Week 4 HW: Protein Design Part 1
Objective: Learn basic concepts of amino acid structure, 3D protein visualization, variety of ML-based design tools
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
There’s 20% of protein so there’s 100g of protein in 500g of meat. Knowing on average amino acid is 100 daltons (g/mol) so 100g divided by 100g/mol comes out to 1 mole of amino acids. 1 mole has about 6 x 10^23 molecules of AA (total in 500g of meat).
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Our body digests the proteins into amino acids and the process becomes molecular building blocks that are reassembled through our genetic instructions. Diet supplies us with energy and does not affect our DNA sequence.
Why are there only 20 natural amino acids?
This core batch was stabilized early on through evolution that was determined for adequate chemical diversity. They span a variety of characteristics such as flexibility, reactivity, acidity that is most optimized for use.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, you can make other non-natural AA. Examples such as photoreactivity, flurescence, and other sensing characteristics that allows for a potential applications for biosensors or electronic materials.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids were likely formed by natural chemical processes in earth. The Miller-Urey experiment in 1953 tested that idea whether building blocks of life formed naturally on early Earth without biology. Miller used water vapor, methane, ammonia, and hydrogen in a closed system and used electrical sparks to simulate lightning. The resulted solution were amino acids which successfully proved that they could be formed from gases and environmental energy.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
This would form a left-handed helix since most are using L-amino acids
Why are most molecular helices right-handed?
Most molecular helices are right-handed since L-amino acids (determined early in evolution) because they are considered the lowest-energy repeating structure available. The stereochemsitry of L-amino acides restricts backbone bond angles that makes right-handed helices more sterically favorable structure.
Why do β-sheets tend to aggregate?
They tend to aggregate due to their “sticky” structure and tend to seek hydrogen bonding neighbors.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
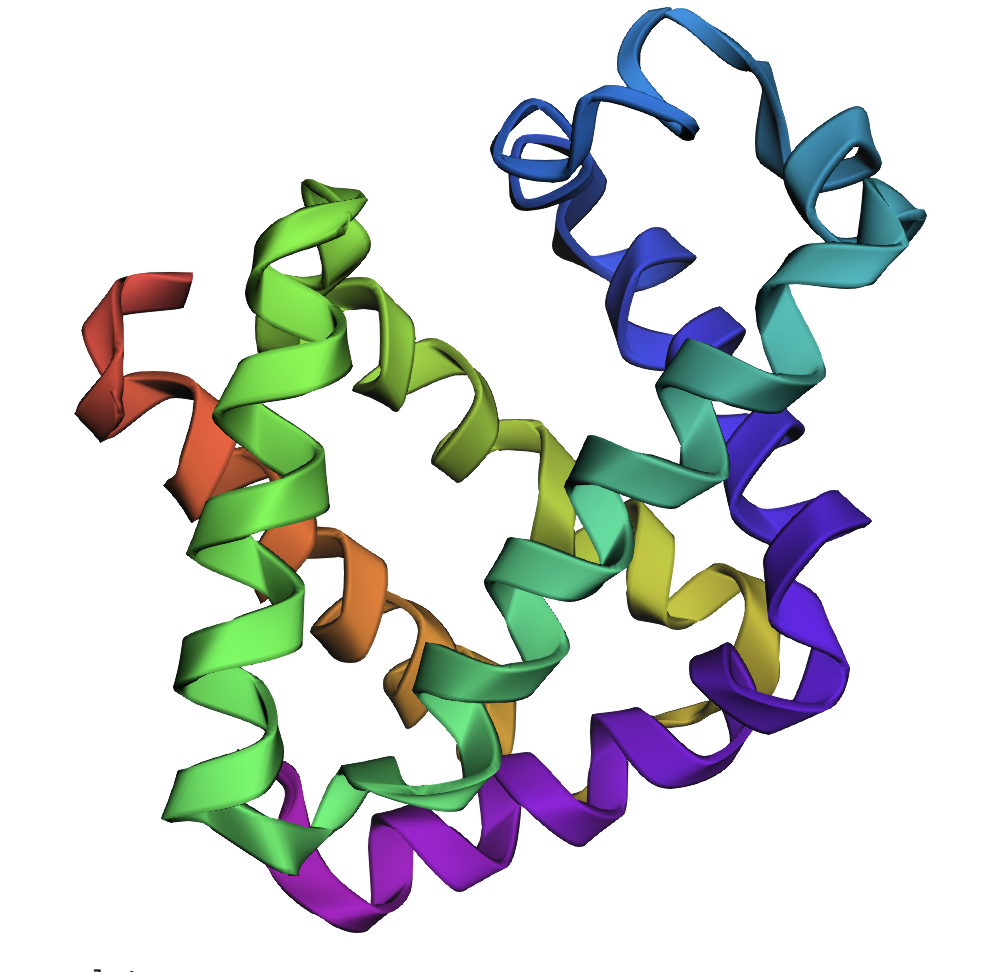
I chose Hydrophobin HFBI (Protein Data Bank ID 2FZ6) protein that is from a filamentous fungus, Trichoderma reesei. Unlike Class I hydrophobins that are insoluable once formed (like a permanent coating) whereas HFBI belongs to class II that is more dynamic and can dissemble. This protein is interesting from a design standpoint since there is potential to interpret is as a tunable surface as they are known to be highly surface-active to adapt surface tension between air-water which is important for fungal growth and spore disperal.
2. Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family?
This protein belongs to the Hydrophobin family and has a 76 AA long sequence:
2FZ6_1|Chains A, B, C, D|Hydrophobin-1|Hypocrea jecorina (51453)
SNGNGNVCPPGLFSNPQCCATQVLGLIGLDCKVPSQNVYDGTDFRNVCAKTGAQPLCCVAPVAGQALLCQTAVGA
The most frequence AA is C (Cysteine) and V (Valine) that has 8 residue count each.
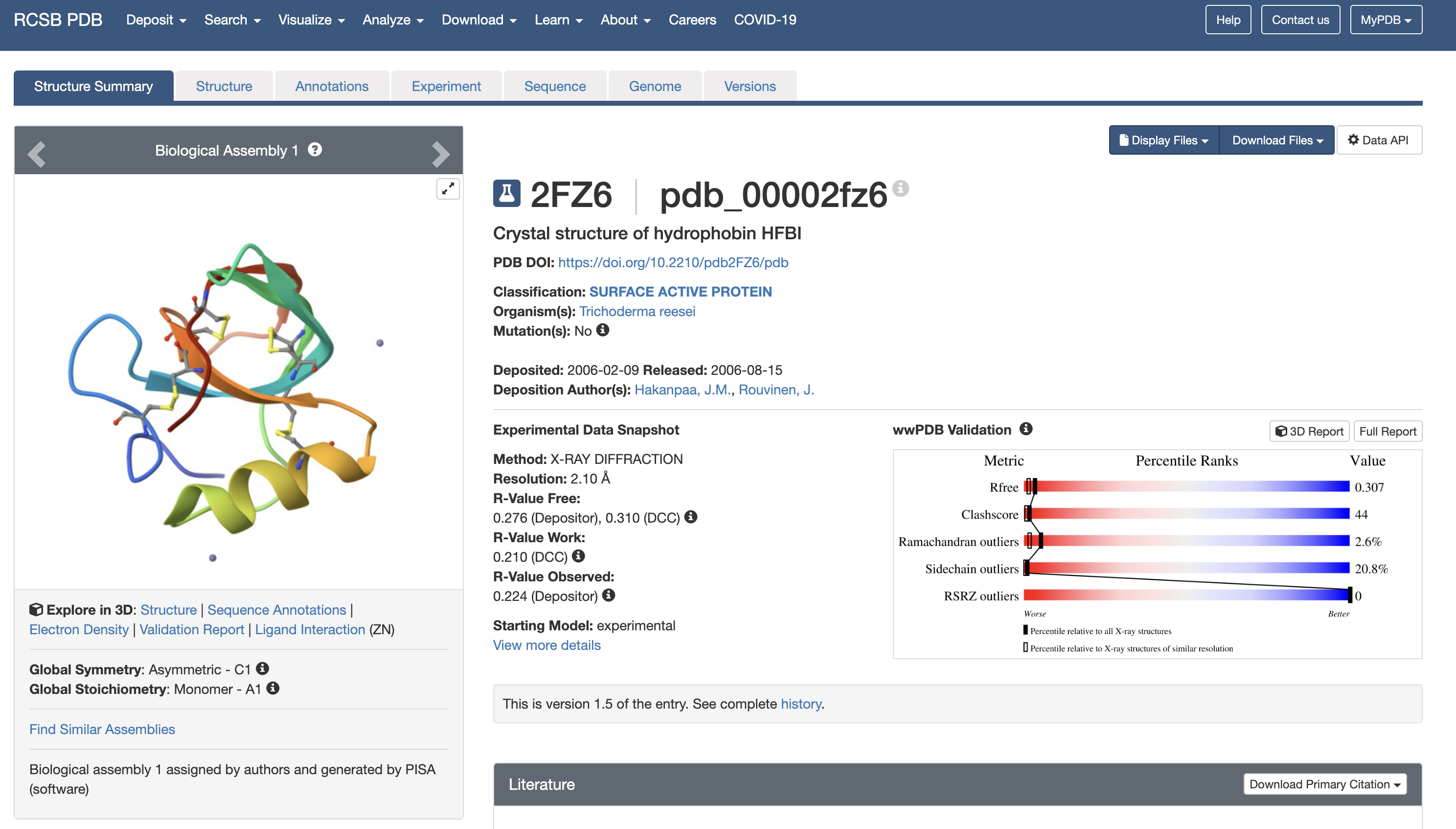
3. Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å). Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
This is the crystal structure of hydrophobin HFBI that was solved in 2006. With a resolution of 2.10 Å, this is a good quality structure. Zinc ion (Zn) is listed as a non-protein molecule which indicates they help with crystal packing or stabilization. The RCSB classifies 2FZ6 as a “surface active protein” in Class II of hydrophobins.
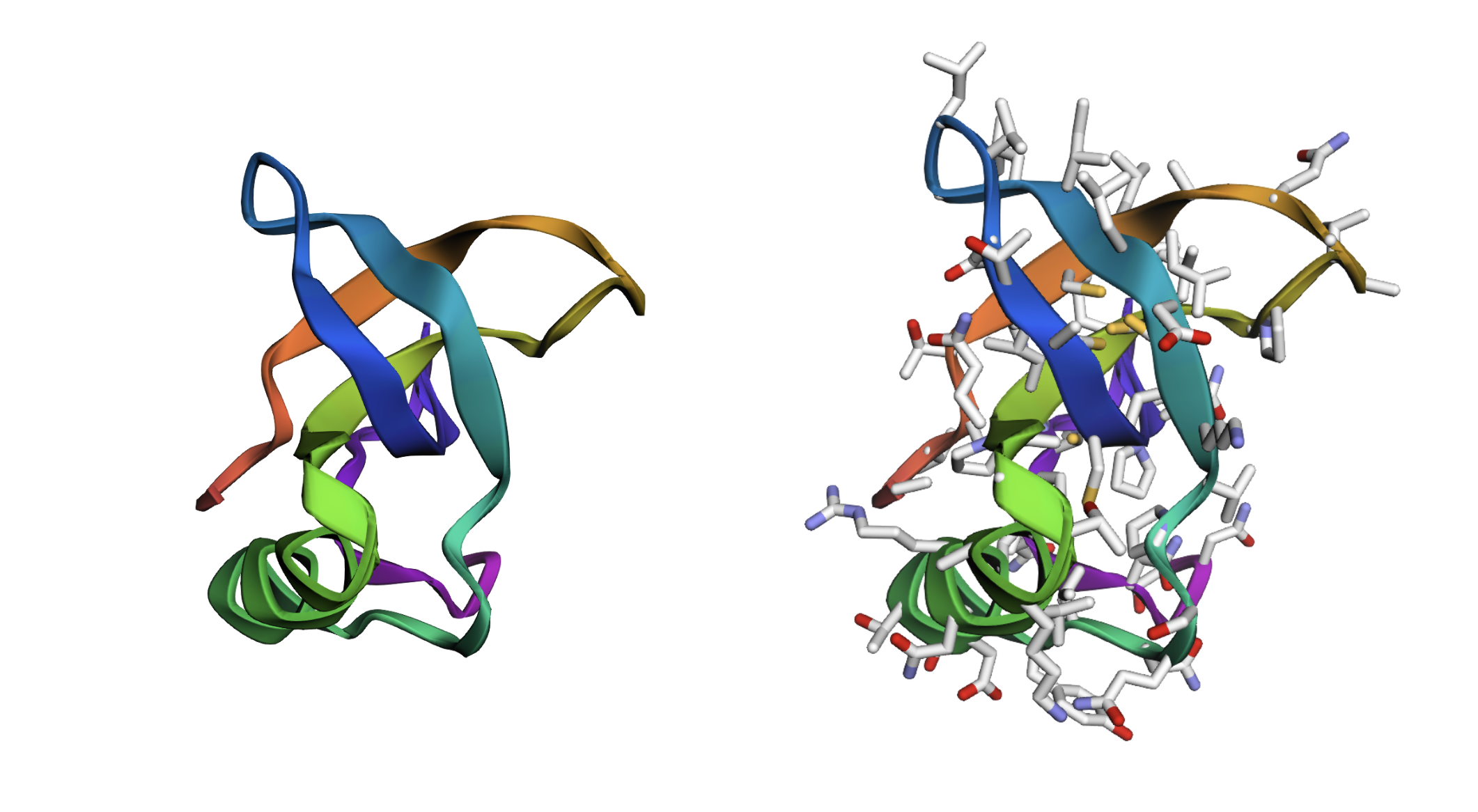
4. Open the structure of your protein in any 3D molecule visualization software: Visualize the protein as “cartoon”, “ribbon” and “ball and stick”. Color the protein by secondary structure. Does it have more helices or sheets? Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Crystal structure of hydrophobin HFBI from RCSB
It has more sheets and clear amphiphilic distinction with hydrophobic patch on one side. There’s no binding pockets apparent.
PyMOL wip - cartoon, ribbon, ball and stick

Part C. Using ML-Based Protein Design Tools
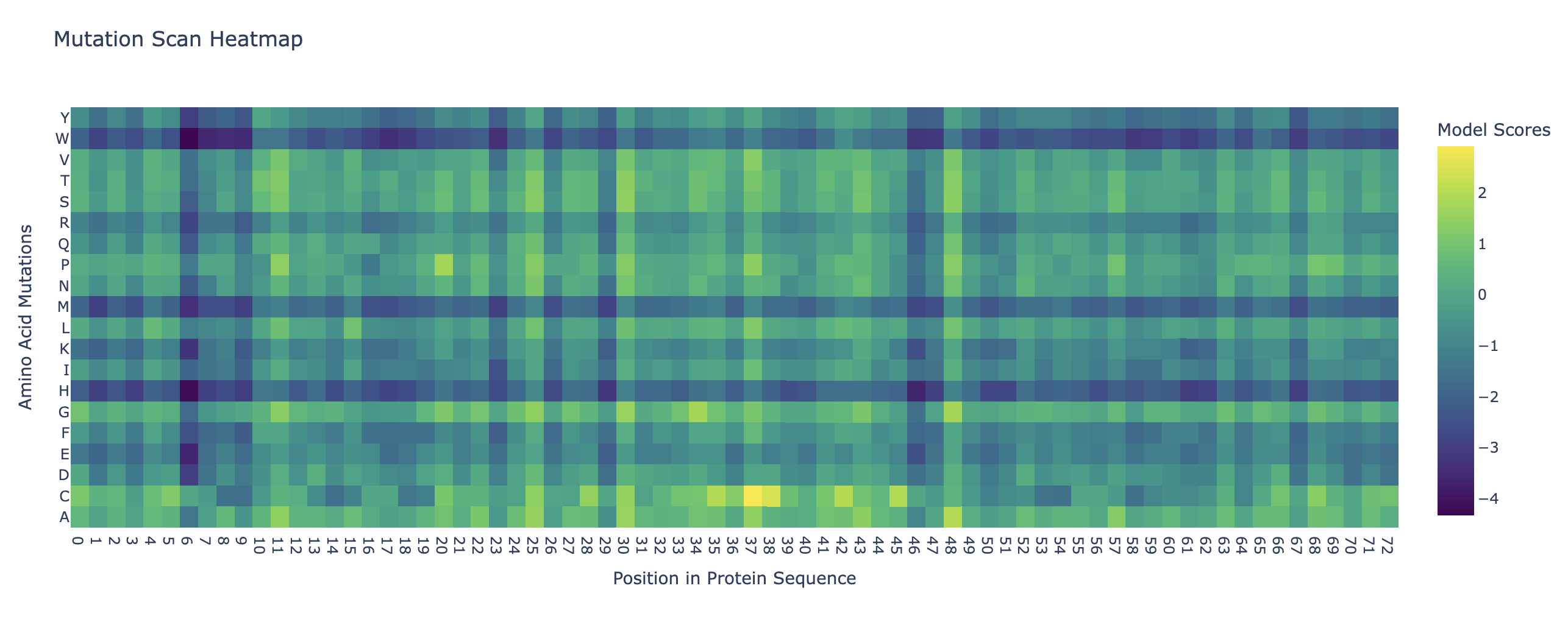
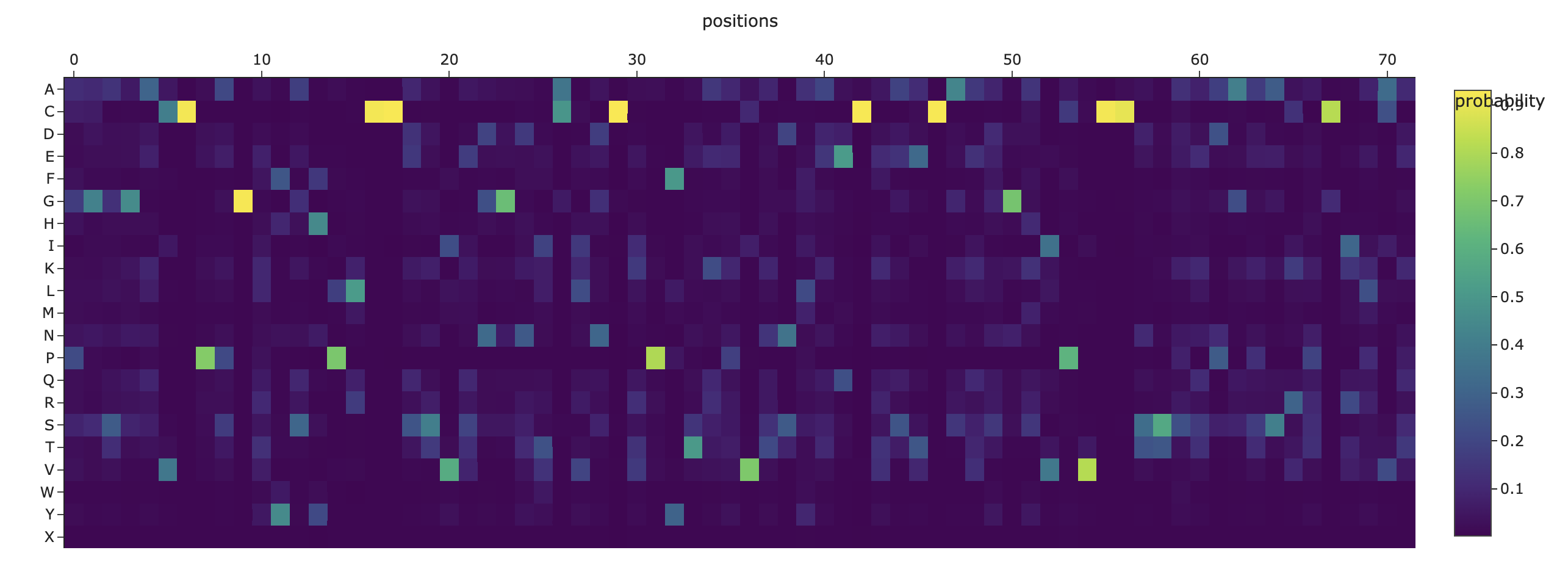
C1. Protein Language Modeling Deep Mutational Scans Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The heat map demonstrates a pattern at position 6 in the protein sequence that has a negative mutation score. It is for V (Valine) which is a nonpolar, hydrophobic AA. The substutution is rarely observed in the evolution since protein like HFBI depends on specific hydrophoc interactions and if there’s a change then it will distrupt the function in the patch.
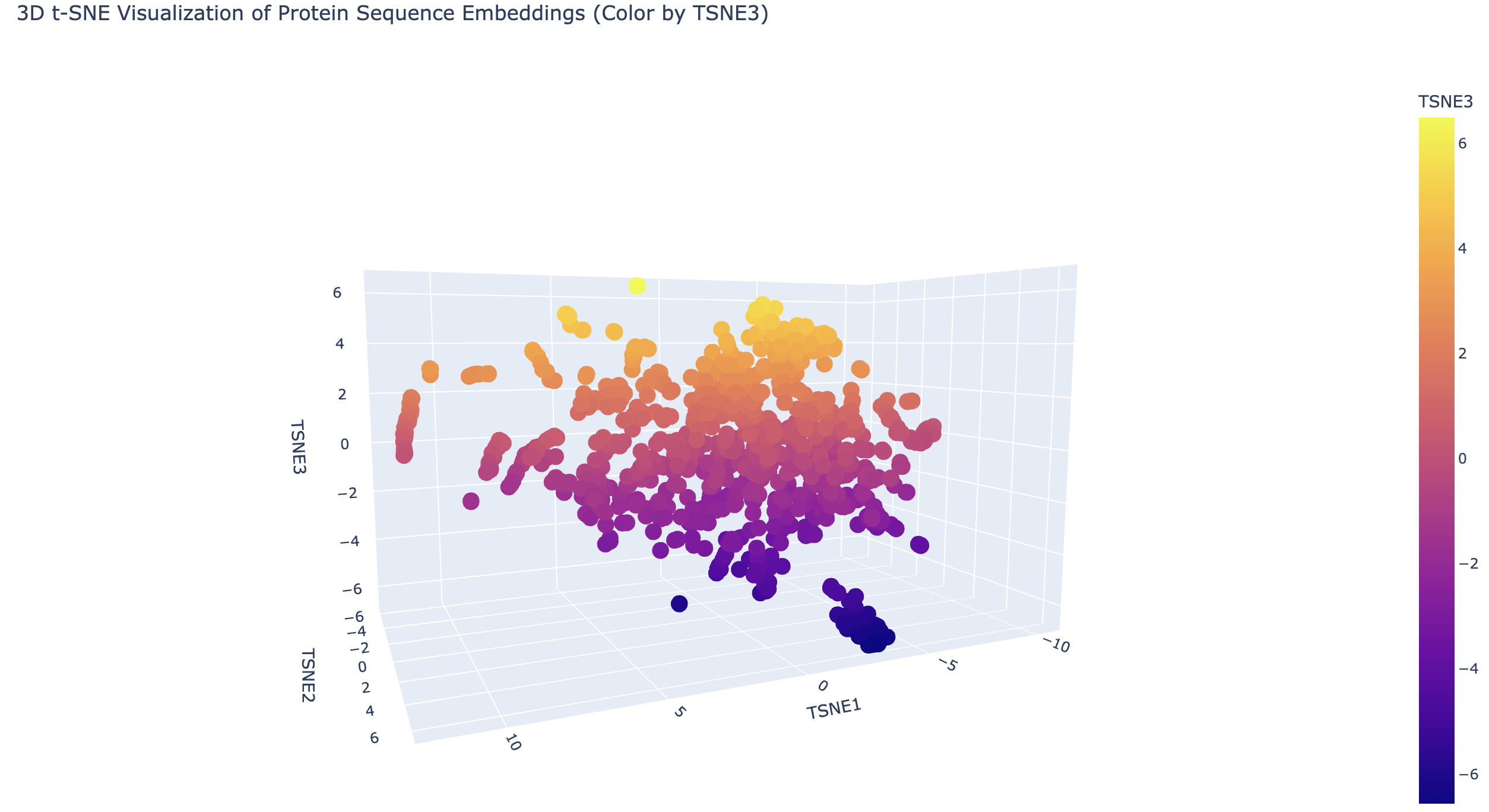
Latent Space Analysis Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins? Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Folding a protein. Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. Input this sequence into ESMFold and compare the predicted structure to your original.
Inverse fold:
New sequence: ALTPEEAALLRAAWAPVAADREANGRAFMLRLFAEYPELREYFPEFKGKSLEEIAASPKLAAFSTAVFDGLERLVATADDAAAMATLLADLAKAHVAKGIGAEHVEKIRAIHPAFVASVAPPPPGADAAWDRLFGLVIAALKAAGA
EMS fold: