week-02-hw-dna-read-write-and-edit
Part 0: Basics of Gel Electrophoresis
I watched the lectures by George Church, Joe Jacobson, and Emily Leproust, as well as the recitation by Ice Kiattisewee on DNA Gel, restriction enzymes, Benchling, and Twist.
Key takeaways from lectures:
Gel electrophoresis works by taking advantage of the negative charge of the DNA molecule due to the phosphate group. DNA fragments being placed in the agarose gel (which is heated and then cooled) move according to their size under the effect of the electric field applied, separating based on their size. The pore size of the agarose gel is determined by the concentration of agarose - (with higher concentrations –> decreased pore size –> smaller fragments/finer fragments being separated easily. The ladder containing the weight of the DNA in kDa helps in estimating the size of the DNA based on its position in the gel. Ethidium bromide is used for staining resulting in the DNA to glow under the UV light.
I found the concept of using gel electrophoresis to create art interesting . The artwork done by the previous students was visually stunning. I felt that the benchling software is a really elegant tool to edit , add DNA sequences and simulate digestion of the genomes using restriction enzymes. I believe that I have not fully explored the potential it holds.
Part 1: Benchling & In-Silico Gel Art
Restriction Enzyme Digestion of Lambda DNA (48,502 bp)
I created a Benchling account and imported the Lambda phage DNA sequence (48,502 bp). I then simulated restriction enzyme digestion with each of the seven enzymes specified:
| Enzyme | Recognition Site | # Cut Sites | Fragment Sizes (bp) |
|---|---|---|---|
| EcoRI | GAATTC | 5 | 21,226; 7,421; 5,804; 5,643; 4,878; 3,530 |
| HindIII | AAGCTT | 7 | 23,130; 9,416; 6,557; 4,361; 2,322; 2,027; 564; 125 |
| BamHI | GGATCC | 5 | 16,841; 7,233; 6,770; 6,527; 5,626; 5,505 |
| KpnI | GGTACC | 2 | 29,942; 17,053; 1,507 |
| EcoRV | GATATC | 4 | 13,286; 10,316; 8,453; 7,656; 5,583; 3,208 |
| SacI | GAGCTC | 3 | 21,060; 14,584; 8,430; 4,428 |
| SalI | GTCGAC | 2 | 32,745; 15,258; 499 |
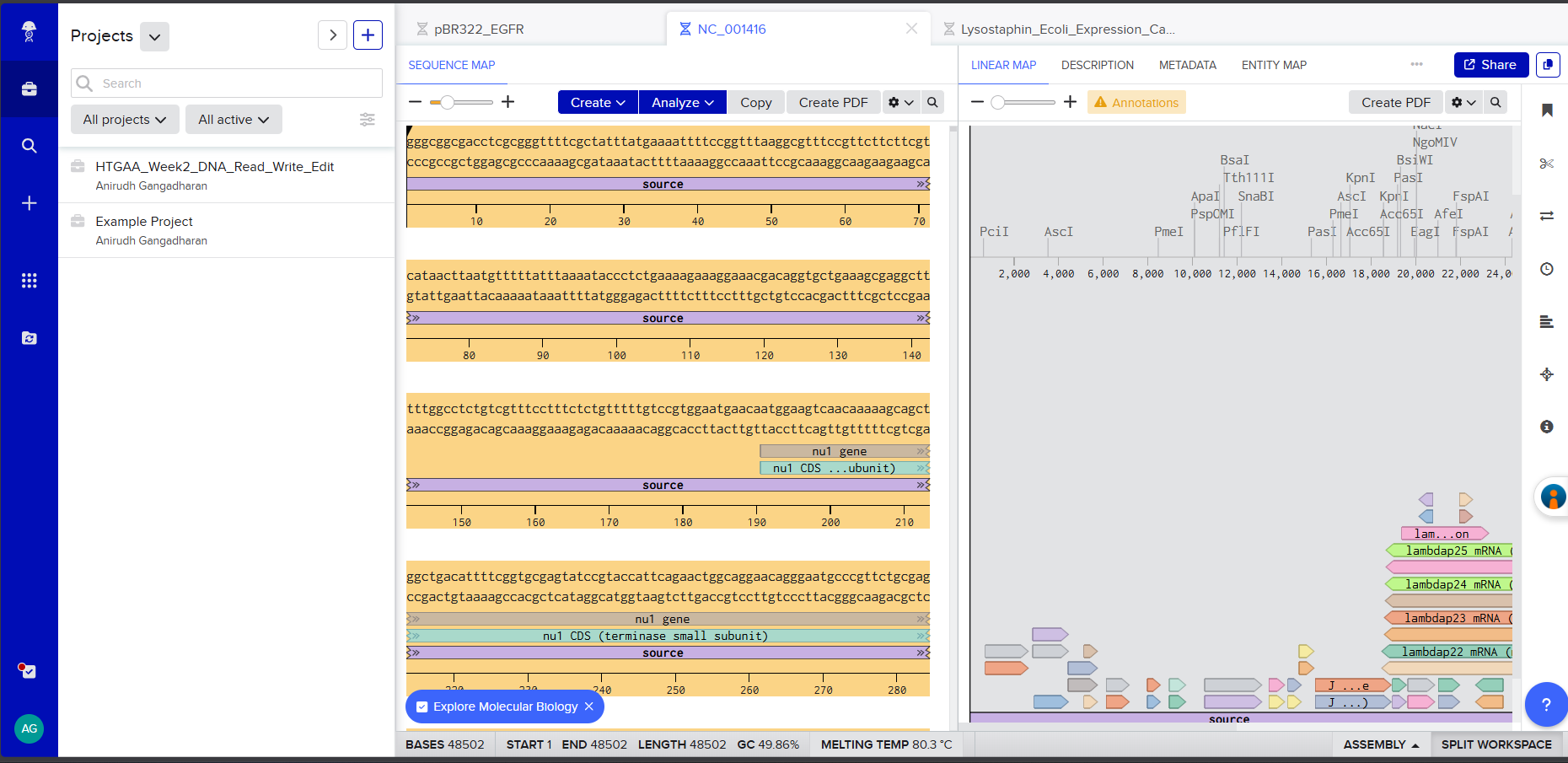 Screenshot 1: The imported Lambda DNA sequence in Benchling showing the circular/linear map.
Screenshot 1: The imported Lambda DNA sequence in Benchling showing the circular/linear map.
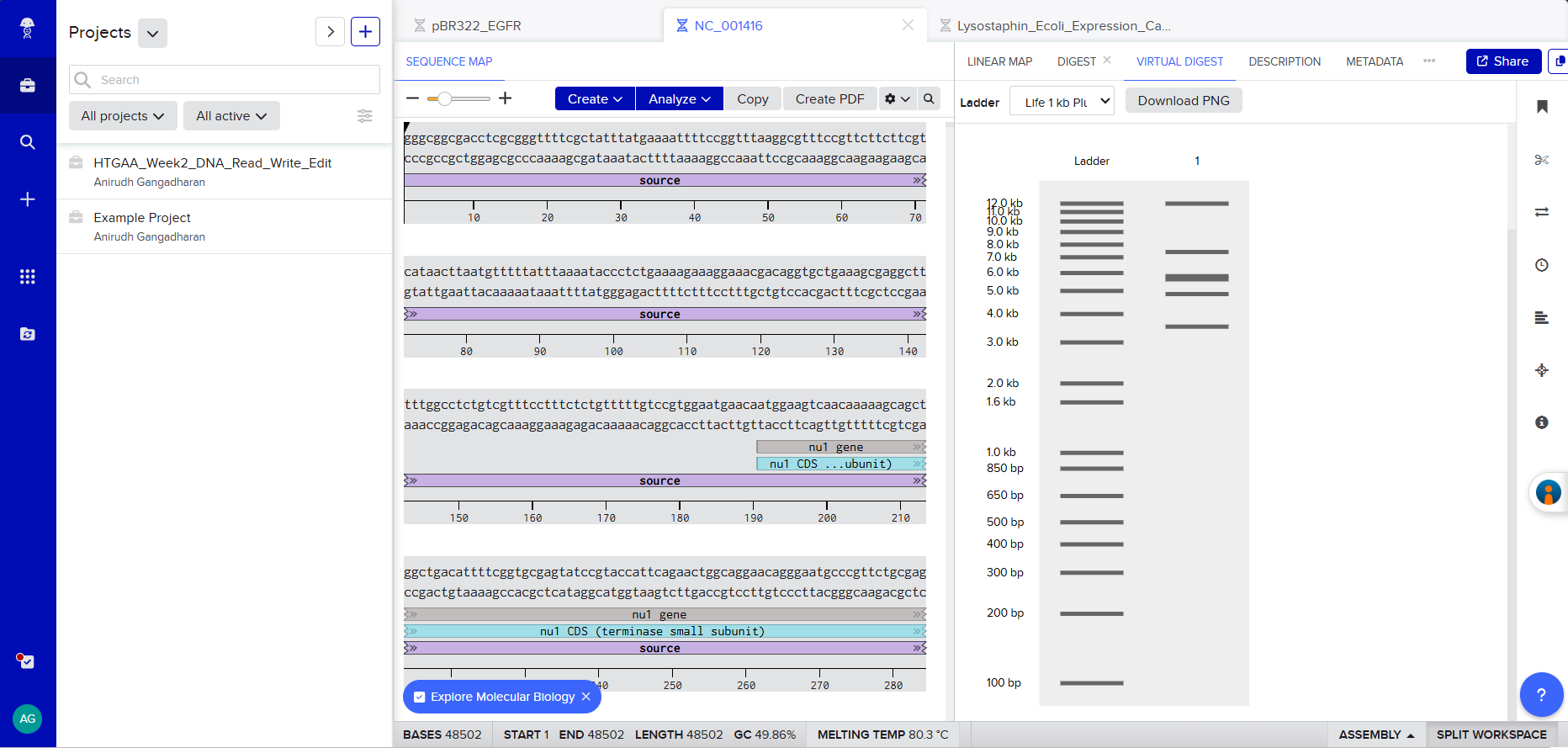 Screenshot 2: The EcoRI digest result showing the gel simulation + fragment table.
Screenshot 2: The EcoRI digest result showing the gel simulation + fragment table.
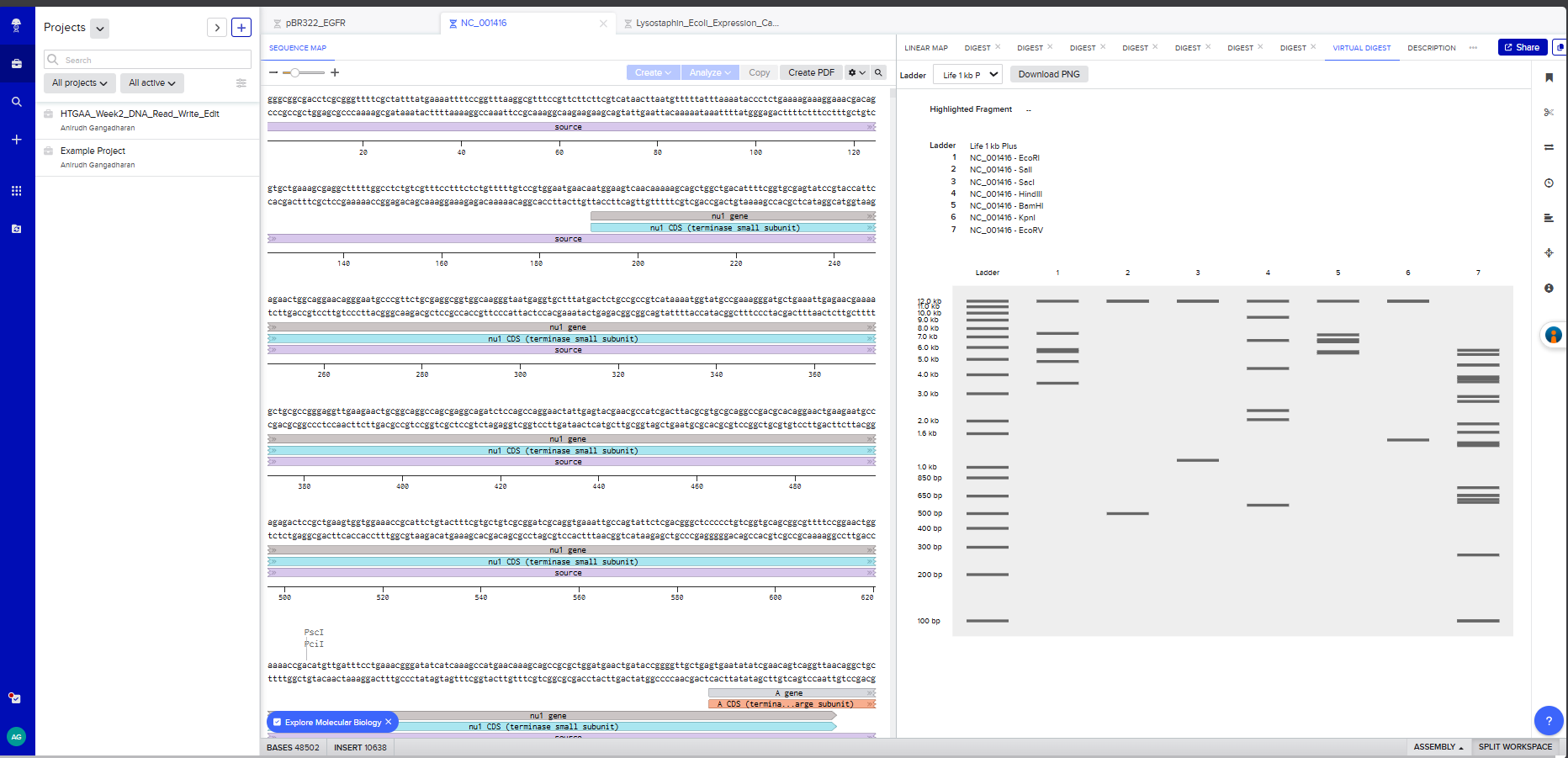 Screenshot 3: A multi-enzyme digest showing all 7 enzymes side by side on the simulated gel
Screenshot 3: A multi-enzyme digest showing all 7 enzymes side by side on the simulated gel
Gel Art Design
On benchling , I virtually digested the DNA in 7 lanes with the following enzymes - EcoRI, SalI, SacI, HindIII, BamHI, KpnI, and EcoRV. I used Ronan’s website to iterate a few, but couldn’t effectively find a way to create the “HAI” pattern that I had envisioned.
Note: As a committed listener without wet-lab access, I completed this as an in-silico exercise.
Part 2: Gel Art - Wet Lab
As a committed listener at the London HTGAA node without direct wet-lab access for this week’s protocol, Part 2 is optional. The in-silico design in Part 1 serves as my documentation.
Part 3: DNA Design Challenge
3.1. Protein Choice: Lysostaphin
Protein: Lysostaphin (EC 3.4.24.75)
Source organism: Staphylococcus simulans biovar staphylolyticus
UniProt ID: P10547
Gene name: lss
Lysostaphin is a zinc metalloenzyme derived from S. simulans acting against S. aureus. The gene for production of lysostaphin can be inserted via a plasmid vector into B. subtilis which is a commonly used chassis organism by biotechnologists. (https://www.mdpi.com/1424-8247/3/4/1139)
The other proteins I had in mind were PlyKp104 and PlyCD(1-174). PlyKp104 is an endolysin derived from Klebsiella pneumoniae phage which can kill Klebsiella pneumoniae and Pseudomonas aeuroginosa. (https://pubmed.ncbi.nlm.nih.gov/37098944). Similarly PlyCD (1-174) has endolytic activity on Clostridium difficile species. It’s derived from a prophage targeting the same. (https://pmc.ncbi.nlm.nih.gov/articles/PMC4649177.)
Why I chose this protein: For my project that I had in mind - engineering living surfaces or biofilms to kill the common causative organisms causing nosocomial/HAIs (Hospital acquired infections) – the expression of these proteins I mentioned above (Lysostaphin, PlyKp104 and PlyCD) by Bacillus subtilis (chassis organism) are needed for the bactericidal action the bacteria causing HAIs.
S. aureus, including methicillin-resistant strains (MRSA), is among the most dangerous nosocomial pathogens. In the medicine wards of my medical college in India, I have seen patients admitted for routine conditions develop life-threatening staphylococcal secondary infections due to inadequate surface sterility. Lysostaphin is a potent weapon against precisely this pathogen.
Also, lysostaphin has already been successfully expressed in B. subtilis WB600 (the expression host relevant to my project) (https://www.aimspress.com/article/doi/10.3934/microbiol.2021017)
Mature lysostaphin protein sequence (246 amino acids, from UniProt P10547, residues 248–493):
The mature enzyme has two functional domains: an N-terminal catalytic domain (residues 1–148, M23 family zinc metalloendopeptidase) responsible for pentaglycine cleavage, and a C-terminal cell wall targeting domain (CWT, residues 149–246, SH3b family) that directs the enzyme to the staphylococcal surface by specifically recognizing the pentaglycine crosslinks.
3.2. Reverse Translation: Protein to DNA
Using the Central Dogma, I worked backwards from the protein sequence to determine the corresponding DNA sequence. I retrieved the native coding sequence for mature lysostaphin from the lss gene on plasmid pACK1 of S. simulans (GenBank Accession: M15686).
Native DNA sequence encoding mature lysostaphin (738 bp):
Note: The final three nucleotides (TAA) represent the stop codon.
3.3. Codon Optimization
Why codon optimization is necessary:
Different organisms have different levels of tRNA for a specefic codon. If the tRNA for a specific codon isn’t abudant enough, the translation process can get stalled.
Target organism for optimization: Escherichia coli (K12 strain)
I chose E. coli because it is the standard workhorse for recombinant protein production and the most well-characterized expression system. The initial cloning, expression validation, and protein characterization will be performed in E. coli BL21(DE3) before eventually transferring the validated construct into B. subtilis for the final project application. E. coli also has the highest density of codon usage tables and optimization tools available.
Codon-optimized lysostaphin DNA sequence for E. coli expression (738 bp):
Using the Twist Bioscience Codon Optimization Tool, avoiding Type IIs enzyme recognition sites (BsaI, BsmBI, BbsI):
Retrospective note: Since the deployment chassis for my final project is B. subtilis, direct codon optimization for B. subtilis would have been more strategically aligned. For the final project construct, a B. subtilis-optimized variant will be generated using the same Twist optimization pipeline.
3.4. From DNA Sequence to Protein: Production Methods
With the codon-optimized lysostaphin sequence in hand, there are two main routes to produce the functional protein:
Method 1: Cell-dependent expression in E. coli (Primary approach)
The codon-optimized DNA sequence is cloned into an expression vector (e.g., pET21 or pTwist Amp High Copy) downstream of a strong inducible promoter such as T7. The construct is transformed into E. coli BL21(DE3) competent cells. The process follows the Central Dogma:
Transcription: Upon induction with IPTG, T7 RNA polymerase binds the T7 promoter and synthesizes mRNA from the lysostaphin DNA template. The RNA polymerase reads the template strand 3’→5’ and produces mRNA 5’→3’, with each T in the DNA transcribed as U in the mRNA.
Translation: Ribosomes bind the mRNA at the ribosome binding site (Shine-Dalgarno sequence), recognize the AUG start codon, and begin translating the mRNA into protein. Each three-nucleotide codon in the mRNA specifies one amino acid via tRNA adaptor molecules. Translation proceeds until the ribosome encounters the UAA stop codon.
Folding and purification: The translated polypeptide folds into its native conformation. With a His-tag appended at the C-terminus, the protein can be purified using nickel affinity chromatography (IMAC), followed by size-exclusion chromatography to obtain pure, active lysostaphin.
Method 2: Cell-free expression (Alternative approach)
Cell-free transcription-translation systems (e.g., PURExpress or E. coli S30 extract) can produce lysostaphin without living cells. The DNA template (linear or circular) is added directly to the cell-free reaction mix containing ribosomes, tRNAs, amino acids, RNA polymerase, and energy sources (ATP, GTP). Transcription and translation occur simultaneously in a single tube within 2–4 hours.
Cell-free is advantageous for rapid prototyping - testing whether the construct produces active protein before committing to cell-based production. It is particularly relevant to my project because cell-free expression could eventually enable on-demand antimicrobial peptide production in resource-limited settings without cold chain requirements.
3.5. How It Works in Nature
How a single gene codes for multiple proteins:
At the transcriptional level, a single gene can produce multiple protein variants through several mechanisms:
Alternative splicing (in eukaryotes): Different exons are included or excluded from the mature mRNA, generating distinct protein isoforms from one gene. The DSCAM gene in Drosophila can produce over 38,000 splice variants.
Alternative promoters: Different promoters upstream of the same gene can initiate transcription at different points, producing mRNAs with different 5’ exons and therefore different N-terminal protein sequences.
Polycistronic mRNAs (in prokaryotes): A single operon produces one mRNA encoding multiple proteins, each translated from its own RBS. The lysostaphin operon itself contains both lss and the immunity factor lif gene.
Central Dogma alignment for the first 30 nucleotides of lysostaphin:
Note: All thymine (T) bases in DNA are transcribed as uracil (U) in mRNA. Each 3-nucleotide codon in the mRNA corresponds to exactly one amino acid in the protein.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Account Setup
I created accounts on both Benchling (benchling.com) and Twist Bioscience (twistbioscience.com).
4.2. Building the DNA Insert Sequence (Expression Cassette)
In Benchling, I created a new linear DNA sequence named “Lysostaphin_Expression_Cassette” and assembled the following expression cassette by concatenating the components in order:
Components:
| Component | Sequence | Length |
|---|---|---|
| Promoter (BBa_J23106) | TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC | 35 bp |
| RBS (BBa_B0034 + spacers) | CATTAAAGAGGAGAAAGGTACC | 22 bp |
| Start Codon | ATG | 3 bp |
| Lysostaphin CDS (codon-optimized) | (738 bp sequence from 3.3, excluding start ATG and stop TAA) | 735 bp |
| 7x His Tag | CATCACCATCACCATCATCAC | 21 bp |
| Stop Codon | TAA | 3 bp |
| Terminator (BBa_B0015) | CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA | 129 bp |
| Total | ~948 bp |
Each component was annotated in Benchling by right-clicking and creating annotations with appropriate labels (Promoter, RBS, Start Codon, CDS: Lysostaphin, His-tag, Stop Codon, Terminator).
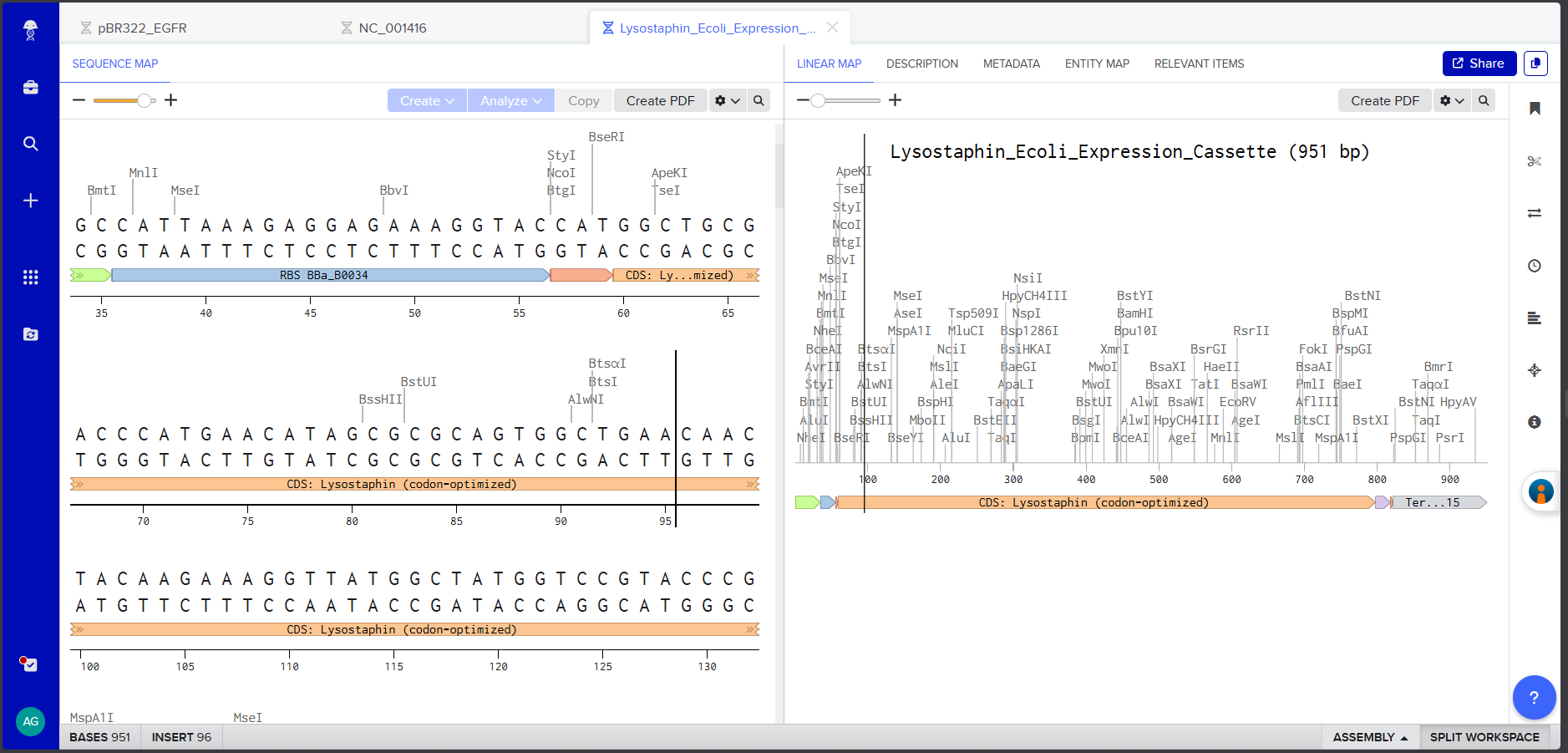 Screenshot 4: The annotated Linear Map showing all components labeled and color-coded.
Screenshot 4: The annotated Linear Map showing all components labeled and color-coded.
I verified the complete assembly using Benchling’s Linear Map view to confirm all annotations are correctly positioned and non-overlapping. The link for review: https://benchling.com/s/seq-QorL0JNRMSvmklWTiDkE?m=slm-138RcqLoOew0hHCOHJtK The FASTA file was downloaded for upload to Twist.
4.3–4.5. Twist Order Setup
 Screenshot 6: The Twist Genes product page.
Screenshot 6: The Twist Genes product page.
On Twist Bioscience, I selected Genes → Clonal Genes and uploaded the FASTA file using the Nucleotide Sequence → Upload Sequence File option.
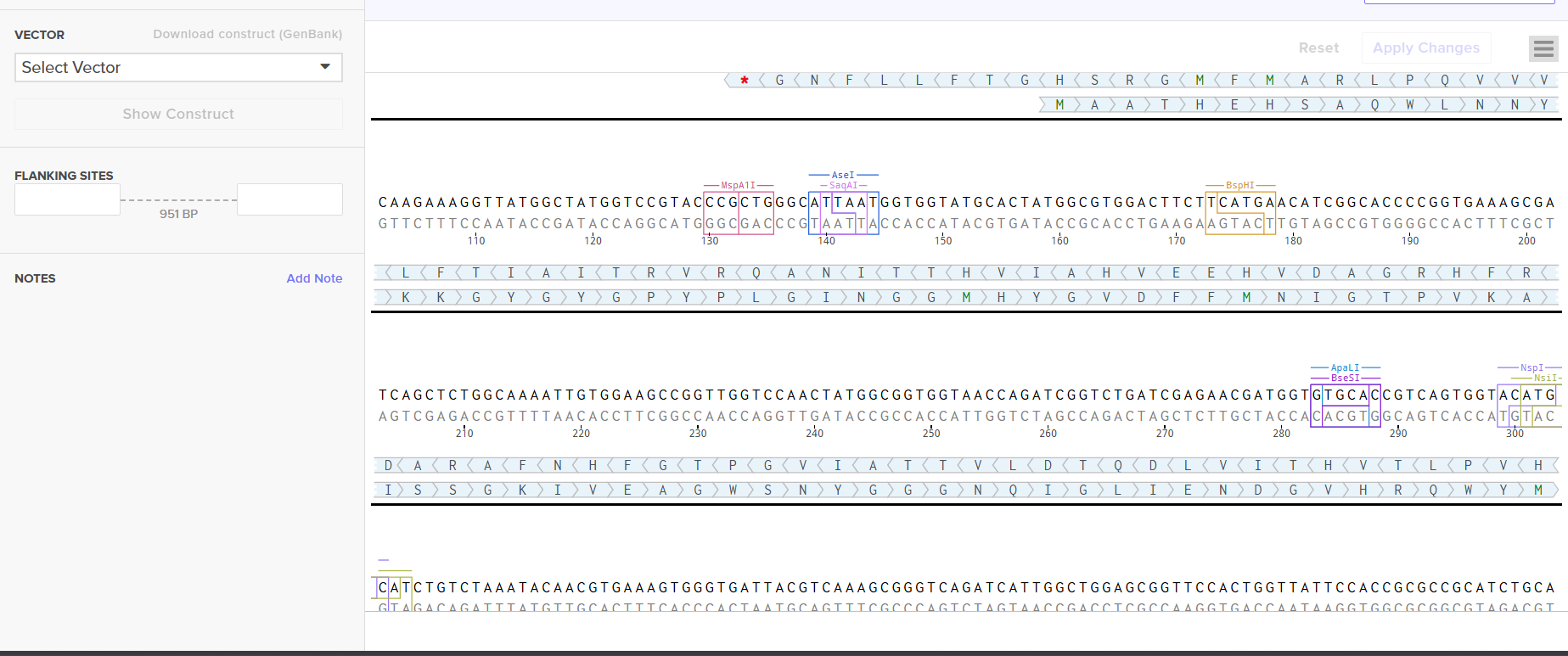 Screenshot 7: The uploaded sequence in Twist’s interface
Screenshot 7: The uploaded sequence in Twist’s interface
I chose clonal genes over gene fragments because clonal genes arrive as circular plasmid DNA that can be directly transformed into E. coli without an additional assembly step - saving 1–2 weeks of experimental time, as noted in the course materials.
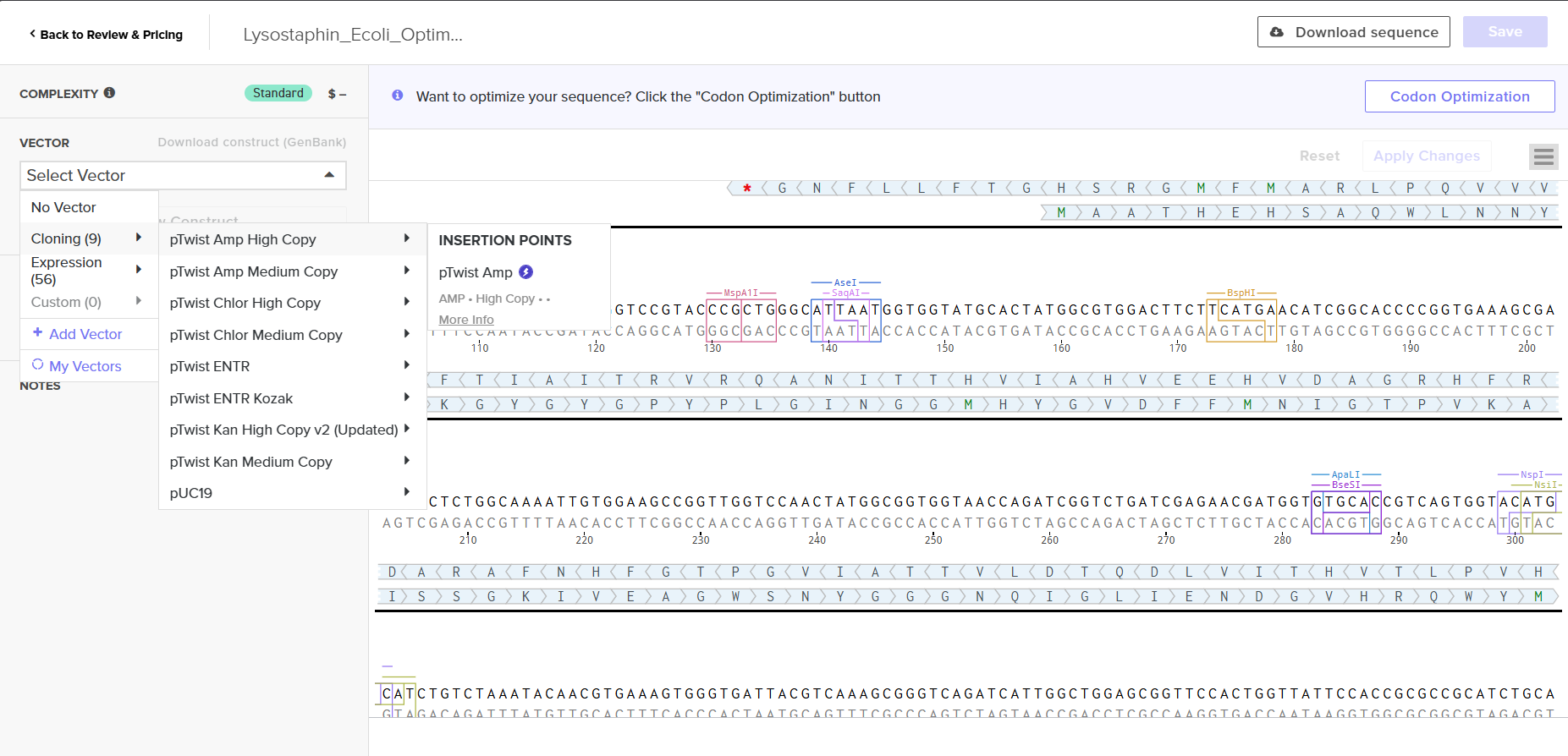 Screenshot 8: The vector selection showing pTwist Amp High Copy
Screenshot 8: The vector selection showing pTwist Amp High Copy
4.6. Vector Selection
I selected pTwist Amp High Copy as the backbone vector from Twist’s vector catalog. Rationale:
- Ampicillin resistance - compatible with standard lab antibiotic stocks
- High copy number - maximizes plasmid yield per miniprep, important for downstream cloning and sequence verification
- ColE1 origin of replication - well-characterized, stable in E. coli
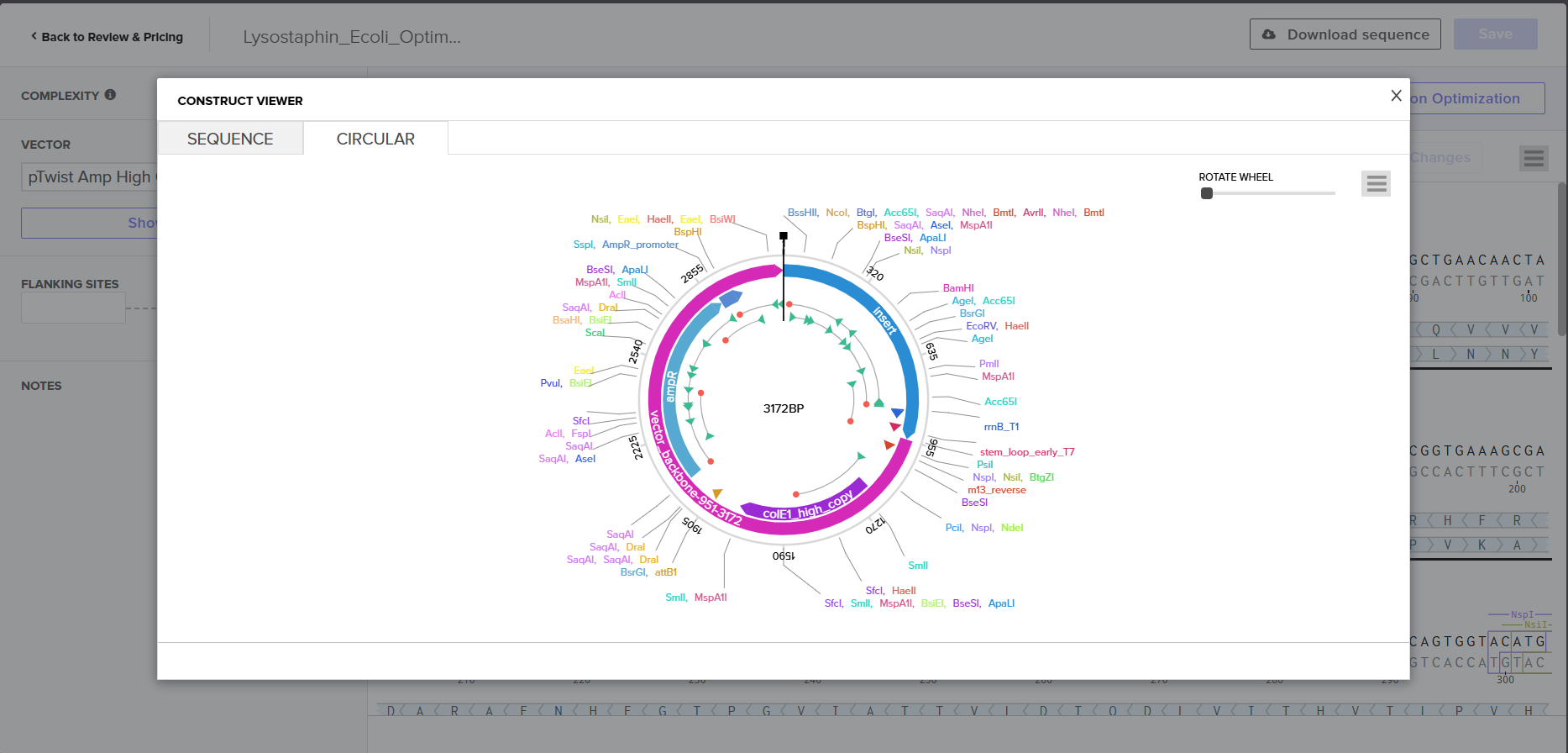
I downloaded the complete construct (GenBank format) from Twist and imported it into Benchling, confirming the circular plasmid map shows the lysostaphin expression cassette correctly inserted into the pTwist backbone.
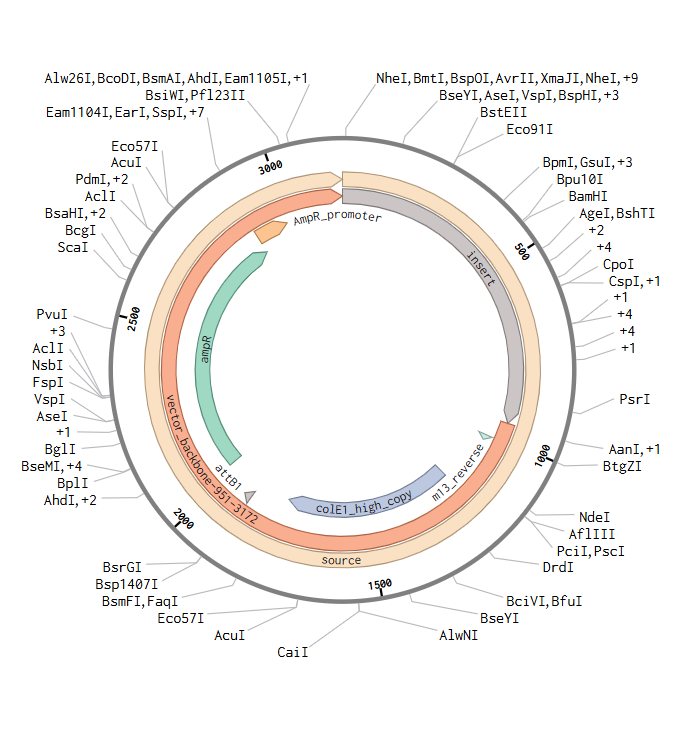 Screenshot 9: Showing the plasmid uploaded to benchling.
Screenshot 9: Showing the plasmid uploaded to benchling.
For the final project, my construct will include:
- Fully annotated Benchling insert fragment (lysostaphin expression cassette)
- pTwist Amp High Copy as the Twist cloning vector
- Flanking restriction sites for future subcloning into B. subtilis shuttle vectors
Part 5: DNA Read/Write/Edit
5.1. DNA Read (Sequencing)
(i) What DNA would I want to sequence and why?
I would want to sequence the resistomes of hospital surfaces in Indian government hospitals - specifically, performing metagenomic sequencing of microbial communities on high-touch surfaces (bedrails, IV poles, doorknobs, ventilator tubing) in the medicine and post-surgical wards where I have worked.
Why this matters: The WHO priority pathogen list includes organisms like MRSA, vancomycin-resistant Enterococcus (VRE), carbapenem-resistant Acinetobacter baumannii, and extended-spectrum beta-lactamase (ESBL)-producing Klebsiella pneumoniae - all of which I have encountered clinically in my wards. Sequencing hospital surface microbiomes would reveal: which resistance genes (AMR genes) are circulating on surfaces, the taxonomic composition of the surface biofilm community, whether these surface communities match the organisms causing actual patient infections (proving the transmission chain), and how the resistome changes over time and in response to cleaning protocols.
This data would directly inform the design of my engineered B. subtilis colonizer by identifying which antimicrobial peptides the system needs to produce. Without knowing precisely what organisms inhabit these surfaces, the engineering is blind.
(ii) Sequencing Technology: Oxford Nanopore MinION
Generation: Third-generation sequencing technology. Unlike first-generation (Sanger, chain termination) and second-generation (Illumina, sequencing by synthesis with short reads), nanopore sequencing reads single DNA molecules in real-time without requiring amplification or fluorescent labeling.
Input preparation - essential steps:
- DNA extraction: Use a bead-beating + column-based kit (e.g., Qiagen DNeasy PowerSoil) to lyse both Gram-positive and Gram-negative organisms from surface swabs.
- Size selection: Optional - use SPRI beads or gel extraction to enrich for fragments >1 kb if long reads are desired.
- Library preparation: Using the Rapid Sequencing Kit (SQK-RAD004): a transposase fragments the DNA and simultaneously attaches sequencing adapters in a single 10-minute step. Alternatively, the Ligation Sequencing Kit (SQK-LSK114) provides higher throughput with more steps: end repair/dA-tailing → adapter ligation → SPRI bead cleanup.
- No PCR amplification required - native DNA is sequenced directly, preserving epigenetic modifications (methylation).
How it decodes bases (base calling):
A single strand of DNA is ratcheted through a biological nanopore (CsgG protein pore) embedded in a synthetic membrane. As each nucleotide passes through the constriction of the pore, it causes a characteristic disruption of the ionic current flowing through the pore. A neural network-based base caller (e.g., Dorado or Guppy) decodes the raw electrical signal (squiggle) into nucleotide sequences. The current signal is influenced by ~5–6 nucleotides simultaneously (the k-mer in the pore), so the base caller uses sequence context to make predictions.
Output:
- FASTQ files containing long reads (N50 typically 5–20 kb, with individual reads >100 kb possible)
- Real-time streaming output - reads are available within minutes of starting the run
- Per-read quality scores (Q-scores), with current chemistry (R10.4.1) achieving median Q20 (~99% accuracy)
- Methylation calls from the same raw signal (no bisulfite conversion needed)
Why MinION specifically: It costs ~$1,000 for the device (or free through Oxford Nanopore’s starter program), runs on a laptop via USB, requires no large infrastructure, and can be used at point-of-care - making it deployable in Indian hospitals where I work. For metagenomic surveillance, long reads resolve species-level taxonomy and link AMR genes to their host organisms on single reads, which short-read sequencing cannot do.
5.2. DNA Write (Synthesis)
(i) What DNA would I want to synthesize and why?
I would want to synthesize a modular antimicrobial peptide expression cassette library for Bacillus subtilis - specifically, a set of genetic constructs each encoding a different antimicrobial effector under the control of a quorum-sensing-responsive promoter. The library would include:
- Lysostaphin (lss) - targets S. aureus including MRSA (the protein designed in Parts 3–4 above)
- Dispersin B (dspB) - a glycoside hydrolase from Aggregatibacter actinomycetemcomitans that degrades poly-N-acetylglucosamine (PNAG) biofilm matrix, disrupting biofilms formed by S. aureus and S. epidermidis
- Art-175 - an engineered endolysin-antimicrobial peptide fusion (artilysin) effective against Gram-negative pathogens including Acinetobacter baumannii and Pseudomonas aeruginosa
Each construct would use the same standardized architecture: [Quorum-sensing promoter] → [RBS] → [Signal peptide for secretion] → [Antimicrobial CDS] → [Terminator]. This modularity allows swapping effectors in and out depending on the pathogen landscape of a specific hospital - a “pharmacological palette” for engineered living surfaces.
The specific genetic sequences are: lysostaphin as described in Part 3.3, and I would design and order the others through Twist.
(ii) Synthesis Technology: Twist Bioscience Silicon-Based Phosphoramidite Synthesis + Enzymatic Assembly
Essential steps of Twist’s synthesis platform:
Oligo synthesis on silicon chips: Short oligonucleotides (up to ~200 nt) are synthesized in parallel on miniaturized wells etched into silicon wafers using phosphoramidite chemistry. Each cycle: (a) detritylation (remove 5’-DMT protecting group), (b) coupling (add next phosphoramidite monomer), (c) capping (block failed couplings), (d) oxidation (stabilize the new phosphodiester bond).
Oligo pool harvesting and error removal: Completed oligos are cleaved from the chip, and error-containing sequences are removed using enzymatic mismatch cleavage or hybridization-based selection.
Hierarchical assembly: Overlapping oligos are assembled into longer gene fragments through PCR-based assembly (e.g., polymerase cycling assembly or Gibson Assembly). Multiple fragments are then joined into the complete gene.
Clonal isolation: Assembled genes are cloned into the selected vector, transformed into E. coli, and individual clones are Sanger-sequenced to confirm 100% sequence accuracy. Only sequence-verified clones are shipped.
Limitations:
- Speed: Typical turnaround is 2–3 weeks for clonal genes, which limits rapid iteration cycles
- Accuracy: While final clonal products are sequence-perfect, the per-base error rate during oligo synthesis (~1 in 200–500 bases) necessitates the error-correction and clonal selection steps, adding time
- Scalability: Individual gene synthesis scales well, but whole-genome synthesis (>100 kb) remains prohibitively expensive and slow - the “gene writing gap” described by Hoose et al. (2023)
- Sequence constraints: Certain sequences are difficult to synthesize: extreme GC content (<25% or >75%), long homopolymer runs, extensive secondary structure, and repetitive sequences can cause synthesis failures
- Cost: While cost per base has dropped dramatically (~$0.07/bp for gene fragments), synthesizing entire pathways or genomes remains expensive
5.3. DNA Edit (Genome Editing)
(i) What DNA would I want to edit and why?
I would want to edit the genome of Bacillus subtilis 168 to create an optimized chassis for my engineered living surface colonizer. Specifically, three categories of edits:
Edit 1: Knockout of sporulation genes (spo0A, sigF).
B. subtilis forms endospores that are extremely resistant to disinfection and could spread the engineered organism beyond hospital surfaces. Deleting key sporulation regulators prevents spore formation, ensuring the organism remains in vegetative form and is susceptible to standard decontamination - a critical biocontainment feature.
Edit 2: Introduction of synthetic auxotrophies.
As I described in my Week 1 governance analysis, I would engineer dependence on a non-canonical amino acid (ncAA) not found in nature. Specifically, I would knock out genes in the biosynthetic pathway for an essential amino acid and replace them with an engineered aminoacyl-tRNA synthetase that charges tRNA with a synthetic analog. The organism can only survive when the synthetic nutrient is supplied - true containment beyond the “Jurassic Park lysine contingency.”
Edit 3: Integration of the quorum-sensing antimicrobial peptide circuits into the chromosome.
Rather than maintaining the constructs on plasmids (which can be lost and impose a fitness burden), I would integrate the engineered circuits directly into the B. subtilis chromosome at neutral loci (e.g., amyE, lacA). Chromosomal integration provides stable, single-copy expression without antibiotic selection - essential for real-world deployment where you cannot maintain antibiotic pressure.
(ii) Editing Technology: CRISPR-Cas9 combined with MAGE (Multiplex Automated Genome Engineering)
For targeted knockouts and insertions, I would use CRISPR-Cas9 as the primary editing tool, supplemented by recombineering for multiplex edits.
How CRISPR-Cas9 edits DNA - essential steps:
Guide RNA design: Design a 20-nt spacer sequence complementary to the target site in the B. subtilis genome, adjacent to a 5’-NGG-3’ PAM (protospacer adjacent motif) recognized by SpCas9. I would use Benchling’s CRISPR guide designer to score guides for on-target efficiency and minimize off-target sites.
Construct assembly: Clone the sgRNA (spacer + scaffold) into an expression vector under a constitutive promoter. Co-deliver or separately provide: (a) Cas9 protein or expression cassette, (b) the sgRNA, and (c) a repair template - a DNA fragment with 500–1000 bp homology arms flanking the desired edit (deletion, insertion, or substitution).
Delivery into B. subtilis: B. subtilis is naturally competent - it can take up exogenous DNA from the environment. Transform competent cells with the CRISPR plasmid and repair template simultaneously.
Double-strand break and repair: The Cas9-sgRNA complex binds the target DNA, verifies PAM recognition, and creates a blunt double-strand break (DSB) 3 bp upstream of the PAM. The cell’s homology-directed repair (HDR) machinery uses the supplied repair template to fix the break, incorporating the desired edit.
Selection and verification: Plate transformants on selective media. Screen colonies by PCR across the edited locus and confirm by Sanger sequencing.
Preparation required:
- Input materials: Cas9 expression plasmid (or purified Cas9 protein), sgRNA expression construct, repair template DNA (synthesized by Twist or generated by PCR), competent B. subtilis cells, selective antibiotics
- Design steps: Target site selection → off-target analysis → guide scoring → repair template design with homology arms → cloning and sequence verification
Limitations:
- Efficiency: In B. subtilis, CRISPR-Cas9 editing efficiency varies (50–95%) depending on the locus, guide quality, and repair template design. Unlike E. coli, B. subtilis has efficient HDR, but Cas9 toxicity can reduce viable transformants.
- Off-target effects: SpCas9 can tolerate mismatches at positions distal from the PAM, potentially causing unintended edits. This can be mitigated by using high-fidelity Cas9 variants (e.g., eSpCas9) and whole-genome sequencing of final strains.
- Multiplexing complexity: Editing multiple loci simultaneously (sporulation knockouts + auxotrophy + circuit integration) requires sequential rounds of editing with plasmid curing between rounds, which is time-consuming. MAGE-like approaches (Wannier et al., 2021) using ssDNA oligonucleotides can accelerate multiplex editing but are less established in B. subtilis than in E. coli.
- PAM restriction: The requirement for an NGG PAM limits targetable sites to ~1 in every 8 bp on average. This can be overcome by using Cas9 orthologs with different PAM preferences (e.g., SaCas9 recognizes NNGRRT, CjCas9 recognizes NNNNRYAC) or engineered PAM-relaxed variants (e.g., SpCas9-NG, SpRY).
- Large insertions: Integrating multi-kb constructs (full antimicrobial circuits) via HDR is less efficient than small edits. For large insertions, combining CRISPR-mediated counterselection with traditional B. subtilis integrative vectors at established loci (amyE, thrC) may be more reliable.
References
- Sabala, I. et al. “Crystal structure of the antimicrobial peptidase lysostaphin from Staphylococcus simulans.” FEBS J. 281, 4112–4122 (2014).
- Nazari, R. et al. “Cloning and expression of Staphylococcus simulans lysostaphin enzyme gene in Bacillus subtilis WB600.” Mol. Biotechnol. 63, 1043–1052 (2021).
- Shendure, J. et al. “DNA sequencing at 40: past, present, and future.” Nature 550, 345–353 (2017).
- Hoose, A. et al. “DNA synthesis technologies to close the gene writing gap.” Nat. Rev. Chem. 7, 144–161 (2023).
- Wannier, T. et al. “Recombineering and MAGE.” Nat. Rev. Methods Primers 1, 7 (2021).
- Wang, J.Y. & Doudna, J.A. “CRISPR technology: A decade of genome editing is only the beginning.” Science 379, eadd8643 (2023).
- Bastos, M.C.F. et al. “Lysostaphin: A Staphylococcal Bacteriolysin with Potential Clinical Applications.” Pharmaceuticals 3, 1139–1161 (2010).
- Recsei, P.A. et al. “Cloning, sequence, and expression of the lysostaphin gene from Staphylococcus simulans.” Proc. Natl. Acad. Sci. USA 84, 1127–1131 (1987).
Citation: I used Claude to help refine the codon-optimized sequence generation and to verify technical details of sequencing/editing technologies. All project framing, protein choice rationale, clinical observations, and design decisions connecting to my final project are my own work.