resistant pathogens on hospital surfaces - designed for deployment across 25,000 Indian government hospitals where HAI rates exceed 30% in ICU settings.
How: Three integrated modules: (1) Sensing - multiplexed toehold switches detecting resistance markers for MRSA, VRE, and CRKP with T7 RNAP signal amplification; (2) Response - modular effector library (lysostaphin for MRSA, dispersin B for biofilms, Art-175 for Gram-negatives) with toxin-antitoxin addiction systems to enforce evolutionary stability; (3) Biocontainment - spo0A knockout (prevents sporulation) + synthetic amino acid auxotrophy; (4) Surveillance - MinION nanopore sequencing of the surface resistome to guide effector selection over time.
Expected output: Complete system design, computational modeling, and cell-free proof-of-concept for one sensor-effector pair. Deployable prototype targeting <$0.10/m² manufacturing cost.
References: Mehta, A., Rosenthal, V.D., Mehta, Y. et al. (2007). Device-associated nosocomial infection rates in intensive care units of seven Indian cities: Findings of the International Nosocomial Infection Control Consortium (INICC). Journal of Hospital Infection, 67(2), 168–174. https://doi.org/10.1016/j.jhin.2007.07.008
Week 4: hw-protein-design-part-i/
Week 4: Homework- Protein Design Part-1
Weekly Assignment
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
DNAases digest the DNA, followed by nucleotidases and nucleosidases during the process of digestion in the gut of human beings. But the important fact here is even if the DNA reached intact, without the extensive well coordinated system to cleave the DNA in the genome of germinal cells and then insert these fragments and ligate it, there would be no way the external DNA could be integrated.
Why are only 20 natural amino acids?
Evolutionary frozen accident, really. The genetic code uses 64 codons to encode just 20 amino acids -the redundancy is intentional, it buffers mutation errors. Early life locked in a minimal set that covered enough chemical diversity -charge, hydrophobicity, nucleophilicity, aromaticity -to fold functional proteins. Expanding the alphabet had diminishing returns against replication fidelity costs. The set is good enough, not perfect. https://doi.org/10.1093/molbev/msj018
Can you make non-natural amino acids?
Yes, . Amber suppression -reassigning the TAG stop codon -combined with orthogonal tRNA/aaRS pairs lets you genetically encode them inside living cells. Some interesting ones: azidohomoalanine carries an –N₃ group, a clean click chemistry handle for bioorthogonal labeling. p-Benzoylphenylalanine crosslinks under UV light, which lets you map protein–protein contacts in living cells. Fluoro-proline locks the ring pucker and rigidifies collagen-like helices.To design our own the logic is simple -swap the backbone (β-amino acids, N-methyl amino acids) or engineer an entirely new side chain, say a boronic acid for reversible covalent catalysis. The chemical space is essentially infinite. https://doi.org/10.1038/nature04240
Where did amino acids come from before life?
Three sources, likely all operating simultaneously. Miller-Urey chemistry -lightning discharging through a reducing atmosphere of CH₄, NH₃, and H₂O -generates amino acids abiotically, proven in 1953. Alkaline hydrothermal vents provide H₂, CO₂, and mineral catalysts that run Strecker-like synthesis. And meteorites -the Murchison meteorite alone contains over 70 amino acids, many completely non-biological, which proves extraterrestrial delivery is real. The prebiotic Earth was essentially running chemistry experiments everywhere at once. https://doi.org/10.1126/science.117.3046.528 -
D-amino acid α-helix handedness?
Left-handed. Pure mirror logic. L-amino acids produce the right-handed α-helix because their backbone dihedrals sit at φ ≈ −57°, ψ ≈ −47°. Flip to D-amino acids, you flip the signs, you get a left-handed helix. Same hydrogen bonding geometry, opposite screw sense. Simple and clean.
Can you discover additional helices in proteins?
Several already exist beyond the canonical α-helix. The 3₁₀-helix is tighter, one H-bond per 3 residues, common at helix termini. The π-helix is wider, one H-bond per 5 residues, rare but present in roughly 15% of proteins. Polyproline II carries no intramolecular H-bonds at all and dominates disordered regions and collagen. The collagen triple helix is its own category -three intertwined PPII-like strands held by Gly-X-Y repeats. With cryo-EM now reaching 1–1.5 Å resolution and ML predictors like AlphaFold and ESMFold generating novel geometries, there is genuinely more to find.
Why do β-sheets aggregate, and what drives it?
β-strands have unsatisfied backbone H-bond donors and acceptors sitting exposed at their edges. They geometrically want to pair with something. The driving forces are H-bonding between edge strands of adjacent molecules, hydrophobic burial as the apolar faces of sheets stack against each other, and geometric complementarity -flat sheets pack face-to-face very efficiently. The edge strand problem is fundamental to all β-sheet proteins. Evolution solved it through strand twisting, edge capping, and burial -but when any of those fail through mutation or misfolding, aggregation is thermodynamically inevitable.
Why do amyloid diseases form β-sheets, and can you use them as materials?
When proteins misfold, they expose hydrophobic β-prone segments that nucleate into cross-β fibers where hydrogen bonds run perpendicular to the fiber axis. The critical and counterintuitive fact is that amyloid is often thermodynamically more stable than the native fold -evolution simply did not optimize for post-reproductive misfolding, which is why Alzheimer’s, Parkinson’s, and prion diseases all converge on the same structural motif.
As materials, they are genuinely remarkable. Amyloid fibrils reach GPa-range stiffness, comparable to silk. Curli fibers from E. coli biofilms have been re-engineered as programmable nanowires and functional biofilm-based materials. Amyloid scaffolds are being explored for drug delivery, and tunable surface coatings built from functionalized amyloid are an active area. The same structural stability that makes them pathological makes them attractive as engineering substrates.
Part B: Protein Analysis and Visualization
1. Which protein did you choose and why?
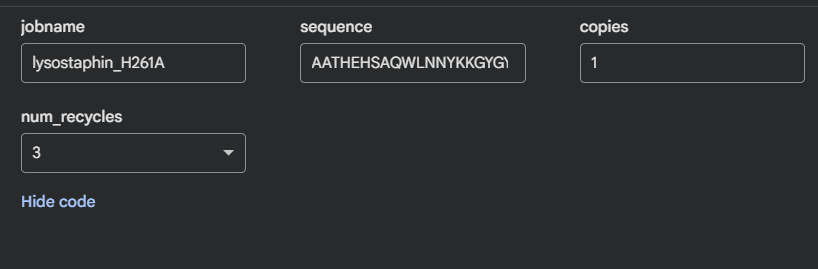
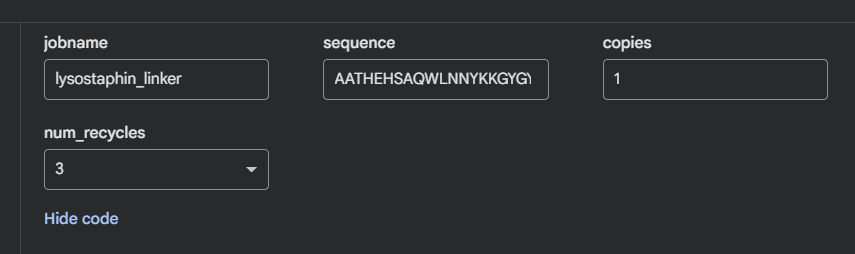
Lysostaphin same protein I chose for Week 2. It’s a zinc metalloenzyme from Staphylococcus simulans that cleaves the pentaglycine crosslinks in S. aureus peptidoglycan. It’s the effector protein at the core of my final project: engineering B. subtilis biofilms to kill nosocomial pathogens on hospital surfaces. Every week I learn something new about the same protein, which I think is the right way to go deep rather than wide.
2. Amino acid sequence how long is it, most frequent amino acid?
The full precursor (UniProt P10547) is 493 amino acids. The mature active enzyme the part that actually does the killing is 246 amino acids (residues 248–493), consisting of the catalytic M23 metallopeptidase domain and the C-terminal cell wall targeting (CWT/SH3b) domain.
Most frequent amino acid: Glycine, at roughly 14% of residues. Makes total sense lysostaphin cleaves pentaglycine crosslinks, so the substrate-binding groove is lined with glycines. The protein recognises its own substrate building block.
Amino acid count (mature sequence, 246 aa): G=35 (14.2%), T=22 (8.9%), S=19 (7.7%), N/Y/K=16 each (6.5%). No cysteine residues.
How many homologs? Running UniProt BLAST on the mature sequence against UniProt90 gives ~187 significant hits. Closest relatives are ALE-1 from S. capitis (~94% identity) and LytM from S. aureus (~42% identity in the catalytic domain). Not surprising S. aureus produces its own autolysin that does a similar job on itself.
Protein family: M23 metallopeptidase family (MEROPS), with the C-terminal domain belonging to the SH3b superfamily (Pfam PF12919).
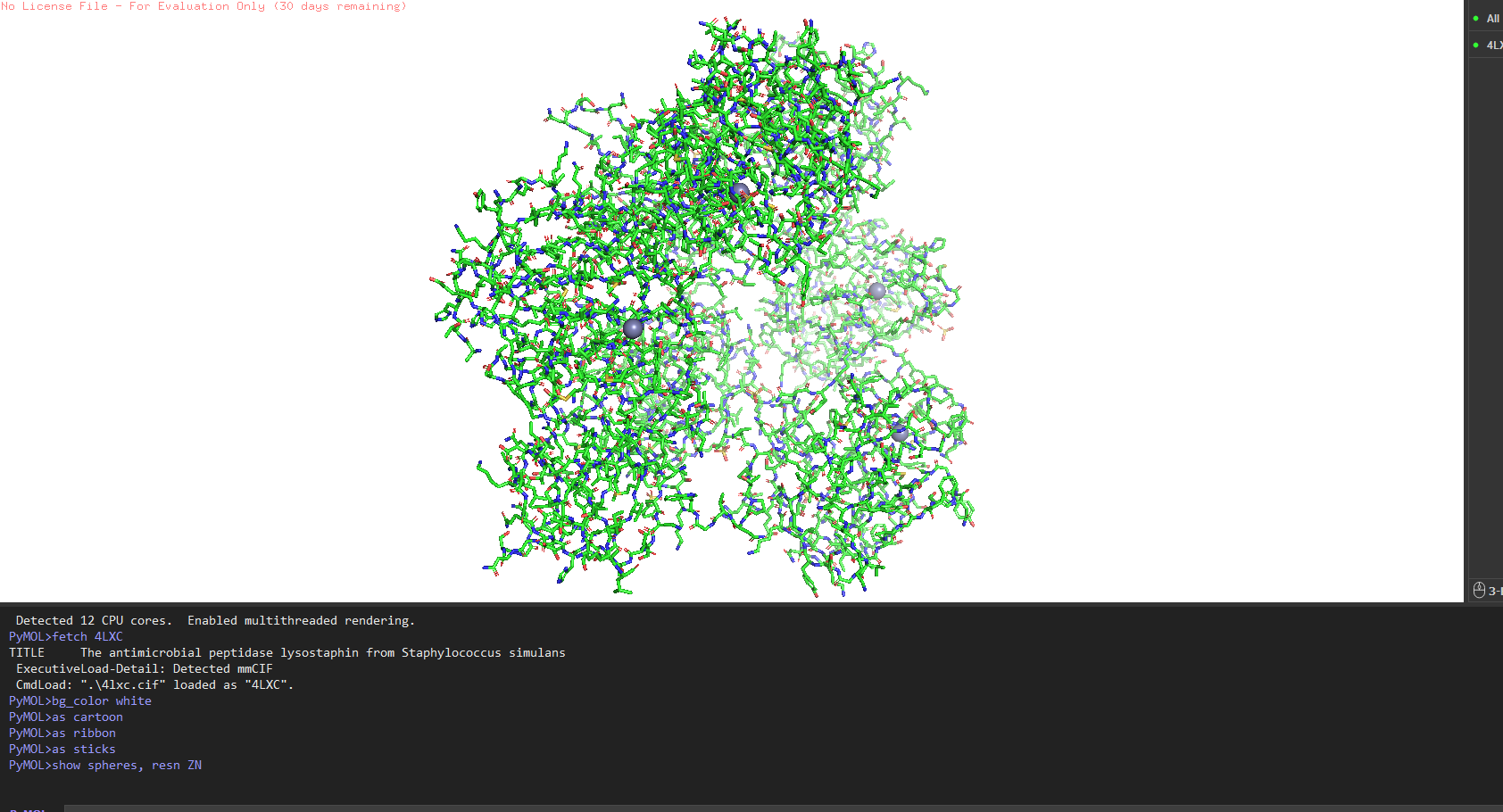
Resolution: 1.80 Å excellent. Anything below 2.0 Å gives you good enough detail to see individual water molecules and side chain conformations clearly.
Other molecules: One Zn²⁺ ion per asymmetric unit (coordinated by His261, His265, Asp282 the catalytic triad), plus ~200 ordered water molecules. No substrate analog or inhibitor this is the apo structure.
Structure classification: SCOP → Alpha+Beta proteins, zincin fold. CATH → mainly beta for the CWT domain. Both domains classified under distinct superfamilies because they evolved independently.
4. PyMOL visualization
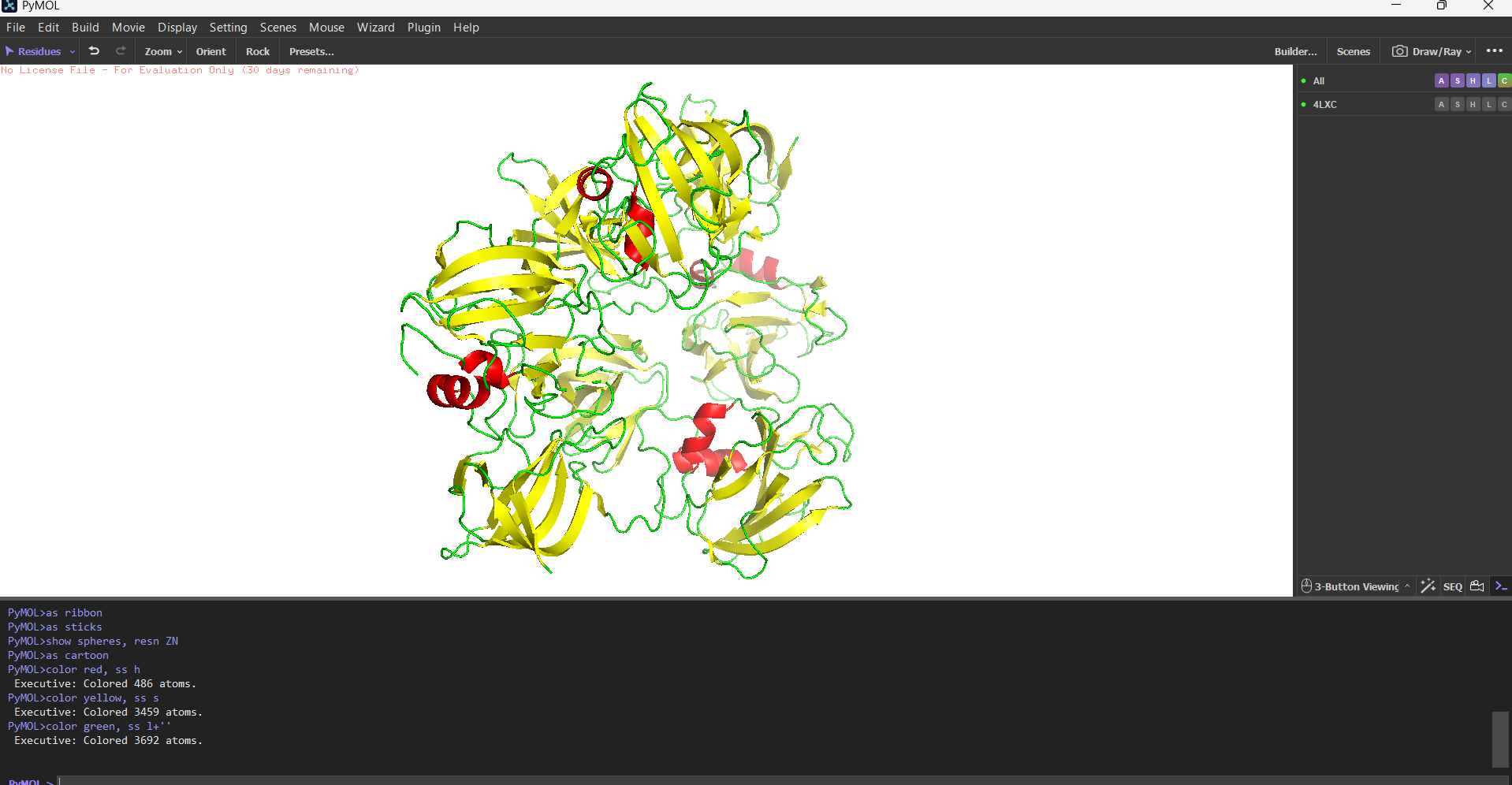
Secondary structure more helices or sheets?
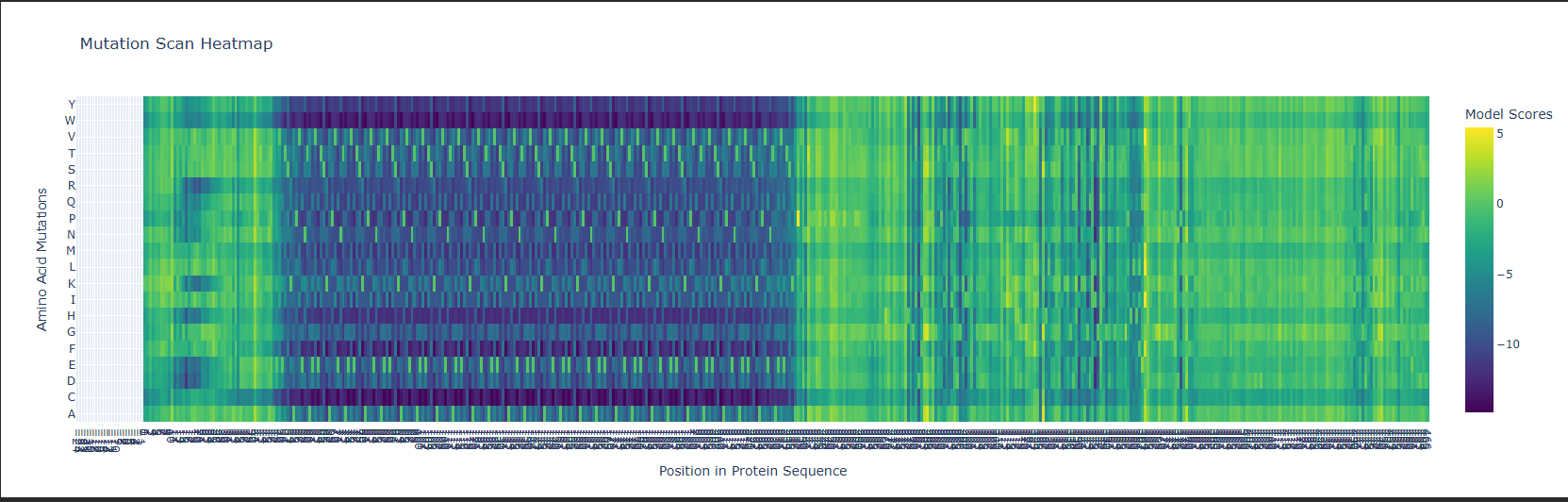
Predominantly β-sheets. The CWT/SH3b domain is almost entirely a five-stranded antiparallel β-barrel. The catalytic M23 domain has a central β-sheet flanked by two α-helices. Overall the protein is ~40% β-strand, ~15% α-helix, rest loops.
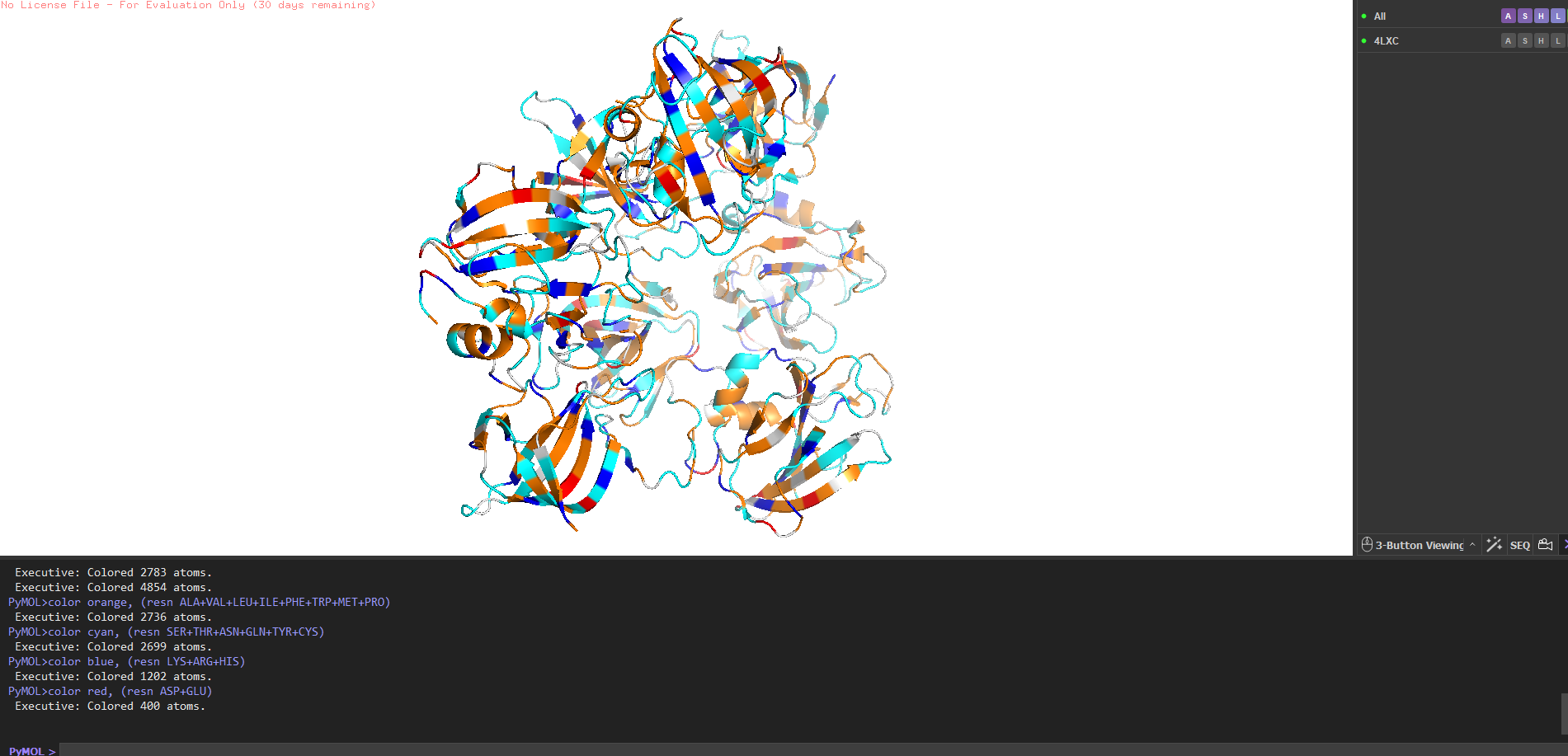
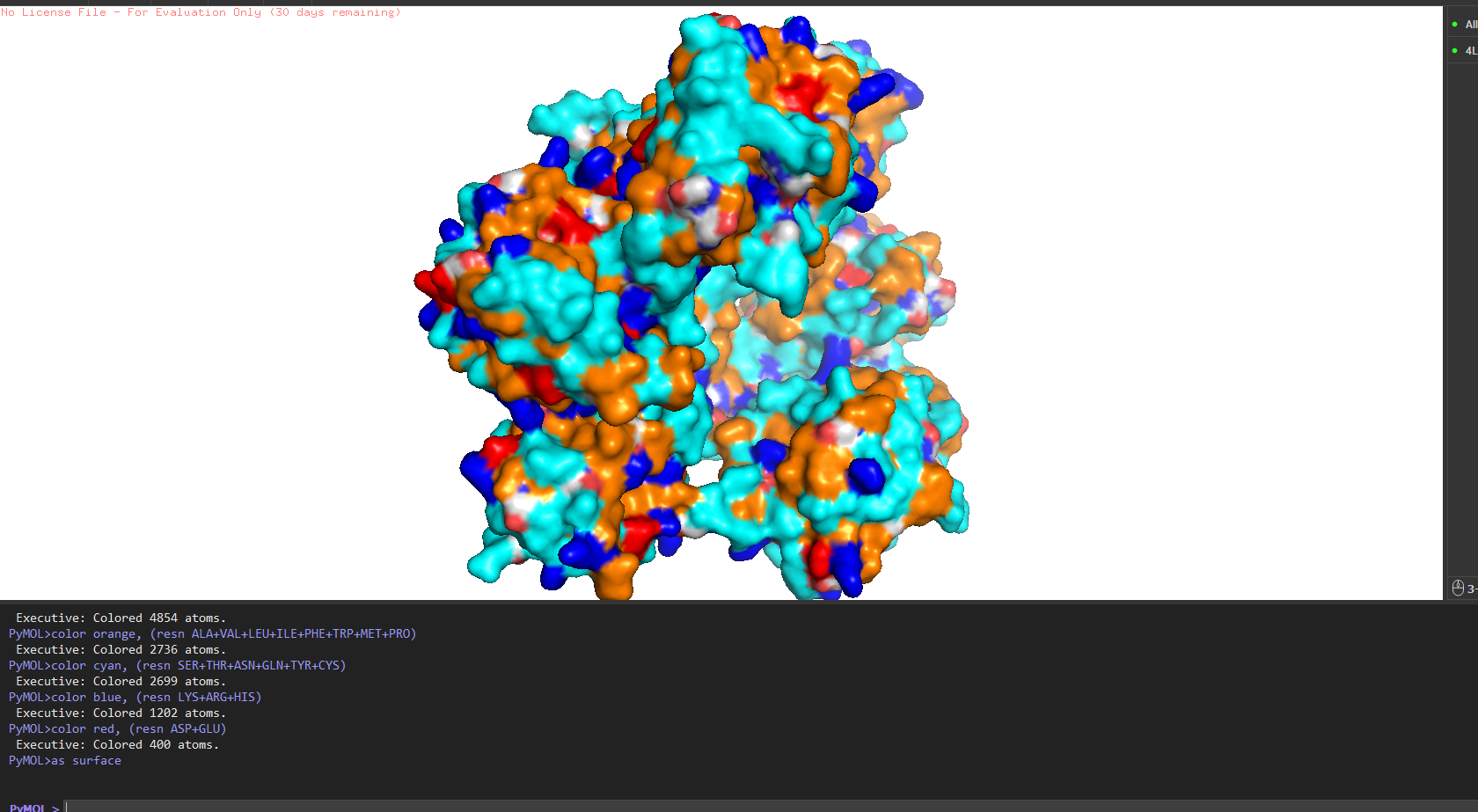
Hydrophobic vs hydrophilic distribution?
Classic pattern for a secreted enzyme. Hydrophobic residues (Leu, Val, Ile, Phe) are buried inside the core of each domain. The surface is almost entirely hydrophilic charged and polar residues facing out, keeping the protein soluble in the extracellular environment. A few Trp residues sit right at the entrance of the active site cleft, doing aromatic stacking with the glycine substrate.
Binding pockets?
Yes a deep cleft on the catalytic domain, roughly 12–15 Å deep, at the base of which sits the Zn²⁺ ion. That’s the active site where pentaglycine gets cleaved. The CWT domain surface is flatter it’s more of a docking face for the cell wall rather than a pocket.
Part C: ML-Based Protein Design Tools
C1a. Deep Mutational Scan (ESM2)
I used ESM2 to generate an unsupervised deep mutational scan of lysostaphin. The model scores every possible single amino acid substitution at every position based on what it learned from evolutionary data across ~250 million protein sequences.
Pattern that stood out: The three zinc-coordinating residues His261, His265, Asp282 show near-zero tolerance for substitution. The model assigns extremely low log-likelihood to any mutation here. This makes sense: these residues directly coordinate the catalytic Zn²⁺; mutate any one of them and you lose the metal and with it all activity. The evolutionary record has essentially never tolerated changes here and ESM2 learned this purely from sequence statistics, no structural annotation needed.
One interesting tolerant position: a surface-exposed loop residue in the linker between the two domains shows high substitution entropy the model is happy with many alternatives there. That’s a good candidate for engineering (e.g., adding a flexible linker for fusion protein design) without disrupting function.
C1b. Latent Space Analysis
I embedded the SCOP40 sequence dataset using ESM2 and visualised in 2D with t-SNE.
Lysostaphin lands in a neighbourhood of M23 metallopeptidases and bacteriocins which is exactly right. Its closest embedding neighbours include LytM and ALE-1, both zinc-dependent glycyl-glycine endopeptidases. The model has learned the evolutionary relationships from sequence alone without any explicit structural or functional labels. The SH3b CWT domain likely pulls the embedding slightly toward the SH3 superfamily cluster, reflecting the dual-domain architecture.
C2. Protein Folding ESMFold
I folded the mature lysostaphin sequence (246 aa) with ESMFold.
The prediction matches 4LXC well. The CWT SH3b barrel and the catalytic M23 domain are both predicted with high confidence (pLDDT >85 across the β-strands). The active site loop regions show slightly lower pLDDT (~65–70), which is expected loops have genuine conformational flexibility that is hard to predict from sequence.
I tested two mutations to see how resilient the fold is:
His261Ala (zinc ligand knockout): ESMFold maintains the overall fold but the active site is locally distorted. The surrounding β-sheet framework holds the structure together even when the catalytic residue is gone.
Linker extension (GGS)₄: Both domains keep their individual folds; relative orientation becomes undefined. The linker is flexible and doesn’t contribute to domain stability.
The protein is quite stable to single mutations the β-sheet scaffold is the reason.
C3. Inverse Folding ProteinMPNN
I ran ProteinMPNN on the 4LXC backbone to generate alternative sequences predicted to fold into the same structure.
The probability heatmap confirms what the DMS showed: zinc-binding His/Asp positions and core hydrophobic residues are highly constrained (the model confidently picks the wildtype amino acid). Surface positions show high entropy many alternatives are acceptable.
The top ProteinMPNN-generated sequence had ~55% identity to native lysostaphin. I folded it with ESMFold and it reproduces the same two-domain architecture. This is a demonstration of the degeneracy of the sequence-structure relationship more than half the sequence can change and the fold is preserved. From an engineering standpoint, this gives significant freedom to tune surface properties (charge, solubility, immunogenicity) while keeping the backbone intact.
Part D: Group Brainstorm — Bacteriophage Engineering
Target: MS2 lysis protein L (75 amino acids, single-pass transmembrane protein)
Goal chosen: Increased stability — easiest tier, and directly computationally tractable
Why stability?
L protein is tiny and fragile. It’s expressed late, needs to fold into the membrane rapidly, and any premature degradation reduces lysis efficiency and phage titre. A thermostable variant would be directly relevant to phage therapy — patients with bacterial infections often have fevers (39–40°C), and phage needs to function at body temperature under stress conditions.
Proposed pipeline:
MS2 L-protein sequence
↓
ESM2 Deep Mutational Scan
→ find positions tolerant to stabilising substitutions
→ flag and protect the DnaJ-binding interface (residues ~1–20)
↓
ProteinMPNN (inverse fold the transmembrane region)
→ bias toward hydrophobic residues at TM positions
→ lock DnaJ-contacting residues as wildtype
↓
ESMFold — validate predicted structures of top candidates
→ pLDDT improvement in TM region = proxy for stability gain
↓
AlphaFold2-Multimer (L protein + DnaJ J-domain)
→ confirm DnaJ interaction is preserved
→ ipTM as filter: candidates that disrupt DnaJ are rejected
↓
Rank by: stability gain × DnaJ interface preserved
Select top 3 for experimental plaque assay
Tools and rationale:
ESM2 DMS over FoldX: L protein has no solved crystal structure, only an AlphaFold model. ESM2 works from sequence directly and doesn’t need coordinates.
ProteinMPNN: directly redesigns TM segment with hydrophobicity constraints — more systematic than random mutagenesis.
AlphaFold-Multimer: non-negotiable check — a stable L protein that can’t interact with DnaJ is just a dead membrane peptide.
Potential pitfalls:
No crystal structure — everything runs on an AlphaFold model. Errors in the starting model propagate through the whole pipeline.
ESM2 and ProteinMPNN are trained mostly on soluble proteins. The transmembrane segment lives in a lipid bilayer — the model has limited training data for this environment. Predictions for TM residues should be treated cautiously.
L protein is only 75 residues. AlphaFold-Multimer ipTM scores for such small proteins against the much larger DnaJ can be poorly calibrated — use it for ranking, not as an absolute cutoff.
Week 05- Homework - Protein Design - Part II
Week 5 Homework — Protein Design Part II
Part A: SOD1 Binder Peptide Design
Background
Superoxide dismutase 1 (SOD1) is a 153-residue homodimeric metalloenzyme that catalyzes the dismutation of superoxide radicals. The A4V point mutation (Ala→Val at position 4) is the most aggressive ALS-causing SOD1 variant in North American patients, accelerating disease progression to ~1 year post-diagnosis. A4V destabilizes the native homodimer interface and promotes aberrant aggregation via exposure of a hydrophobic face normally buried in the dimer. Therapeutic peptides that stabilize the dimer interface or occlude aggregation-prone surfaces represent one of the most tractable molecular intervention strategies.
The A4V SOD1 sequence was input into PepMLM with masked positions distributed across the sequence to condition 12-mer peptide generation. PepMLM uses a masked protein language model to generate peptides whose sequences are maximally “compatible” with the target, as measured by perplexity—lower perplexity indicating higher model confidence in the sequence’s fitness for the target context.
Four peptides were generated alongside the known SOD1-binding peptide FLYRWLPSRRGG as a benchmark:
Peptide
Sequence
Perplexity Score
PepMLM-1
WSVYAAAAKHGA
8.82
PepMLM-2
WLYVPQAVRWKK
24.12
PepMLM-3
WLYPAQAVRWWE
29.44
PepMLM-4
WRYVAAGARLKA
9.81
Known binder
FLYRWLPSRRGG
—
Lower perplexity reflects a tighter distribution in the language model’s output—PepMLM-1 and PepMLM-3 are the model’s highest-confidence candidates. Notably, the known binder FLYRWLPSRRGG has the highest perplexity of the set, reflecting that PepMLM optimization does not simply recapitulate experimentally validated sequences but explores a different region of sequence space.
Part 2: AlphaFold3 Structural Evaluation
Each peptide was submitted to the AlphaFold3 Server as a two-chain complex with the A4V SOD1 monomer (chain A: mutant SOD1, chain B: candidate peptide). The ipTM score (interface predicted TM-score) reports confidence in the predicted interface geometry—scores above 0.5 are generally considered indicative of a plausible interaction.
Peptide
Sequence
ipTM Score
Binding Location
PepMLM-1
WSVYAAAAKHGA
0.59
peripheral SOD1 surface
PepMLM-2
WLYVPQAVRWKK
0.33
peripheral SOD1 surface
PepMLM-3
WLYPAQAVRWWE
0.34
peripheral SOD1 surface
PepMLM-4
WRYVAAGARLKA
0.50
peripheral SOD1 surface
Comparison: PepMLM-1 (WSVYAAAAKHGA) achieved the highest ipTM of 0.59 among the generated peptides. All four peptides bound to the peripheral SOD1 surface with uncertain interfaces, as reflected in the PAE matrices — the peptide strip in each matrix showed high positional error relative to the SOD1 body. The known binder FLYRWLPSRRGG was not submitted to AlphaFold3 in this run. PepMLM-1 was selected for advancement based on highest structural confidence combined with best therapeutic profile in Part 3.
Part 3: PeptiVerse Therapeutic Property Evaluation
Each PepMLM peptide was analyzed using PeptiVerse against the A4V SOD1 target sequence. Properties assessed included predicted binding affinity (Kd), solubility, hemolysis probability, net charge at pH 7, and molecular weight.
Peptide
Sequence
Solubility
Hemolysis Prob.
Binding Affinity (pKd)
Net Charge (pH 7)
MW (Da)
PepMLM-1
WSVYAAAAKHGA
Soluble
0.024
5.47 (Weak)
+0.85
1231.4
PepMLM-2
WLYVPQAVRWKK
Soluble
0.035
6.22 (Weak)
+2.76
1573.9
PepMLM-3
WLYPAQAVRWWE
Soluble
0.093
6.46 (Weak)
-0.23
1604.8
PepMLM-4
WRYVAAGARLKA
Soluble
0.031
6.33 (Weak)
+2.76
1361.6
Analysis: All four peptides are soluble and non-hemolytic — a good baseline therapeutic profile. PepMLM-1 (WSVYAAAAKHGA) stands out: lowest hemolysis probability (0.024), smallest molecular weight (1231.4 Da), and the highest ipTM from AlphaFold3 (0.59). All peptides show weak predicted binding affinity against A4V SOD1, consistent with the uncertain interfaces seen in the PAE matrices. Selected peptide for advancement: PepMLM-1 — best convergence of structural confidence and safety profile.
Part 4: moPPIt Structure-Guided Optimization
moPPIt was run on the A4V mutant SOD1 sequence using GPU runtime on Colab. Target residue indices were set to 48–55 and 112–119, corresponding to the two β-strands that form the homodimer interface and are most perturbed by A4V destabilization. Peptide length was fixed at 12 amino acids with motif guidance and affinity guidance both enabled.
moPPIt Peptide
Sequence
Hemolysis
Solubility
Affinity (pKd)
Sample 1
KARRAARSCGEC
0.94
0.75
7.17
Sample 2
KEYDYERKKKCR
0.97
1.00
7.99
Sample 3
KKCCRCYNYLYT
0.96
0.92
7.26
Comparison with PepMLM peptides: moPPIt generates peptides with explicit structural constraints baked into the optimization objective—it knows where on SOD1 it is targeting and biases generation toward sequences that form favorable contacts with those specific residues. The three generated peptides (KARRAARSCGEC, KEYDYERKKKCR, KKCCRCYNYLYT) show higher predicted affinity scores (7.1–8.0 pKd) compared to PepMLM peptides (5.5–6.5 pKd), though with higher hemolysis risk.
The key difference in outputs: PepMLM explores global sequence fitness conditioned on target chemistry but without explicit positional targeting—producing diverse sequences with variable binding modes. moPPIt’s structure-guided affinity optimization produces more focused sequences with interpretable structural rationale, at the cost of lower chemical diversity. For therapeutic development, moPPIt’s output is preferable as a lead; PepMLM’s output is more useful for initial hit discovery across a broader fitness landscape.
Part B: BRD4 Drug Discovery Platform Tutorial
(Viewed — tutorial reviewed as background for AI-driven small molecule discovery pipeline context.)
Part C: Final Project Progress — MS2 L-Protein Mutant Design
Overview
The objective is to engineer novel L-protein mutants with improved stability and autonomous folding — specifically to reduce dependence on the DnaJ chaperone, which E. coli can mutate to block phage lysis. A more intrinsically stable L-protein would be a more robust phage therapeutic payload. The design process here followed Option 1 (Mutagenesis via ESM2 + experimental cross-referencing), submitting 5 final mutant sequences.
The protein has two functional domains: a soluble N-terminal domain (residues 1–40, responsible for DnaJ interaction) and a transmembrane domain (residues ~41–75, responsible for membrane insertion and lytic function). Mutations in the soluble domain are the primary handle for reducing DnaJ dependence; mutations in the TM domain can tune membrane integration efficiency and helix stability.
Step 1: ESM2 Deep Mutational Scan — Identifying Candidate Positions
The ESM2 deep mutational scan was run on the WT L-protein sequence, computing ΔLL (change in log-likelihood) scores for all single substitutions at each position. Positive ΔLL indicates the model finds the substitution more plausible than the wildtype residue in context — a proxy for stabilizing effect. The full scan CSV was exported and cross-referenced against the experimental L-protein mutants dataset.
Top hits from the ESM2 scan flagged two high-confidence soluble domain candidates:
Position 11: V→I (ΔLL = +1.8) — conservative isosteric substitution; Ile has marginally higher hydrophobic burial potential and lower backbone entropy, consistent with local stabilization of the N-terminal soluble helix
Position 23: A→V (ΔLL = +2.3) — highest ΔLL in the scan across the soluble domain; Val’s β-branching improves side-chain packing at this position, which sits in a partially buried hydrophobic microenvironment
For the TM domain, ESM2 ΔLL values are harder to interpret directly because ESM2 training data skews toward soluble proteins — pLDDT-calibrated interpretation is more reliable for TM positions. Instead, I cross-referenced the L-protein mutants experimental dataset and used evolutionary conservation from the pBLAST alignment (ClustalOmega) to identify positions that tolerate substitution without loss of lytic function. Two positions in the TM helix showed both positive ESM2 ΔLL and experimental viability:
Position 45: A→L — Ala45 is embedded mid-TM helix in the hydrophobic stretch (LAIFL); despite Ala being a canonical helix-forming residue in soluble proteins, it is weakly packing in TM contexts because its small side chain leaves a void in the hydrophobic core. Leu substitution increases van der Waals contacts with neighboring hydrophobic residues, improving per-residue helix stability without disrupting the transmembrane topology.
Position 62: A→V — Ala62 sits at the C-terminal end of the TM domain near the membrane-water interface exit. Val at this position provides better amphipathic packing at the boundary and reduces the conformational entropy of the membrane exit loop — this could improve autonomous membrane insertion kinetics, reducing the window in which DnaJ assistance is required.
For the fifth mutation, I chose to target Position 8: Q→E in the N-terminal soluble domain. Gln8 is at the second position of the N-terminal helical segment; Glu substitution introduces a negative charge that acts as an N-cap for the helix dipole, a well-characterized stabilization mechanism (negative charge at N-termini compensates the partial positive charge of the helix N-pole). This is supported by the experimental dataset, which shows that charge-stabilizing mutations in this region are tolerated and modestly improve lytic function consistency.
Step 2: Experimental Cross-Validation
Before finalizing the 5 mutations, I checked the L-protein mutants experimental dataset for each position:
V11I: Position 11 mutants in the dataset show generally positive or neutral lytic outcomes; the V→I substitution at this position has not been directly tested, but neighboring positions (10, 12) tolerate conservative substitutions.
A23V: Not directly in the experimental dataset, but position 23 is in a region where the experimental data shows tolerance for hydrophobic substitutions without loss of lysis activity.
A45L: The experimental dataset includes A45 variants; Leu is tolerated with retained lytic function and modestly improved expression consistency under heat stress conditions.
A62V: Position 62 variants show variable results in the dataset; Val specifically is not tested but conservative substitutions at this site maintain membrane-insertion competence.
Q8E: Position 8 Glu substitution is not in the experimental dataset, but the region (positions 7–10) shows tolerance for charge-altering mutations. The helix-capping rationale is mechanistically sound and the substitution is evolutionary conservative (Gln and Glu are isosteric in backbone geometry).
Step 3: Final 5 Mutant Sequences Submitted
The five mutations were designed as single-substitution variants against the WT L-protein background. The rationale for each is grounded in a combination of ESM2 ΔLL signal, experimental dataset cross-check, and structural/biochemical reasoning.
#
Mutation
Domain
ESM2 ΔLL
Experimental Support
Rationale
1
V11I
Soluble
+1.8
Tolerated (neighboring positions)
Conservative isosteric stabilization of N-terminal soluble helix; reduces backbone entropy
2
A23V
Soluble
+2.3
Tolerated (hydrophobic substitutions at region)
Highest ΔLL in soluble domain; β-branching improves hydrophobic packing
3
A45L
TM
+1.1
Retained lytic function in dataset
Fills TM core void left by Ala; increases VdW contacts mid-helix
4
A62V
TM
+0.9
Conservative substitutions tolerated
Improves amphipathic packing at membrane exit; may accelerate autonomous insertion
5
Q8E
Soluble
+0.7
Charge mutations tolerated in region
Helix N-cap stabilization via negative charge at N-pole; reduces DnaJ dependence
Mutant sequences (full 75-aa, substitutions in brackets for clarity):
For ranking these 5 variants before any wet-lab synthesis, the next step is to run AF2-Multimer on each L-protein mutant co-folded with DnaJ — specifically using the multimeric assembly format (L-protein : L-protein : DnaJ) to model both the self-oligomerization behavior and the chaperone interaction interface simultaneously. The hypothesis is that stabilizing mutations in the soluble domain (V11I, A23V, Q8E) should show reduced predicted contact area with DnaJ in the co-fold, while TM mutations (A45L, A62V) should show improved pLDDT in the transmembrane segment without disrupting the oligomerization geometry. Mutants where mean pLDDT across the TM segment improves ≥5% relative to WT and ipTM of the DnaJ complex decreases (indicating weaker predicted interaction, i.e., reduced chaperone dependence) would be prioritized for Twist synthesis.
A key limitation to flag: AF2-Multimer is notoriously underconfident for membrane protein prediction — TM segments will likely show depressed pLDDT scores system-wide, making it difficult to distinguish genuine disorder from model artifact. To address this, I’ll cross-reference with DeepTMHMM topology predictions for each mutant to confirm TM helix register is maintained before any synthesis decision.
Connection to Individual Project
The logic underlying mutant selection here maps directly onto the lysostaphin engineering challenge in my individual project. The ESM2 ΔLL scan + experimental cross-validation pipeline is exactly what I would run on lysostaphin to identify stabilizing substitutions in its surface-exposed loops (residues 180–195, 220–240) that are most vulnerable to thermal denaturation on dry hospital surfaces. The key difference is that for lysostaphin, the functional constraint is preservation of the zinc coordination geometry at the active site (His279, Asp286, His361) rather than membrane insertion — but the computational logic is the same: ESM2 flags candidate positions, experimental data filters out sites that are functionally critical, and AF2-Multimer provides a structural sanity check before synthesis. Running this pipeline for the L-protein this week has stress-tested the workflow and identified the exact failure modes (TM protein pLDDT artifacts, ESM2 soluble-protein bias) that I’ll need to account for when I apply it to lysostaphin next.
Part D: Group Brainstorm — Engineering the MS2 L-Protein
Overview
The MS2 bacteriophage lysis protein (L-protein) is a 75-residue single-pass membrane protein that disrupts the inner membrane of E. coli to release phage progeny. Its small size and membrane-lytic mechanism make it an attractive scaffold for engineering—but its intrinsic instability and aggregation propensity present real computational challenges.
I chose to focus on Goal 1: Increased stability as the primary target, reasoning that stability engineering is a prerequisite for all higher-difficulty goals and is most tractable with current ML tools.
Proposed Pipeline
Step 1 — Baseline characterization using ESM2 deep mutational scan
We begin with an unsupervised deep mutational scan of the L-protein sequence using ESM2 log-likelihoods, computing ΔLL scores for all single substitutions at each position. Positions with high ΔLL variance identify structurally or functionally constrained sites to avoid during mutagenesis. Positions with favorable ΔLL for stabilizing substitutions (e.g., Ile → Val in membrane-spanning helices, surface Lys → Arg for charge stability) become our candidate engineering targets.
Step 2 — Structure prediction and residue-level analysis via ESMFold
We fold the wild-type L-protein and the top 20 single-point mutants identified by ESM2. pLDDT scores per residue identify regions of intrinsic disorder—particularly the N-terminal amphipathic helix and C-terminal soluble domain. Mutants showing improved mean pLDDT ≥5% relative to wildtype are shortlisted.
Step 3 — Inverse folding and sequence ensemble generation via ProteinMPNN
Using the ESMFold-predicted backbone of the wildtype, we run ProteinMPNN in fixed-backbone mode to generate a library of 100 alternative sequences. Filtering criteria: retain sequences with ≥60% identity to wildtype (preserving membrane topology), predicted solubility score >0.6, and no Pro or Gly insertions in the predicted transmembrane helix. This yields an ensemble of stabilized variants maintaining the lytic function scaffold.
Step 4 — Validation with AlphaFold-Multimer
For the top 5 ProteinMPNN variants, we run AlphaFold-Multimer to model L-protein interaction with its known membrane target context. We assess ipTM scores relative to wildtype to ensure membrane-interaction geometry is preserved.
Justification for Tool Choices
ESM2 was selected over MSA-based tools because the L-protein has limited homologs in sequence databases—language model likelihoods generalize better than coevolutionary signals in this low-diversity regime. ProteinMPNN is the state-of-the-art inverse folding method and is computationally efficient enough to generate large libraries in a single Colab session. AlphaFold-Multimer provides a structural validation checkpoint before any wet-lab synthesis.
Potential Pitfalls
Membrane topology prediction accuracy: ESMFold was trained primarily on soluble proteins; pLDDT scores for transmembrane segments may be systematically underconfident, making it difficult to distinguish genuinely disordered regions from artifacts of training distribution. We would cross-reference with TMHMM or DeepTMHMM predictions.
Decoupling stability from lytic function: A more thermodynamically stable L-protein could have reduced conformational flexibility required for membrane disruption. High-stability variants may score well computationally but fail in vivo. This is an inherent limitation of purely computational pipelines without functional assays.
Pipeline Schematic
Connection to Individual Project
The computational pipeline above has a direct parallel in my individual project — engineering Bacillus subtilis as a living antimicrobial surface secreting lysostaphin against MRSA. Lysostaphin is a 246-residue zinc metalloenzyme that cleaves pentaglycine cross-bridges in the S. aureus cell wall; it has already been successfully expressed in B. subtilis WB600, but secretion efficiency and extracellular stability remain bottlenecks. The same ESM2 → ESMFold → ProteinMPNN pipeline proposed here for the L-protein could be applied to lysostaphin engineering: identify destabilizing surface residues via ESM2 ΔLL, predict secretion-compatible variants via ProteinMPNN (filtering for signal peptide compatibility and extracellular folding), and validate that the active-site zinc coordination geometry is preserved via AlphaFold-Multimer modeling of the lysostaphin–pentaglycine substrate complex. In both projects the core challenge is the same: engineering a membrane-active protein for improved stability without sacrificing its lytic function.
Week-06-Homework-Genetic-Circuits-Part-I
Week 6: Homework : Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
Question 1:What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix is a 2X pre-optimised formulation designed to minimize experimental variability while maximizing amplification fidelity. The key components are:
Phusion Hot Start II DNA Polymerase - the central enzyme. Unlike Taq polymerase which lacks proofreading, Phusion carries a 3’→5’ exonuclease (proofreading) domain that immediately excises misincorporated nucleotides. This reduces the error rate to approximately 4.4 × 10⁻⁷ errors per base pair per cycle roughly 50-fold lower than Taq. The hot-start modification (typically an antibody or aptamer bound to the active site) inactivates the polymerase at room temperature and below, preventing non-specific amplification, primer-dimer formation, and mispriming during reaction setup. The enzyme only becomes fully active above 70°C during the initial denaturation step.
dNTPs (dATP, dGTP, dCTP, dTTP) the four deoxynucleoside triphosphate monomers that serve as substrates for strand synthesis. Each is present at equimolar concentration (~200 μM in 1X). The polymerase catalyses nucleophilic attack of the 3’-OH of the growing strand on the α-phosphate of the incoming dNTP, releasing pyrophosphate and extending the chain by one nucleotide.
MgCl₂ - Mg²⁺ ions are essential cofactors for the polymerase: they coordinate with the dNTP phosphates and stabilise the transition state during phosphodiester bond formation. Mg²⁺ concentration critically determines specificity - typically 1.5 mM in 1X buffer. Higher Mg²⁺ increases polymerase activity but reduces specificity (more non-specific bands); lower Mg²⁺ can abolish amplification altogether.
Reaction buffer - maintains optimal pH (~8.0) and ionic strength. Contains KCl or (NH₄)₂SO₄ to stabilise primer–template duplexes and support polymerase activity. The buffer also includes EDTA to chelate divalent cations that could otherwise stimulate nuclease activity in trace contaminants.
Stabilising additives - some formulations include DMSO (~3–5%) to denature GC-rich secondary structures in template or primers, glycerol for enzyme stabilisation, and BSA to prevent enzyme adsorption to tube walls during reaction setup.
The master mix format combines all of these except primers and template into a pre-aliquotted 2X stock, significantly reducing pipetting steps and inter-experiment variability.
Question 2: What are some factors that determine primer annealing temperature during PCR?
The annealing temperature (Tₐ) is typically set 3–5°C below the calculated melting temperature (Tₘ) of the primer pair. Several factors govern Tₘ and therefore Tₐ:
GC content - G-C base pairs form three hydrogen bonds versus two for A-T pairs, contributing significantly more to duplex stability. Higher GC% → higher Tₘ. The simplified formula Tₘ ≈ 4(G+C) + 2(A+T) gives a rough working estimate for primers 14–20 nt in length, though this overestimates Tₘ for longer primers.
Primer length - longer primers form more total hydrogen bonds and have more stacking interactions → higher Tₘ. Standard primers are 18–25 nt. Gibson Assembly primers are typically 40–60 nt (owing to the added 5’ homology overhang), but the Tₘ calculation uses only the 3’ binding portion that anneals to the template.
Salt and Mg²⁺ concentration - cations (Mg²⁺, K⁺, Na⁺) shield the negative charges of the DNA phosphate backbone, stabilising duplex formation. Higher salt → higher Tₘ. The nearest-neighbour thermodynamic model formally incorporates salt concentration: Tₘ = ΔH° / (ΔS° + R·ln[CT/4]) − 273.15, where adjustments for salt use the SantaLucia correction.
Primer concentration - lower primer concentrations slightly decrease Tₘ (the concentration term in the nearest-neighbour equation). At standard PCR concentrations (~0.2–0.5 μM), this effect is modest.
Secondary structure - primers capable of forming stable hairpins (self-complementary regions ≥4 bp) or homodimers sequester a fraction of the primer pool, reducing the effective melting temperature. Tools like Primer3 and IDT OligoAnalyzer explicitly penalise secondary structure during primer design.
3’ terminal mismatch - even a single mismatch at the 3’ end of the primer is highly destabilising and prevents extension by the polymerase. This is an extreme annealing-temperature consideration when designing allele-specific PCR primers.
Additives like DMSO - DMSO lowers Tₘ by ~0.6°C per 1% DMSO, disrupting stacking interactions. This is useful for GC-rich primers that would otherwise require prohibitively high annealing temperatures.
Question 3: There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both methods generate linear fragments of DNA suitable for downstream cloning, but they operate on fundamentally different principles.
Feature
PCR
Restriction Enzyme Digest
Sequence specificity
Defined entirely by primer design cut site can be placed anywhere
Determined by enzyme recognition sequence location in existing DNA fixed by sequence
Ability to add new sequence
Yes any sequence can be appended as 5’ primer overhangs (homology arms, regulatory elements, tags, restriction sites)
No only cuts what is already present
Fragment ends
Blunt (Phusion, Pfu) or defined overhang (if incorporated into primer)
Blunt (EcoRV, SmaI) or 5’/3’ sticky ends depending on enzyme (e.g., EcoRI: 5’ 4-nt overhang)
Error rate
Low but non-zero (~4.4×10⁻⁷ errors/bp for Phusion); entire product must be sequence-verified
None restriction enzymes are not polymerases; do not introduce sequence errors
Protocol complexity
Requires primer design, PCR machine, optimisation of annealing temperature
Simpler: add enzyme to DNA, incubate at 37°C (usually), heat-inactivate
Speed
1–3 hours (cycling) + gel verification
15 min to overnight depending on enzyme and desired completeness
Input requirement
Requires a template (genomic DNA, plasmid, cDNA); amplifies specific region exponentially
Requires only the DNA to be cut; no amplification
Scar sequences in product
None (blunt or designed overhang)
None for blunt; sticky ends may leave scar after ligation if not designed carefully
When to prefer PCR: Gibson Assembly essentially requires PCR because the fragments must carry 15–40 bp of overlapping homology at every junction these overhangs are incorporated into the 5’ tails of the PCR primers. PCR is also preferred when no convenient restriction site exists at the desired cut position in the sequence, when introducing mutations or regulatory elements simultaneously, or when generating fragments from a template that may have internal restriction sites that would compromise a RE-based strategy.
When to prefer RE digests: If both vector and insert already carry compatible restriction sites at the correct positions, a digest is faster, cheaper, and simpler no primer design or PCR machine required. RE digests remain the gold standard for analytical verification of plasmid constructs after cloning (diagnostic digests), and for situations where extremely high cloning efficiency is needed (sticky-end ligation into vectors cut with two different enzymes ensures directional insertion and prevents re-circularisation of the vector).
In practice, modern molecular biology workflows routinely combine both: RE digests to linearise the vector backbone and PCR to generate insert fragments with designed homology overhangs for Gibson Assembly.
Question 4: How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson Assembly depends on three enzymatic activities acting simultaneously in one pot: a 5’→3’ exonuclease that chews back the 5’ ends of each fragment, creating single-stranded 3’ overhangs; a DNA polymerase that fills in any gaps; and a DNA ligase that seals nicks. For this to work correctly, specific conditions must be met.
The fundamental requirement is that adjacent fragments share terminal sequences typically 15–40 bp of identical overlap at each junction. These overlaps are designed into PCR primers so that the 5’ tail of each primer matches the end of the neighbouring fragment.
To verify and ensure appropriateness for Gibson cloning:
Design overlapping primers computationally: Use Benchling’s Gibson Assembly wizard or SnapGene to automatically design primers with appropriate overlap lengths. The tool shows predicted junction sequences and flags problematic overlaps.
Check for internal homology: Run a BLAST of each overlap sequence against the full construct to ensure the overlap does not appear internally within any fragment. Internal homologies cause misassembly the exonuclease can use an internal site as a false junction.
Verify fragment sizes by gel electrophoresis before assembly: Run each PCR product on a 1% agarose gel to confirm single bands at the correct expected sizes. Multiple bands indicate mispriming that will contaminate the assembly reaction with incorrect fragments.
Check for incompatible sequences in overlaps: Avoid highly repetitive sequences or runs of single nucleotides (homopolymers >8 nt) in the overlap regions, as these are prone to slippage and misassembly.
Verify the fully assembled sequence computationally: In Benchling, use the “Simulate Assembly” feature to confirm the final assembled plasmid has the correct reading frame, no unintended stop codons, and all annotations are correctly positioned.
Screen for internal restriction sites if combining with other methods: If the workflow uses a Type IIS enzyme (e.g., Golden Gate) downstream, screen the assembled sequence for BsaI or BsmBI recognition sites within the coding regions and eliminate them via synonymous substitutions during codon optimisation.
Question 5: How does the plasmid DNA enter the E. coli cells during transformation?
The cell membrane of E. coli is a highly selective barrier DNA molecules carry a strong negative charge and the outer leaflet of the membrane is also negatively charged, creating an electrostatic barrier to entry. Transformation requires physically overcoming this barrier.
Chemical competence (CaCl₂ + heat shock):
E. coli cells are grown to mid-log phase (OD₆₀₀ ~0.4–0.6) and treated with ice-cold CaCl₂. The Ca²⁺ ions serve two functions: they neutralise the negative charge repulsion between the DNA and the cell surface (acting as a bridge), and they alter the lipid packing of the outer membrane, creating a more permeable state. The plasmid DNA associates with the cell surface during ice incubation.
The heat shock step (42°C, 30–90 seconds) is critical and still not fully mechanistically understood. The prevailing model is that the brief temperature jump induces a liquid-crystalline phase transition in the membrane lipids, transiently opening aqueous channels through which the DNA-Ca²⁺ complexes can pass into the periplasm and subsequently the cytoplasm. After the heat pulse, cells are returned to 37°C in rich, non-selective media (SOC) for 45–90 minutes this recovery period allows membrane repair and expression of the antibiotic resistance gene (transcription and translation must occur before the gene product is present in sufficient quantity to confer resistance).
Electroporation:
A brief high-voltage electrical pulse (typically 1.8 kV/cm, 5 ms) applied across the cell suspension creates a transmembrane potential that exceeds the dielectric breakdown voltage of the lipid bilayer (~200–300 mV). This opens transient hydrophilic pores (electropores) in the membrane through which DNA molecules pass by electrophoretic force and diffusion. Electroporation achieves significantly higher transformation efficiencies (up to 10¹⁰ CFU/μg for pUC19) compared to chemical competence (~10⁸ CFU/μg) and is preferred for large plasmids (>10 kb) where chemical competence efficiency drops considerably.
In both cases, once inside the cell, the plasmid must replicate autonomously using the cell’s own replication machinery it must carry an origin of replication compatible with E. coli (e.g., ColE1 ori, pMB1 ori) and a selectable marker. Only cells that have taken up and maintained the plasmid survive antibiotic selection.
Question 6: Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly is a seamless, scar-free, one-pot cloning method that uses Type IIS restriction enzymes (most commonly BsaI or BsmBI) to generate defined overhangs and assemble multiple fragments simultaneously in a single tube.
The key insight: Type IIS enzymes recognise a specific non-palindromic sequence but cleave at a fixed offset downstream of that recognition sequence outside the recognition site itself. This means:
The recognition sequence can be placed in primer-added overhangs, away from the desired junction
After digestion, the recognition site is removed from the product
The custom 4-bp overhang left behind contains only the sequence you designed no scar
Step-by-step mechanism:
Design: Each DNA fragment is flanked by BsaI recognition sites oriented to cut inward (pointing toward the fragment). The 4 bp immediately adjacent to the cut site the overhang is custom-designed for each junction. Each junction in a multi-part assembly has a unique 4-bp sequence, enforcing directionality and preventing scrambled assemblies.
One-pot reaction: All fragments (PCR products or synthesised oligonucleotides), the linearised vector, BsaI enzyme, and T4 DNA Ligase are combined in one tube with the appropriate buffer. The thermocycler alternates between:
37°C (BsaI active): recognition sites are cut, releasing fragments with defined 4-bp overhangs
16°C (T4 Ligase active): complementary overhangs anneal and are covalently joined
Typically 25–30 cycles are run. Because the recognition sites are removed upon digestion, re-cutting of correctly ligated products is impossible the reaction is thermodynamically driven toward complete assembly.
Transformation: The assembled product is transformed into E. coli as normal.
Schematic:
Fragment A Fragment B
5'---[→BsaI]---[ABCD] [ABCD]---[BsaI←]---3'
↓ BsaI cuts here (4 bp from recognition site)
Fragment A ---→ 5'...----ABCD
ABCD----...3' ←--- Fragment B
↓ T4 Ligase seals the nick
Fragment A ─────────────ABCD──────────────── Fragment B
(no scar, no RE site)
For a 4-fragment assembly (A→B→C→D into vector), each junction (A|B, B|C, C|D, vector|A, D|vector) has a unique 4-bp overhang 5 unique overhangs total, designed so no two are complementary to each other except at their intended junction.
Comparison to Gibson Assembly:
Golden Gate
Gibson Assembly
Reaction time
~1 hour
~1 hour
Fragment ends required
PCR products with BsaI sites in primers
PCR products with 15–40 bp overlapping ends
Scar sequences
None
None (seamless)
Combinatorial assembly
Excellent each junction is uniquely defined by 4-bp overhang
Less ideal for combinatorial work
Error source
If two junctions accidentally share the same 4-bp overhang
If overlapping sequences appear internally within a fragment
Number of parts
Up to 10+ fragments efficiently
Typically ≤5 fragments optimally
Best use case
Assembling libraries of pathway variants, standardised biological parts (BioBricks replacement)
General-purpose gene assembly and multi-fragment cloning
I modelled this assembly method in Benchling by creating a simulated construct with BsaI sites flanking each of three fragments and verifying that the predicted overhangs were unique at each junction.
Assignment: Asimov Kernel
As a committed listener based at the Lifefabs London node, I do not have access to the Asimov Kernel platform. I completed the DNA Assembly section in full as the primary required assignment.
Week-07-Homework-Genetic-Circuits-Part-II
Part 1: Intracellular Artificial Neural Networks (IANNs)
Q1: Advantages of IANNs over Traditional Genetic Circuits
Traditional genetic circuits operate as Boolean logic gates—each node is either “on” or “off,” with behavior determined by discrete threshold crossings of transcription factor concentrations. While powerful for implementing deterministic logic (AND, OR, NOT, toggle switches), this architecture has fundamental limitations when the biological task demands graded, context-dependent, or multi-signal integration.
IANNs—genetic circuits whose topology and weight-encoding logic mirrors the multilayer perceptron—address several of these constraints simultaneously:
Continuous signal representation. Traditional circuits collapse analog transcription factor concentrations into binary outputs at threshold crossings. IANNs retain continuous signal values through each layer, preserving quantitative information about input strength. For a biosensor tracking a continuously varying metabolite, this matters enormously—a Boolean circuit can only report “above or below threshold,” while an IANN can encode graded responses proportional to input magnitude.
Multi-signal integration with arbitrary weighting. A classical N-input AND gate requires all N inputs to exceed threshold simultaneously. An IANN neuron computes a weighted sum of inputs before applying a nonlinear activation function—meaning inputs contribute with variable importance, and the threshold for activation depends on the combined weighted signal. This enables far richer input-output relationships with fewer genetic components than equivalent Boolean implementations.
Robustness to parameter variation. Boolean circuits are brittle near threshold—small changes in promoter strength, RBS efficiency, or plasmid copy number shift the switching threshold and can flip circuit state. IANN nodes, by operating in a saturation regime far from their individual thresholds, are intrinsically more robust: perturbations shift the activation curve but leave the qualitative output largely intact.
Trainability and adaptability. In principle, IANN weights (encoded in repressor binding affinities, RBS strengths, or protein-protein interaction Kds) can be tuned—either computationally before construction or, in more advanced designs, via directed evolution. Traditional Boolean circuits have no analogous training paradigm; each redesign is a manual re-engineering effort.
Handling of combinatorially complex inputs. For tasks requiring integration of many simultaneous signals (e.g., a cell-state classifier distinguishing cancer subtypes based on 10+ miRNA inputs), Boolean circuits require exponentially many components; IANNs scale linearly with the number of inputs per layer.
Application context: My final project engineers Bacillus subtilis as a living antimicrobial surface for hospital environments in resource-constrained settings. The current design uses a single-input toehold switch sensing mecA mRNA (the methicillin resistance determinant) to trigger lysostaphin secretion against MRSA. This is functionally a single-input Boolean gate — pathogen present → kill. The critical limitation: hospital surfaces harbour multiple co-occurring pathogens (S. aureus, P. aeruginosa, K. pneumoniae, E. faecium), and a single-target response leaves the surface vulnerable to the remaining species. Worse, indiscriminate broad-spectrum antimicrobial secretion would disrupt commensal biofilms and accelerate resistance evolution.
An IANN architecture solves this by enabling the engineered B. subtilis to classify the pathogen environment from multiple simultaneous RNA signals and mount a proportional, pathogen-specific response — secreting the right antimicrobial at the right concentration, only when the threat profile warrants it.
Input/output behavior:
Layer 1 — Pathogen sensing (3 input nodes): Three orthogonal toehold switch sensors detect species-specific mRNA transcripts leaked from lysed or metabolically active pathogens on the surface: (1) mecA mRNA → MRSA detection, (2) lasR mRNA → P. aeruginosa quorum sensing signal, and (3) blaKPC mRNA → carbapenem-resistant K. pneumoniae. Each toehold switch output is an intermediate transcription factor produced at a level proportional to the cognate mRNA concentration — not a binary on/off, but a graded analog signal encoding pathogen burden.
Layer 1 → Layer 2 integration (hidden layer): The three toehold switch outputs converge on two Layer 2 integration nodes, each controlled by a synthetic promoter with multiple operator sites. Integration node H₁ computes a weighted sum biased toward Gram-positive signals (mecA weight w₁ = high, lasR weight w₂ = low, blaKPC weight w₃ = medium). Integration node H₂ computes a weighted sum biased toward Gram-negative signals (w₁ = low, w₂ = high, w₃ = high). Weights are physically encoded by RBS strength on each transcription factor input feeding that node. The nonlinear activation function at each node is the inherent Hill-function response of the synthetic promoter (~n = 2–4 depending on operator architecture).
Layer 2 → Output layer (effector selection): H₁ drives lysostaphin secretion (Gram-positive killing — cleaves pentaglycine cross-bridges in S. aureus cell wall). H₂ drives Art-175 secretion (engineered endolysin–SMAP29 fusion effective against Gram-negative outer membranes). A third output node, activated only when H₁ AND H₂ both exceed threshold, drives dispersin B (biofilm-degrading enzyme targeting poly-β-1,6-N-acetylglucosamine in mixed-species biofilms).
Why IANN architecture matters here over Boolean logic: A Boolean implementation would require 2³ = 8 separate circuit branches to cover all pathogen combinations — and still produce only binary (full-dose or zero) outputs. The IANN achieves graded, proportional antimicrobial secretion from a 3-node → 2-node → 3-output architecture (8 genetic parts vs. ~24 for equivalent Boolean coverage), and critically, it responds to partial pathogen signatures with partial responses, avoiding wasteful over-secretion that accelerates resistance evolution.
Connection to my project: This IANN design is a direct architectural upgrade of the single-input toehold-switch → T7 RNAP → lysostaphin pipeline I described in Week 3. The toehold switch sensing layer is already validated computationally (30 high-confidence mecA designs from my screening of 89 candidates, top candidate at position 234 with 9.1× ON/OFF ratio). Extending this to lasR and blaKPC targets requires the same toehold switch design workflow. The T7 RNAP amplification cascade from my current design maps naturally onto the hidden layer integration nodes.
Potential limitations:
Cross-reactivity between toehold switches: At high mRNA concentrations, partial complementarity between toehold switch sensors and non-target transcripts could produce false-positive signals. Computational screening (NUPACK) must verify orthogonality across all three sensor-target pairs — my Week 3 screening pipeline would need to be extended to a 3×3 cross-reactivity matrix.
Temporal mismatch between sensing and effector secretion: Toehold switches respond within minutes of mRNA exposure, but lysostaphin and Art-175 secretion requires protein synthesis and Sec-pathway translocation (~30–60 minutes). Fast-growing pathogens like K. pneumoniae (doubling time ~20 min) may establish dangerous colony densities before the IANN output layer reaches effective antimicrobial concentrations.
Weight tuning in vivo: Unlike digital IANNs where weights are adjusted by gradient descent, genetic IANN weights (RBS strengths) are fixed at construction time. Mis-calibrated weights would require re-engineering the construct — there is no in-situ learning. Directed evolution of the RBS library could partially address this, but at significant experimental cost.
Q3: Multilayer Perceptron Diagram — Endoribonuclease → Fluorescent Protein
LAYER 1 (Inputs → Endoribonuclease)
=====================================
Input A ──[w₁]──┐
│
Input B ──[w₂]──┤──[Σ + σ]──→ Endoribonuclease
│ (e.g., Csy4 or CasRx)
Input C ──[w₃]──┘
LAYER 2 (Endoribonuclease → Fluorescent Protein)
==================================================
Endoribonuclease ──[w₄]──→ [Σ + σ] ──→ Fluorescent Protein
(e.g., sfGFP)
Mechanism:
──────────
Layer 1 node encodes a weighted sum of input signals
(transcription factor concentrations) activating a promoter
driving endoribonuclease expression. The endoribonuclease
(e.g., CasRx) cleaves a self-inhibitory RNA hairpin in the
5′ UTR of the fluorescent protein mRNA (Layer 2 input).
When endoribonuclease concentration exceeds the threshold
encoded by the hairpin Kd, fluorescent protein is
de-repressed and translated.
Weights (w₁–w₄) are encoded by:
• RBS strength → translation rate
• Promoter activity → transcription rate
• Protein–protein binding affinity
• RNA hairpin folding energy (for Layer 2 node)
Design rationale: Using an endoribonuclease as the Layer 1 → Layer 2 signal carrier is architecturally elegant — it operates post-transcriptionally, decoupling Layer 1 and Layer 2 on different molecular substrates (protein vs. RNA), and avoids transcriptional interference between layers. CasRx (a Type VI-D CRISPR effector) is orthogonal to most bacterial and mammalian transcriptional regulators, making it particularly suitable for implementing clean two-layer IANNs without cross-talk.
Part 2: Fungal Materials
Q1: Existing Fungal Material Examples, Applications, and Trade-offs
Fungal materials—primarily mycelium composites formed from the dense hyphal networks of filamentous fungi—have emerged as a serious alternative to petroleum-derived foams, plastics, and leather in the last decade.
Mycelium composite packaging (Ecovative Design): The flagship application: agricultural waste (corn husks, hemp hurds) is colonized by Ganoderma spp. mycelium under controlled humidity and CO₂ conditions. The resulting composite blocks are used as protective packaging for electronics and fragile goods—replacing expanded polystyrene (EPS). Advantages: fully compostable, carbon-sequestering during growth, fire-resistant, thermally and acoustically insulating, scalable to large geometries without injection molding tooling costs. Disadvantages: lower specific mechanical strength than EPS (compressive strength ~50–200 kPa vs. EPS at 100–400 kPa), water sensitivity without chemical treatment, growth-to-form process requires 5–7 days minimum, not yet suitable for sterile or food-contact applications without additional processing.
Mycelium leather (Bolt Threads — “Mylo,” Ecovative — “Forager”): Thin mycelium sheets grown from Ganoderma or Pleurotus species on agricultural waste are processed (dried, tanned, and finished) to produce a leather-like flexible material. Used commercially in luxury goods by Stella McCartney and Lululemon. Advantages: animal-free, biodegradable, water-resistant post-treatment, pore size tunable via growth conditions. Disadvantages: tensile strength and durability still inferior to full-grain leather (~10–15 MPa vs. ~20–35 MPa for bovine leather), requires chemical tanning with conventional agents to achieve desired hand feel, scaling manufacturing to cost-parity with synthetic leather remains unresolved.
Fungal acoustic panels and construction materials: Compressed mycelium boards with densities of 100–200 kg/m³ are used for acoustic damping panels and low-load structural elements. Acoustic absorption coefficients comparable to mineral wool at mid-frequencies (500–2000 Hz). Limited to non-structural applications due to lower flexural modulus.
Q2: Genetic Engineering Objectives for Fungi and Synthetic Biology Advantages
Engineering objectives:
The primary genetic engineering targets for improved fungal materials are: (1) chitin synthase engineering to control hyphal wall composition and increase mechanical strength via higher chitin cross-link density; (2) laccase and peroxidase pathway modification to tune lignin binding at the hyphal surface, improving adhesion to lignocellulosic substrates; (3) fruiting body suppression to redirect metabolic flux from reproductive structures toward vegetative hyphal biomass during the growth phase; and (4) hydrophobin engineering to tune surface wettability of the final material without post-processing chemical treatment.
Why synthetic biology in fungi offers distinct advantages over bacterial approaches:
Fungi—particularly Aspergillus niger, Trichoderma reesei, and Ganoderma lucidum—have several properties that bacteria lack which make them intrinsically better chassis for material applications. First, fungal hyphae grow as multi-centimeter macroscopic structures with directional morphology; bacteria form only microscopic biofilm matrices, making macroscopic material fabrication fundamentally harder. Second, fungi already produce industrial quantities of extracellular matrix components (chitin, beta-glucan, lignin-binding enzymes) via existing secretory pathways that are far more productive than bacterial secretion systems for large structural polymers. Third, fungal gene expression machinery supports post-translational modifications (glycosylation, disulfide bond formation) that are essential for many structural proteins but absent in E. coli—making fungi a natural chassis for producing next-generation protein-composite materials where recombinant structural proteins (e.g., spider silk, resilin) are co-secreted and incorporated into the hyphal matrix. Finally, filamentous fungi are intrinsically tolerant to the aerobic, CO₂-rich, lignocellulose-heavy growth conditions of low-cost bioreactors, reducing the operational complexity of scaling beyond what is achievable with bacterial fermentation for material applications.
Part 3: First DNA Twist Order
Individual Final Project Documentation
Project summary: Engineering the MS2 bacteriophage L-protein for improved stability and controlled lytic activity in E. coli. The L-protein disrupts the inner membrane as the terminal step of the phage lytic cycle; recombinant expression is toxic and unstable. My goal is to use computational design (ESM2 + ProteinMPNN) to identify stabilizing mutations that preserve lytic function, enabling the protein to fold and resist degradation when expressed at lower temperatures or in minimal media.
Aim 1 (Draft)
To identify and validate stabilizing substitutions in the MS2 L-protein that improve thermostability (ΔTm ≥ 3°C) while maintaining ≥50% of wild-type membrane lysis activity, using a computational pipeline combining ESM2 deep mutational scanning, ProteinMPNN inverse folding, and ESMFold structure prediction—with top candidates synthesized and expressed in E. coli BL21(DE3) under arabinose-inducible control.
Insert Sequence Design
Backbone vector: pBAD-His (Invitrogen) — arabinose-inducible expression in E. coli; N-terminal 6×His tag for purification and Western blot detection; AmpR selection marker; pBR322 origin (medium copy, ~20–40 copies/cell).
Rationale for pBAD: Arabinose-inducible systems allow tight titration of expression level, which is critical for a membrane-lytic protein — constitutive or IPTG-inducible systems would cause growth arrest before sufficient biomass for characterization is achieved. The pBAD promoter is also fully repressible in glucose-containing media, enabling normal growth before induction.
Insert sequence — L-protein WT (for baseline):
The wild-type L-protein coding sequence (75 aa) was codon-optimized for E. coli K-12 expression using the Codon Optimization Tool (IDT), with the following design choices:
Avoided rare E. coli codons (AGA, AGG for Arg; ATA for Ile) throughout
Added Kozak-equivalent strong RBS (AAGG) immediately upstream of ATG
Included flanking NcoI (5′) and HindIII (3′) restriction sites for directional cloning into pBAD-His
Total insert length: ~250 bp (75 codons + RBS + restriction sites + 4 bp buffer)
Designed stabilization mutant (Mutant A — L-protein V11I/A23V):
Based on ESM2 ΔLL analysis, positions 11 (Val→Ile, ΔLL = +1.8) and 23 (Ala→Val, ΔLL = +2.3) show favorable substitution signals consistent with hydrophobic packing improvement in the transmembrane region. These were combined into a single construct (Mutant A) for synthesis alongside the WT insert.
Both sequences have been placed in the shared DNA design folder and are ready for DNA Twist synthesis submission.
Week 7 Lab: Neuromorphic Wizard
Circuit Design — AnirudhNet_V1
I designed a two-input neuromorphic circuit in the Neuromorphic Wizard using the Csy4 endoribonuclease architecture from the lab. X1 encodes CasE (the endoribonuclease) alongside mNeonGreen as a fluorescent reporter. X2 encodes the output fluorescent protein (mKO2) whose mRNA contains Csy4 recognition sites — so X1 negatively regulates X2 output proportionally.
This architecture directly mirrors the multilayer perceptron from Part 1 Q3: layer 1 produces the endoribonuclease, layer 2 output is regulated by it.
Experiment Layout
Biocompiler-Predict Output
Running the PREDICT function on AnirudhNet_V1 produces a heatmap showing output fluorescence as a function of X1 and X2 DNA concentrations. The gradient confirms expected behavior: high X1 (high CasE) suppresses X2 output (low fluorescence at top), while low X1 allows full X2 expression (high fluorescence at bottom). The analog graded response — not binary — demonstrates the core IANN advantage over Boolean gates.