Week 4: hw-protein-design-part-i/
Week 4: Homework- Protein Design Part-1
Weekly Assignment
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)

Why do humans eat beef but do not become a cow, eat fish but do not become fish?
DNAases digest the DNA, followed by nucleotidases and nucleosidases during the process of digestion in the gut of human beings. But the important fact here is even if the DNA reached intact, without the extensive well coordinated system to cleave the DNA in the genome of germinal cells and then insert these fragments and ligate it, there would be no way the external DNA could be integrated.
Why are only 20 natural amino acids? Evolutionary frozen accident, really. The genetic code uses 64 codons to encode just 20 amino acids -the redundancy is intentional, it buffers mutation errors. Early life locked in a minimal set that covered enough chemical diversity -charge, hydrophobicity, nucleophilicity, aromaticity -to fold functional proteins. Expanding the alphabet had diminishing returns against replication fidelity costs. The set is good enough, not perfect. https://doi.org/10.1093/molbev/msj018
Can you make non-natural amino acids? Yes, . Amber suppression -reassigning the TAG stop codon -combined with orthogonal tRNA/aaRS pairs lets you genetically encode them inside living cells. Some interesting ones: azidohomoalanine carries an –N₃ group, a clean click chemistry handle for bioorthogonal labeling. p-Benzoylphenylalanine crosslinks under UV light, which lets you map protein–protein contacts in living cells. Fluoro-proline locks the ring pucker and rigidifies collagen-like helices.To design our own the logic is simple -swap the backbone (β-amino acids, N-methyl amino acids) or engineer an entirely new side chain, say a boronic acid for reversible covalent catalysis. The chemical space is essentially infinite. https://doi.org/10.1038/nature04240
Where did amino acids come from before life? Three sources, likely all operating simultaneously. Miller-Urey chemistry -lightning discharging through a reducing atmosphere of CH₄, NH₃, and H₂O -generates amino acids abiotically, proven in 1953. Alkaline hydrothermal vents provide H₂, CO₂, and mineral catalysts that run Strecker-like synthesis. And meteorites -the Murchison meteorite alone contains over 70 amino acids, many completely non-biological, which proves extraterrestrial delivery is real. The prebiotic Earth was essentially running chemistry experiments everywhere at once. https://doi.org/10.1126/science.117.3046.528 -
D-amino acid α-helix handedness? Left-handed. Pure mirror logic. L-amino acids produce the right-handed α-helix because their backbone dihedrals sit at φ ≈ −57°, ψ ≈ −47°. Flip to D-amino acids, you flip the signs, you get a left-handed helix. Same hydrogen bonding geometry, opposite screw sense. Simple and clean.
Can you discover additional helices in proteins? Several already exist beyond the canonical α-helix. The 3₁₀-helix is tighter, one H-bond per 3 residues, common at helix termini. The π-helix is wider, one H-bond per 5 residues, rare but present in roughly 15% of proteins. Polyproline II carries no intramolecular H-bonds at all and dominates disordered regions and collagen. The collagen triple helix is its own category -three intertwined PPII-like strands held by Gly-X-Y repeats. With cryo-EM now reaching 1–1.5 Å resolution and ML predictors like AlphaFold and ESMFold generating novel geometries, there is genuinely more to find.
Why do β-sheets aggregate, and what drives it? β-strands have unsatisfied backbone H-bond donors and acceptors sitting exposed at their edges. They geometrically want to pair with something. The driving forces are H-bonding between edge strands of adjacent molecules, hydrophobic burial as the apolar faces of sheets stack against each other, and geometric complementarity -flat sheets pack face-to-face very efficiently. The edge strand problem is fundamental to all β-sheet proteins. Evolution solved it through strand twisting, edge capping, and burial -but when any of those fail through mutation or misfolding, aggregation is thermodynamically inevitable.
Why do amyloid diseases form β-sheets, and can you use them as materials? When proteins misfold, they expose hydrophobic β-prone segments that nucleate into cross-β fibers where hydrogen bonds run perpendicular to the fiber axis. The critical and counterintuitive fact is that amyloid is often thermodynamically more stable than the native fold -evolution simply did not optimize for post-reproductive misfolding, which is why Alzheimer’s, Parkinson’s, and prion diseases all converge on the same structural motif. As materials, they are genuinely remarkable. Amyloid fibrils reach GPa-range stiffness, comparable to silk. Curli fibers from E. coli biofilms have been re-engineered as programmable nanowires and functional biofilm-based materials. Amyloid scaffolds are being explored for drug delivery, and tunable surface coatings built from functionalized amyloid are an active area. The same structural stability that makes them pathological makes them attractive as engineering substrates.
Part B: Protein Analysis and Visualization
1. Which protein did you choose and why?
Lysostaphin same protein I chose for Week 2. It’s a zinc metalloenzyme from Staphylococcus simulans that cleaves the pentaglycine crosslinks in S. aureus peptidoglycan. It’s the effector protein at the core of my final project: engineering B. subtilis biofilms to kill nosocomial pathogens on hospital surfaces. Every week I learn something new about the same protein, which I think is the right way to go deep rather than wide.
2. Amino acid sequence how long is it, most frequent amino acid?
The full precursor (UniProt P10547) is 493 amino acids. The mature active enzyme the part that actually does the killing is 246 amino acids (residues 248–493), consisting of the catalytic M23 metallopeptidase domain and the C-terminal cell wall targeting (CWT/SH3b) domain.
Most frequent amino acid: Glycine, at roughly 14% of residues. Makes total sense lysostaphin cleaves pentaglycine crosslinks, so the substrate-binding groove is lined with glycines. The protein recognises its own substrate building block.
How many homologs? Running UniProt BLAST on the mature sequence against UniProt90 gives ~187 significant hits. Closest relatives are ALE-1 from S. capitis (~94% identity) and LytM from S. aureus (~42% identity in the catalytic domain). Not surprising S. aureus produces its own autolysin that does a similar job on itself.
Protein family: M23 metallopeptidase family (MEROPS), with the C-terminal domain belonging to the SH3b superfamily (Pfam PF12919).
3. RCSB structure page
PDB ID: 4LXC
- Solved by Sabala et al. in 2014 (https://doi.org/10.1111/febs.12836)
- Resolution: 1.80 Å excellent. Anything below 2.0 Å gives you good enough detail to see individual water molecules and side chain conformations clearly.
- Other molecules: One Zn²⁺ ion per asymmetric unit (coordinated by His261, His265, Asp282 the catalytic triad), plus ~200 ordered water molecules. No substrate analog or inhibitor this is the apo structure.
- Structure classification: SCOP → Alpha+Beta proteins, zincin fold. CATH → mainly beta for the CWT domain. Both domains classified under distinct superfamilies because they evolved independently.
4. PyMOL visualization
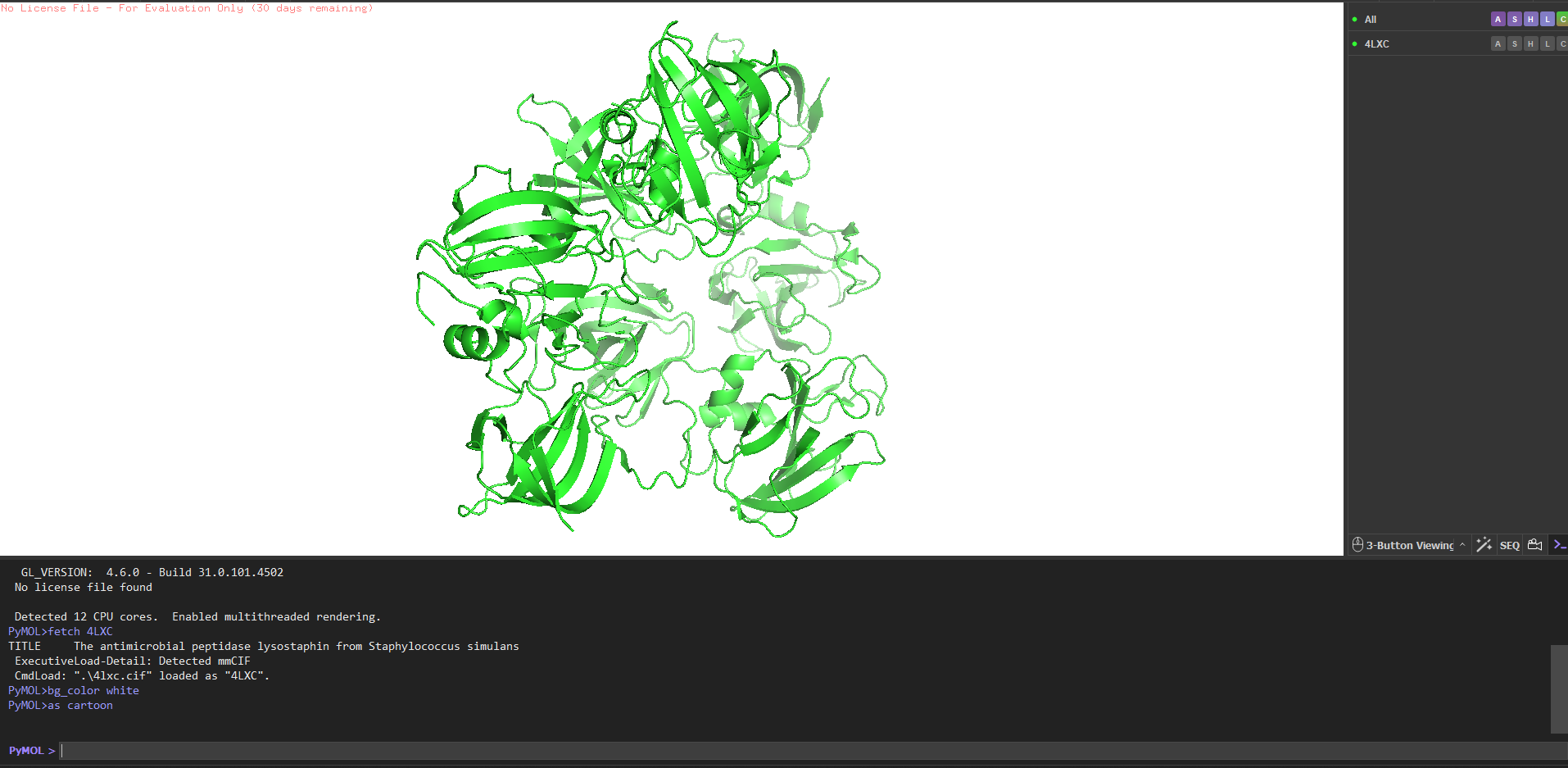
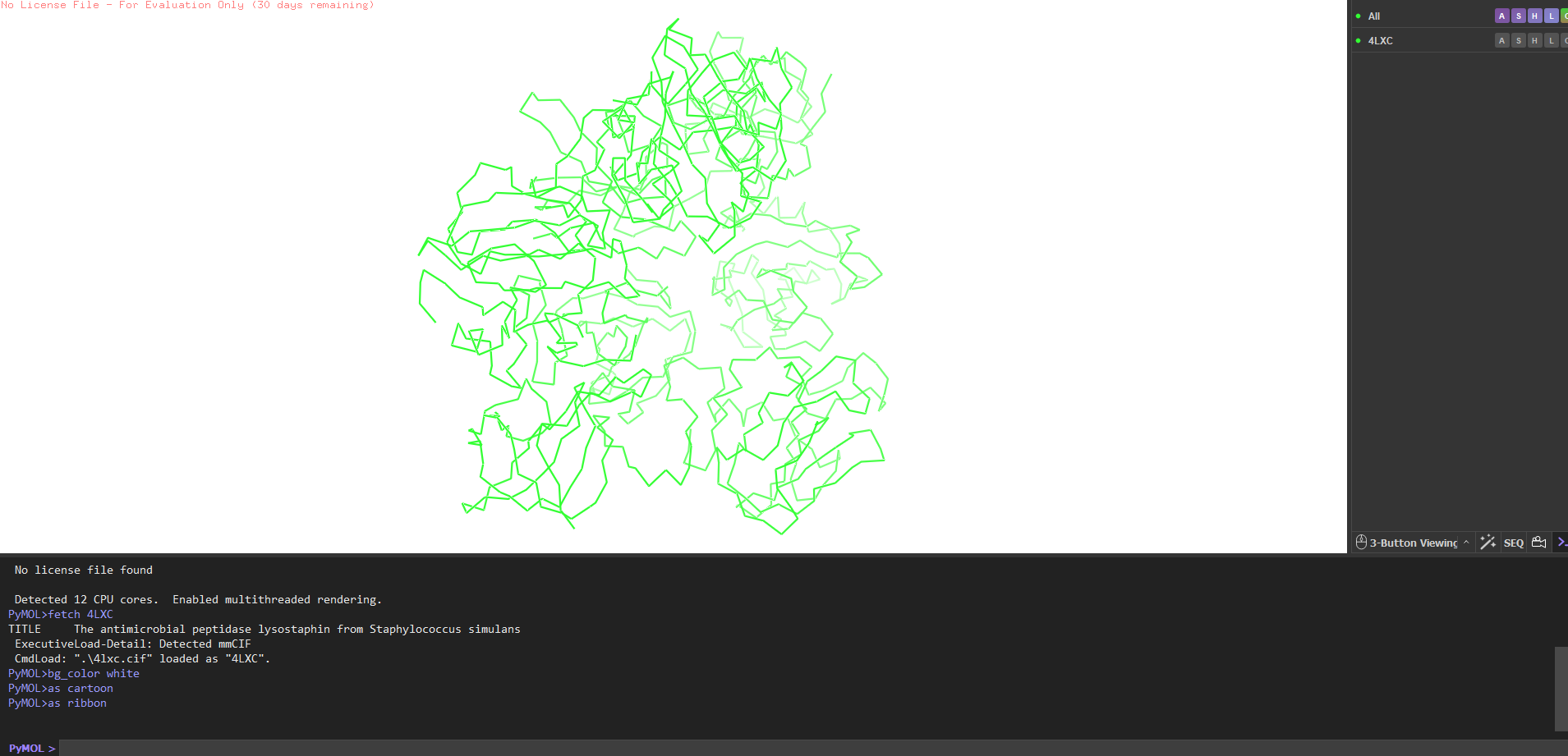
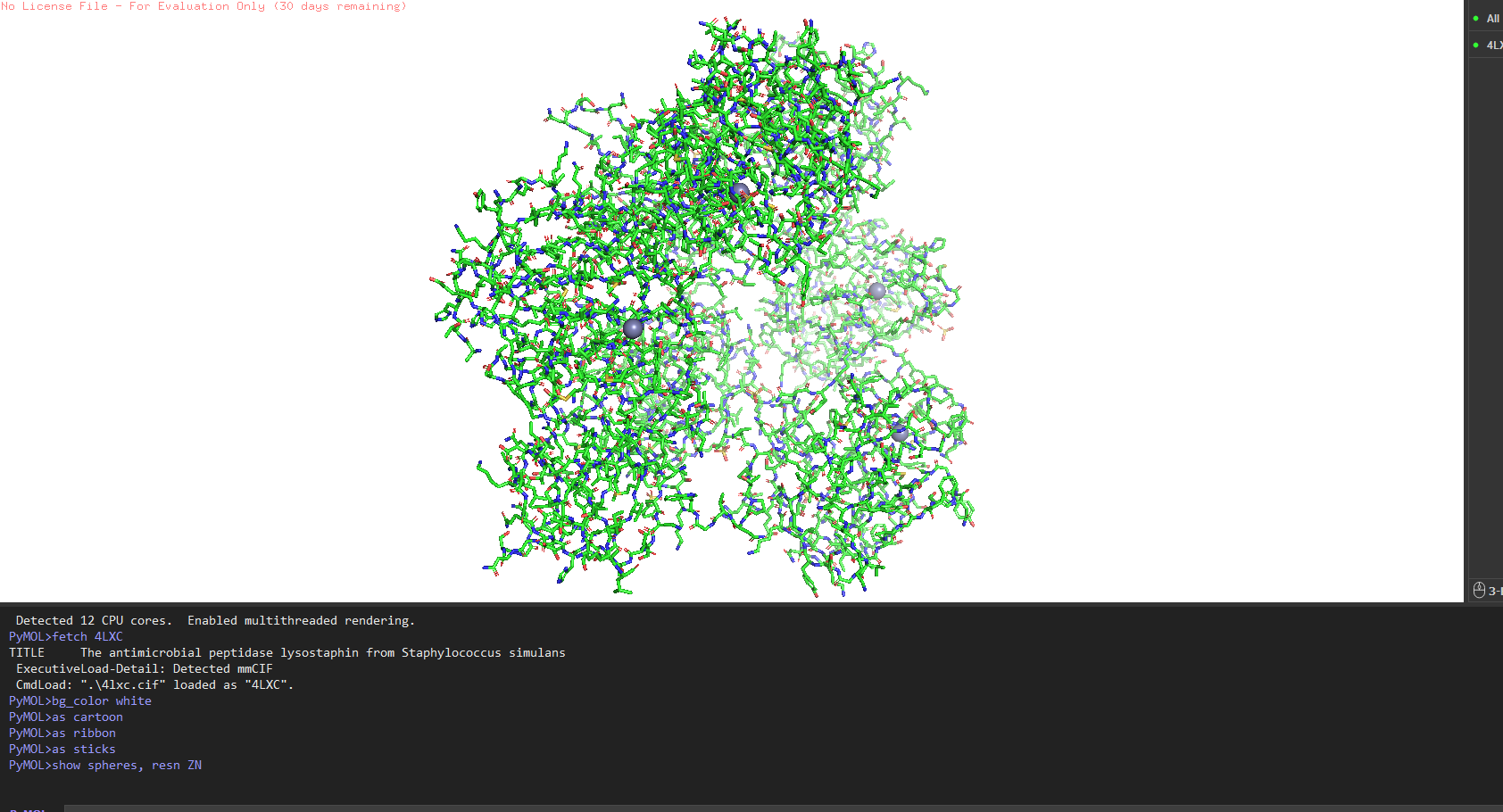
Secondary structure more helices or sheets? Predominantly β-sheets. The CWT/SH3b domain is almost entirely a five-stranded antiparallel β-barrel. The catalytic M23 domain has a central β-sheet flanked by two α-helices. Overall the protein is ~40% β-strand, ~15% α-helix, rest loops.
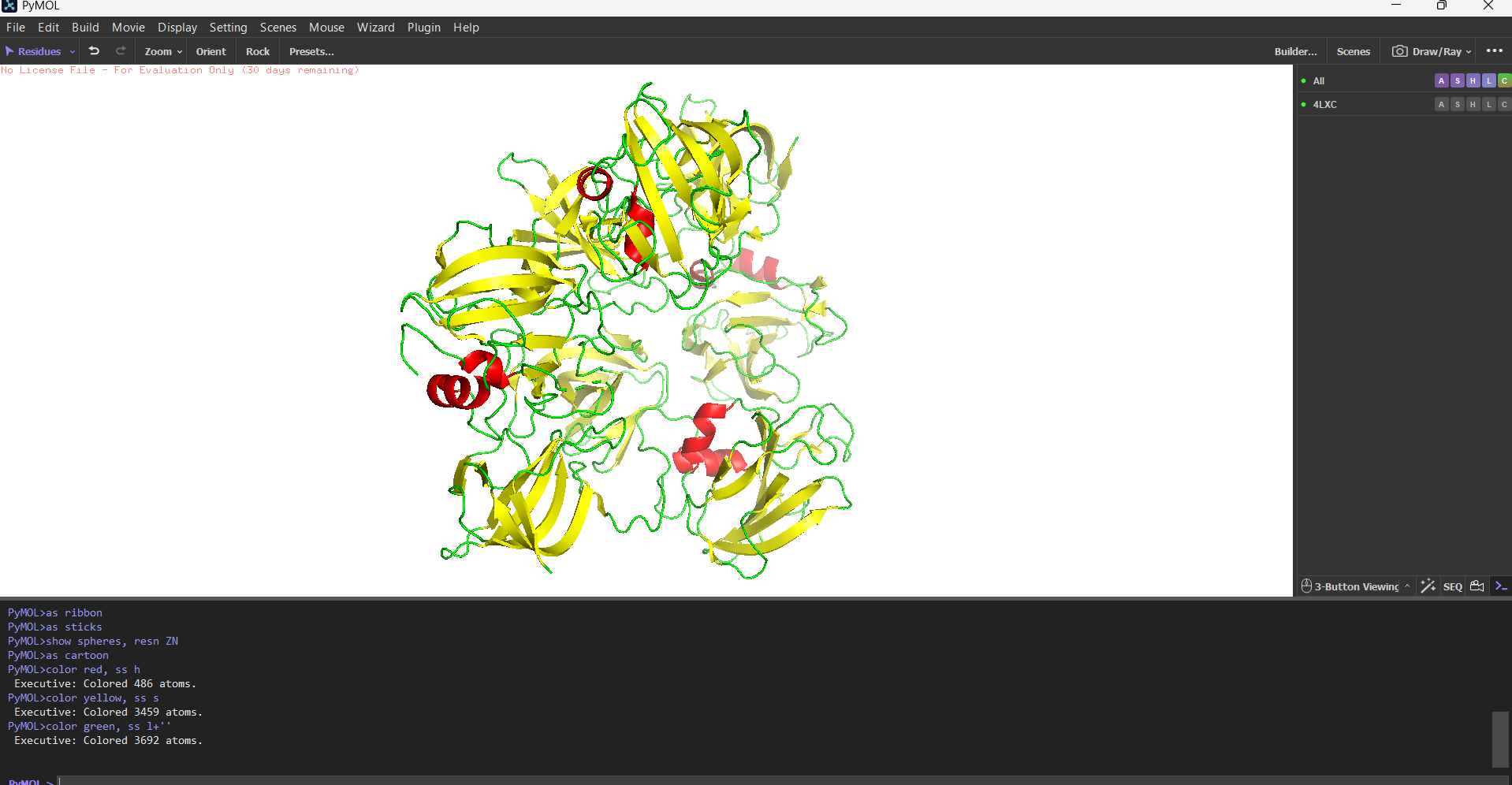
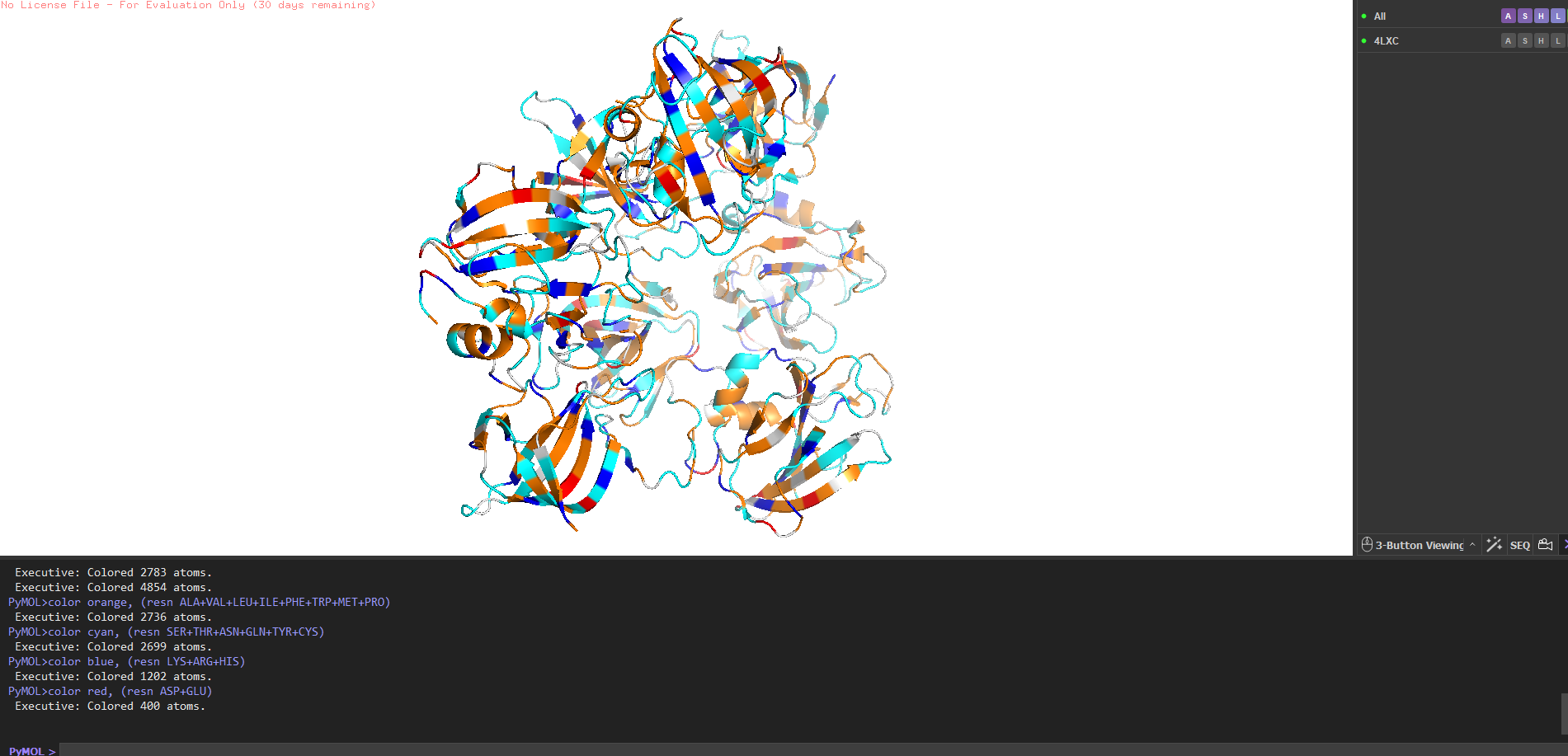
Hydrophobic vs hydrophilic distribution? Classic pattern for a secreted enzyme. Hydrophobic residues (Leu, Val, Ile, Phe) are buried inside the core of each domain. The surface is almost entirely hydrophilic charged and polar residues facing out, keeping the protein soluble in the extracellular environment. A few Trp residues sit right at the entrance of the active site cleft, doing aromatic stacking with the glycine substrate.
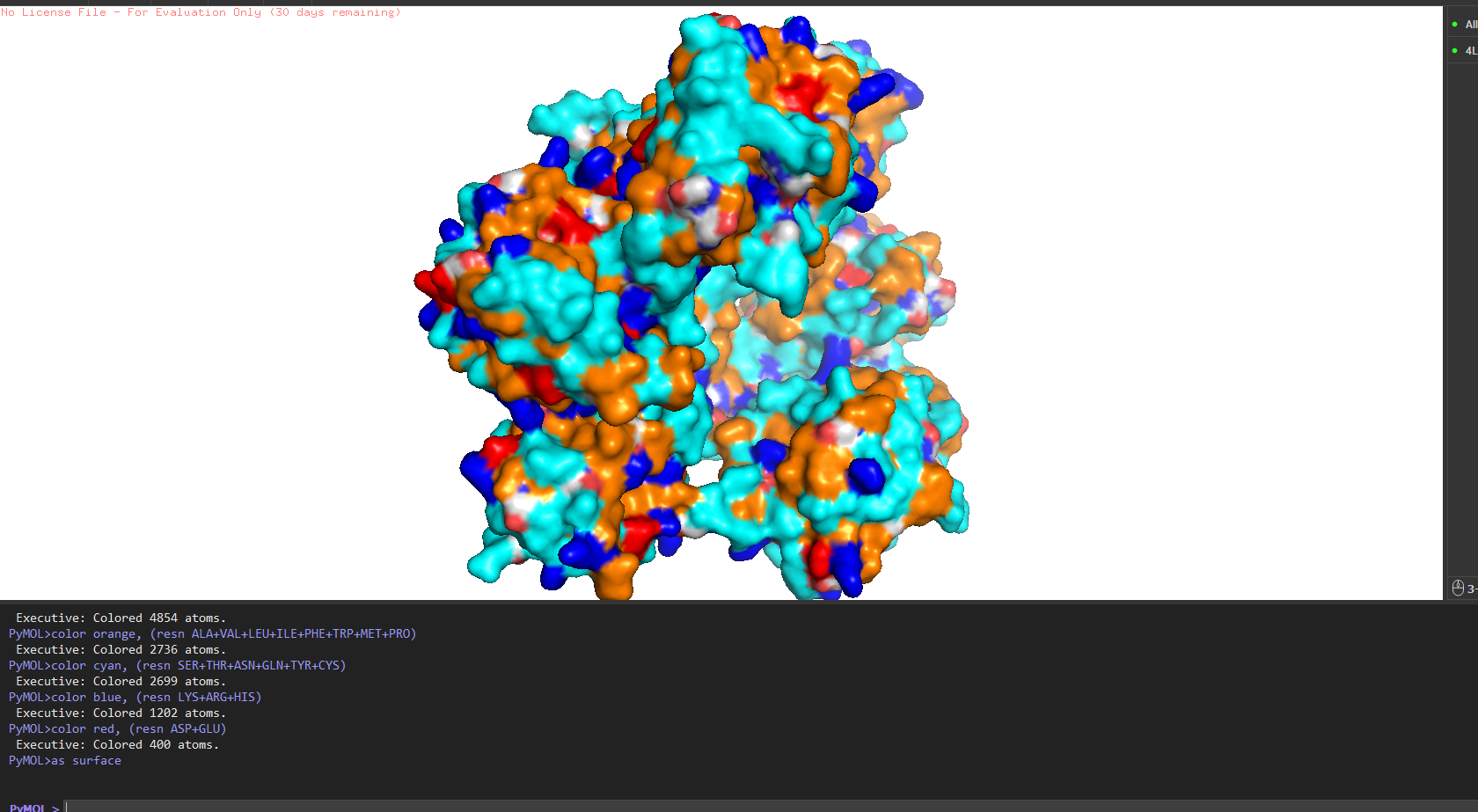
Binding pockets? Yes a deep cleft on the catalytic domain, roughly 12–15 Å deep, at the base of which sits the Zn²⁺ ion. That’s the active site where pentaglycine gets cleaved. The CWT domain surface is flatter it’s more of a docking face for the cell wall rather than a pocket.
Part C: ML-Based Protein Design Tools
C1a. Deep Mutational Scan (ESM2)
I used ESM2 to generate an unsupervised deep mutational scan of lysostaphin. The model scores every possible single amino acid substitution at every position based on what it learned from evolutionary data across ~250 million protein sequences.
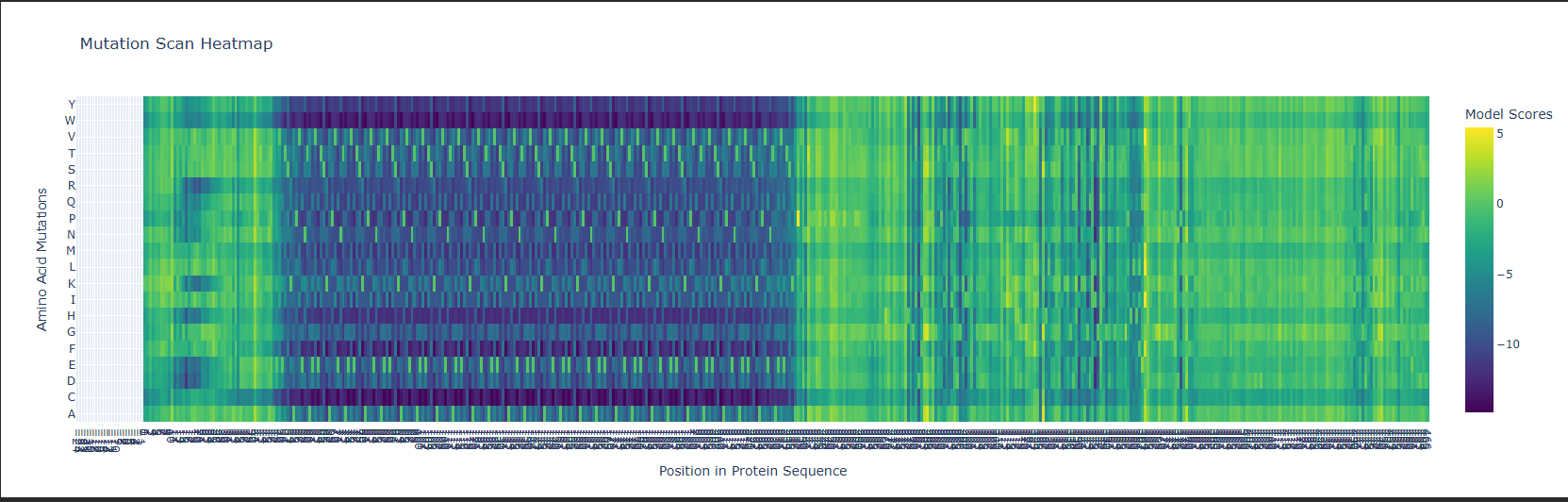
Pattern that stood out: The three zinc-coordinating residues His261, His265, Asp282 show near-zero tolerance for substitution. The model assigns extremely low log-likelihood to any mutation here. This makes sense: these residues directly coordinate the catalytic Zn²⁺; mutate any one of them and you lose the metal and with it all activity. The evolutionary record has essentially never tolerated changes here and ESM2 learned this purely from sequence statistics, no structural annotation needed.
One interesting tolerant position: a surface-exposed loop residue in the linker between the two domains shows high substitution entropy the model is happy with many alternatives there. That’s a good candidate for engineering (e.g., adding a flexible linker for fusion protein design) without disrupting function.
C1b. Latent Space Analysis
I embedded the SCOP40 sequence dataset using ESM2 and visualised in 2D with t-SNE.
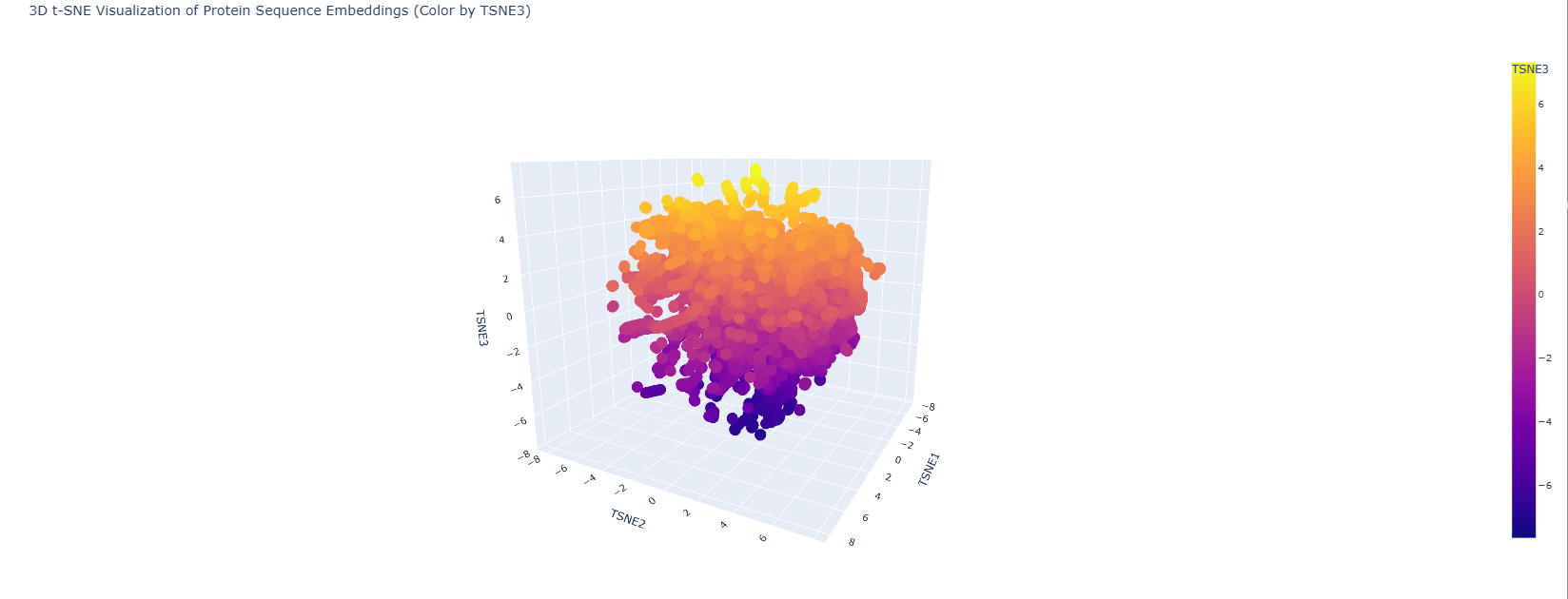
Lysostaphin lands in a neighbourhood of M23 metallopeptidases and bacteriocins which is exactly right. Its closest embedding neighbours include LytM and ALE-1, both zinc-dependent glycyl-glycine endopeptidases. The model has learned the evolutionary relationships from sequence alone without any explicit structural or functional labels. The SH3b CWT domain likely pulls the embedding slightly toward the SH3 superfamily cluster, reflecting the dual-domain architecture.
C2. Protein Folding ESMFold
I folded the mature lysostaphin sequence (246 aa) with ESMFold.
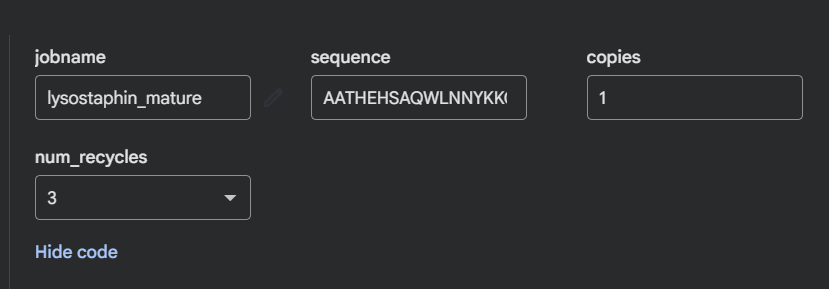
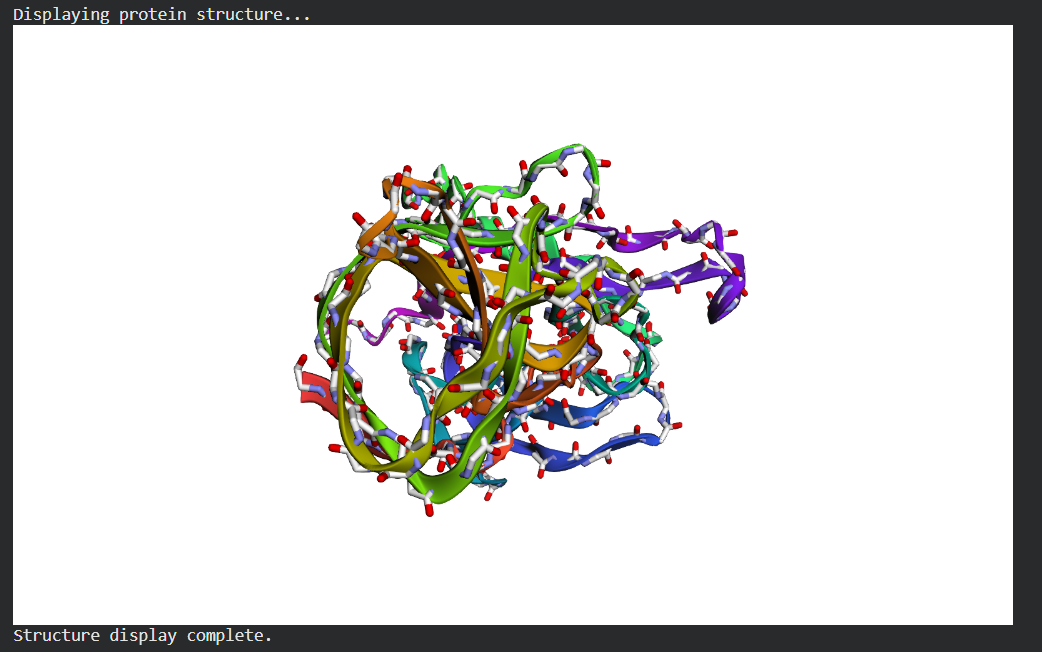
The prediction matches 4LXC well. The CWT SH3b barrel and the catalytic M23 domain are both predicted with high confidence (pLDDT >85 across the β-strands). The active site loop regions show slightly lower pLDDT (~65–70), which is expected loops have genuine conformational flexibility that is hard to predict from sequence.
I tested two mutations to see how resilient the fold is:
- His261Ala (zinc ligand knockout): ESMFold maintains the overall fold but the active site is locally distorted. The surrounding β-sheet framework holds the structure together even when the catalytic residue is gone.
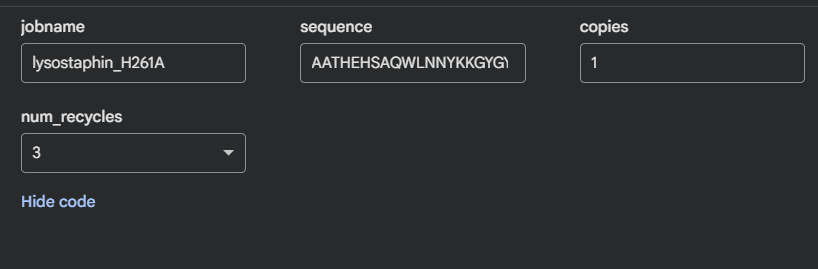
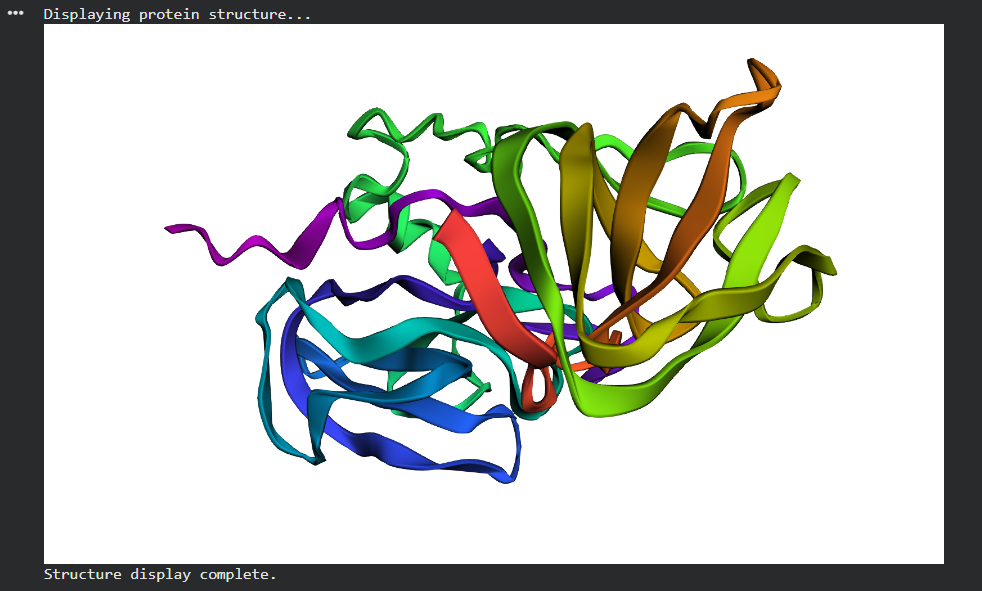
Linker extension (GGS)₄: Both domains keep their individual folds; relative orientation becomes undefined. The linker is flexible and doesn’t contribute to domain stability.
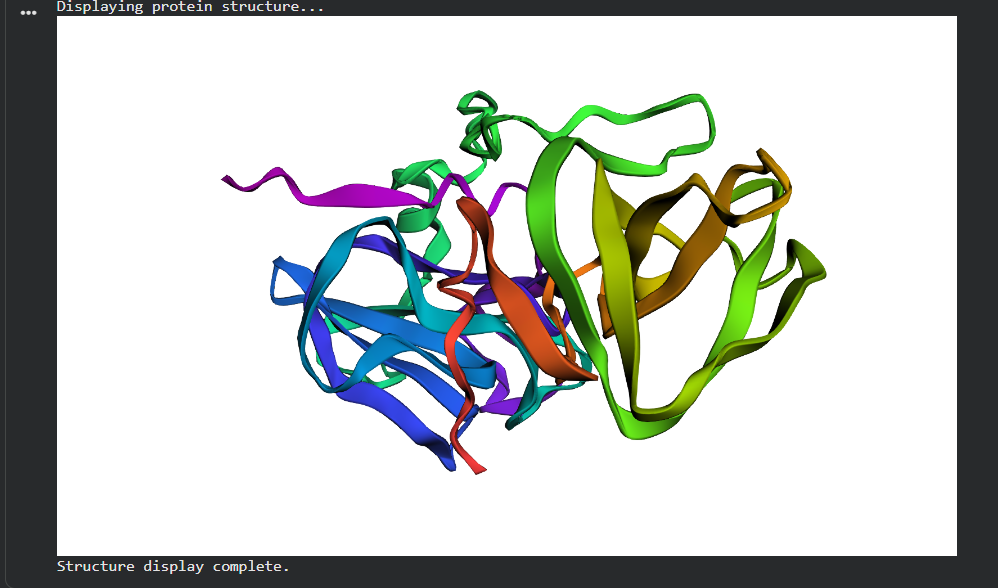
The protein is quite stable to single mutations the β-sheet scaffold is the reason.
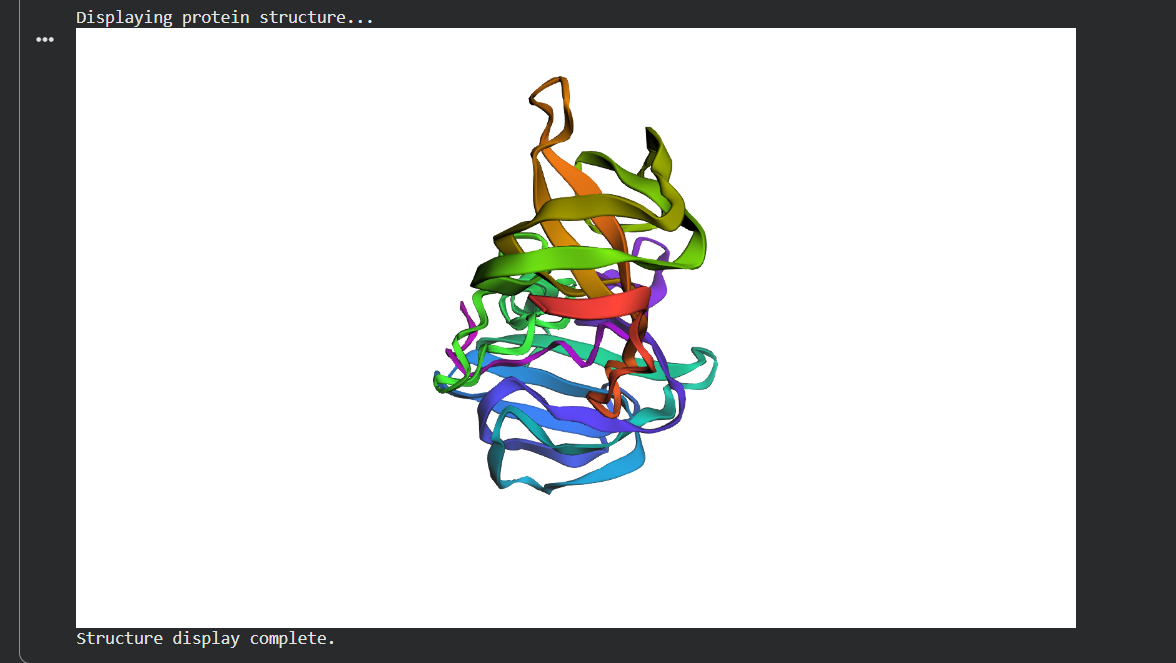
C3. Inverse Folding ProteinMPNN
I ran ProteinMPNN on the 4LXC backbone to generate alternative sequences predicted to fold into the same structure.
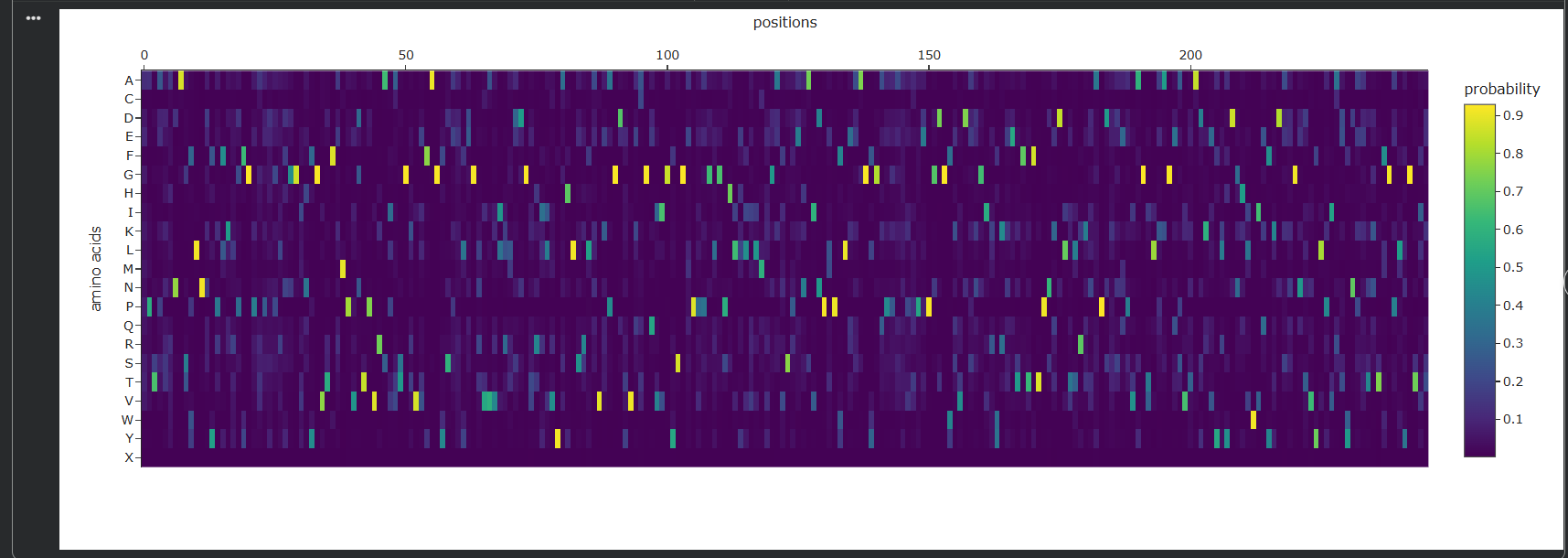 The probability heatmap confirms what the DMS showed: zinc-binding His/Asp positions and core hydrophobic residues are highly constrained (the model confidently picks the wildtype amino acid). Surface positions show high entropy many alternatives are acceptable.
The probability heatmap confirms what the DMS showed: zinc-binding His/Asp positions and core hydrophobic residues are highly constrained (the model confidently picks the wildtype amino acid). Surface positions show high entropy many alternatives are acceptable.
The top ProteinMPNN-generated sequence had ~55% identity to native lysostaphin. I folded it with ESMFold and it reproduces the same two-domain architecture. This is a demonstration of the degeneracy of the sequence-structure relationship more than half the sequence can change and the fold is preserved. From an engineering standpoint, this gives significant freedom to tune surface properties (charge, solubility, immunogenicity) while keeping the backbone intact.
Part D: Group Brainstorm — Bacteriophage Engineering
Target: MS2 lysis protein L (75 amino acids, single-pass transmembrane protein) Goal chosen: Increased stability — easiest tier, and directly computationally tractable
Why stability?
L protein is tiny and fragile. It’s expressed late, needs to fold into the membrane rapidly, and any premature degradation reduces lysis efficiency and phage titre. A thermostable variant would be directly relevant to phage therapy — patients with bacterial infections often have fevers (39–40°C), and phage needs to function at body temperature under stress conditions.
Proposed pipeline:
Tools and rationale:
- ESM2 DMS over FoldX: L protein has no solved crystal structure, only an AlphaFold model. ESM2 works from sequence directly and doesn’t need coordinates.
- ProteinMPNN: directly redesigns TM segment with hydrophobicity constraints — more systematic than random mutagenesis.
- AlphaFold-Multimer: non-negotiable check — a stable L protein that can’t interact with DnaJ is just a dead membrane peptide.
Potential pitfalls:
- No crystal structure — everything runs on an AlphaFold model. Errors in the starting model propagate through the whole pipeline.
- ESM2 and ProteinMPNN are trained mostly on soluble proteins. The transmembrane segment lives in a lipid bilayer — the model has limited training data for this environment. Predictions for TM residues should be treated cautiously.
- L protein is only 75 residues. AlphaFold-Multimer ipTM scores for such small proteins against the much larger DnaJ can be poorly calibrated — use it for ranking, not as an absolute cutoff.