Week 2 HW: DNA read-write-edit
Part 1: Gel Electrophoresis
Due to no access to equipment and space for gel electrophoresis I simulated the same to understand the process on https://www.labxchange.org/library/items/lb:LabXchange:9548bee3:lx_simulation:1?fullscreen=true
Workflow: Design plasmid DNA with protein of interest → Transform bacteria with plasmid DNA → Get many copies of plasmid DNA → Introduction of plasmid DNA to cells
Working in Benchling
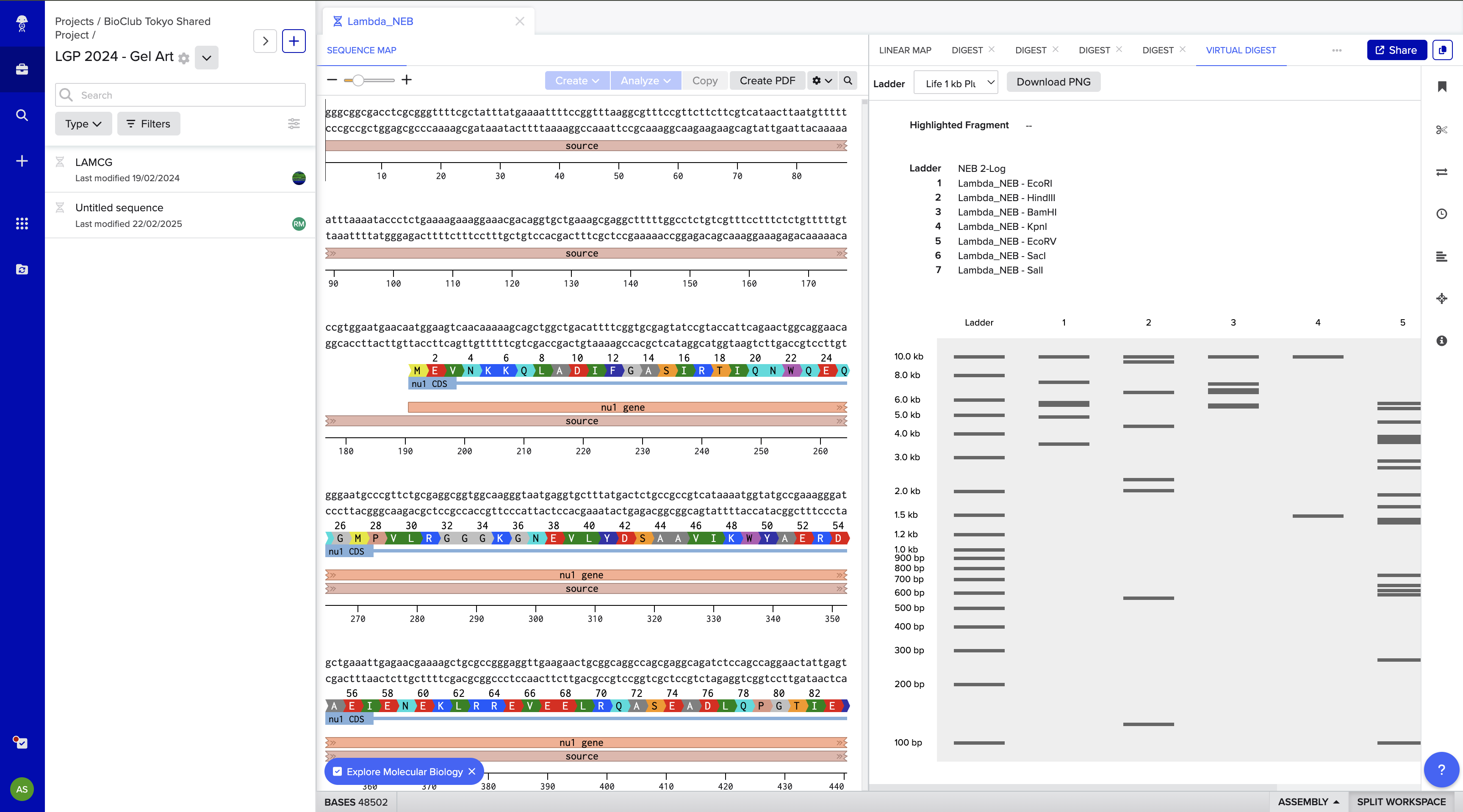
After signing in I imported it into Benchling and ran digests for:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
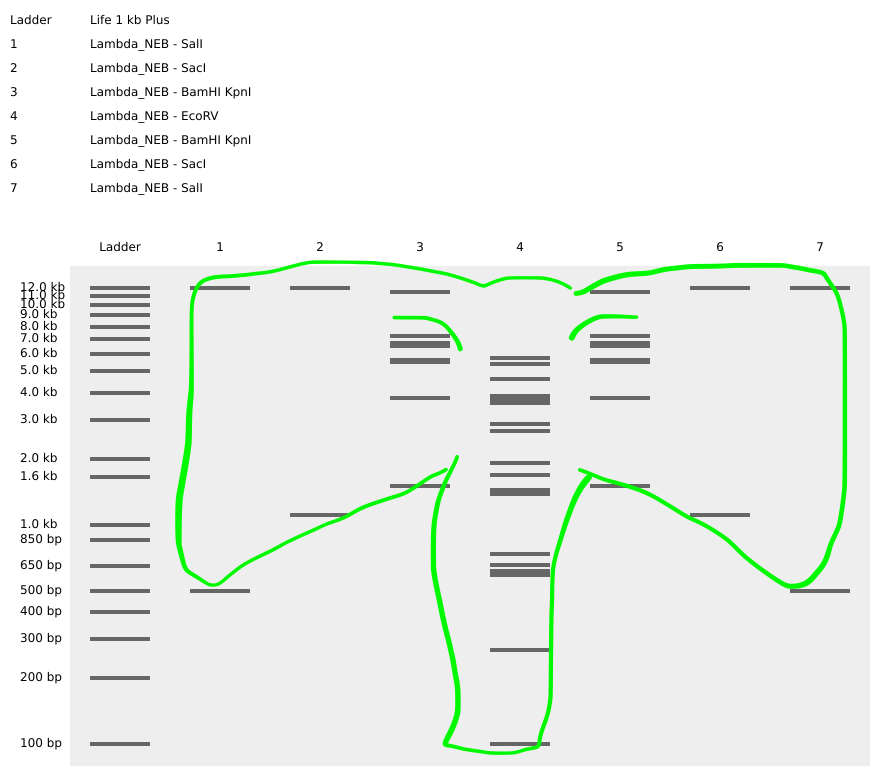
And then ran digests on SalI, SacI, BamHI, KpnI, EcoRV, BamHI, KpnI, SacI, SalI to create an Elephant! 🐘
For this, I referred to an iGEM video to understand how enzyme digesting works: https://www.youtube.com/watch?v=7cGev-SKLao
Part 3: DNA Design Challenge
3.1 Chosen Protein: Actin
tr|D3BD07|D3BD07_HETP5 Actin OS=Heterostelium pallidum (strain ATCC 26659 / Pp 5 / PN500) OX=670386 GN=act10 PE=3 SV=1 MEGEDVQALVIDNGSGMCKAGFAGDDAPRAVFPSIVGRPRHTGVMVGMGQKDSYVGDEAQ SKRGILTLKYPIEHGIVTNWDDMEKIWHHTFYNELRVAPEEHPVLLTEAPLNPKANREKM TQIMFETFNTPAMYVAIQAVLSLYASGRTTGIVMDSGDGVSHTVPIYEGYALPHAILRLD LAGRDLTDYMMKILTERGYSFTTTAEREIVRDIKEKLAYVALDFENEMQTAASSSALEKS YELPDGQVITIGNERFRCPEALFQPSFLGMESAGIHETTYNSIMKCDVDIRKDLYGNVVL SGGTTMFPGIADRMQKELTALAPSTMKIKIIAPPERKYSVWIGGSILASLSTFQQMWISK EEYDESGPSIVHRKCF
3.2 Reverse Translation
ATGGAAGGCGACGTTCAAGCGCTGGTGATCGACAATGGTTCTGGCATGTAAAGCGGGTTTCGCAGGCGACGACGCACCGCGCGCGCGCGTTCTTTCCTTCGATTGTGCGCCGTCGCCGTCATACCGGCGTGATGGTTGTGGGGATGCAGCAAGAGGACTCCTACGTGGGCGACGAGGCGCAGTCGAAAGGTGGGATCCTGACCCTGAAGTACCCGATCGAACACGGGATTGGTGACTAACAATGGGACGATATGAAGGAAATCTGGCACCACACGTTCTTATAACGAATTAAGAGTGGCGCCGGAAGAACCAGTTCCTGTGCTGCTGACCGAGGCGCCGCTGAACCCGAAAGCCAACCGTGAAGAAATGAAGACCAGGATTATGTTTGAACCTTTCAACACGCCGGCGATGTATGTGGCGATTCAAGCGGTGTTGTCGCTGTATGCCTCGGGTCGTACCACCGGTATTGTGATGGATTCTGGCGACGGCGTGTCCCATACGGTGCCCATCTATGAAGGTTATGCCTTACCGCACCGCATCCTCCGCCTGGATCTGGCGGGTCGCGATCTGACTGACTATGATGATGAAGATCCTGACTGAACGTGGTTATTCGTTTACGACCACCGCCGAAAGGGAAATCGTCGACATCAAAGAGAAGCTGGCGTATGTGGCACTTGATTTCGAGAACGAGATGCAAACGGCGGCGTCGTCGTCGTCGCGTTGAAAAGT CGTATGAACTGCCGGACGGCCAGGTCATCACTATCGGTAACGAACGTTTCCGCTGCCCTGCGCTTTCAACCGTCGTTCTTAGGCATGGAAAGCGCGGGCATACACGAAACCACGTACAACAGCATTATGAAATGCGATGTCGACATTCGCAAGGATCTGTATGGTAACGTGGTCCTGGGCGGCACCACGATGTTCCCGGGCATCGCCGAACGCATGCAAGAAACTGACCACCGCGCTGGCGCCGTCGACCATGAAAATCAAGATCATTGCCGCGCCGGAACGTAAGTCTTGGGTCATCGGCGGCTCGTTGGCCTCGTCGACCTTCCAGCAGATGTGGATCAGCAAAGAAGAGTATGACGAAAGCGGTCCTTCGGTGATCCACCGTAAGTTCTTCGCGAAACCGCAAGATTAA
3.3 Codon Optimization
Codon optimization is necessary because different organisms use synonymous codons (DNA triplets encoding the same amino acid) at different frequencies. Even if the amino acid sequence is identical, a coding sequence with rare codons for the host organism causes ribosome stalling, reduced translation efficiency, and lower protein yield. By swapping rare codons for frequently used equivalents in the host, we maximize the probability that the host’s tRNA pool can keep up with translation demand.
I optimized the actin sequence for E. coli expression, since it is the most common and accessible bacterial chassis for initial protein production. E. coli is fast-growing, inexpensive to culture, and has well-characterized expression systems, making it the practical choice for early characterization work even though actin is a eukaryotic protein.
Optimized DNA Sequence (1122 bp):
atggaaggcgatgtccaggcgctggtgatcgacaacggctccggcatgaaggccggcttcgccggcgatgatgcccccagggcggcggtgtcttcccctcgatcgtgggccgtccgcgtcacaccggtgtgatggtggtgggtatgcagcagaaagattcctatgtgggcgacgaagcgcaatcgaaagggcatcctgaccctgaagtatccgatcgagcatggcatcgtgaacaactgggacatggagaagatctggcaccacatgttctacaacgagctgcgtgtggcgccggaagaaccccacgtgctgctgaccgaggcgccgctgaacccgaaggccaaccgtgaacgcaagatgaagaccaggatcatgatgttcgaacagttcaacacgccggcgatgtatgtggcgattcaagcggtgctgtcgctgtatgcctcgggccgtaccaccggcatcgtgatggactccggcgatggcgtttcccacatcgtgcccatctatgaaggctatgcgctgccgcatgccatcctgcgcctggatctggcgggcagggatctgaccgactacatgatgaagatcctgaccgaacgcggttatagcttcaccaccaccgcggagaagatcgtccgggacatcaagaagaaactggcgtatgtggcgctcgatttcgaaaacgaaatgcaagcgaccgcgagctcgagcgccctggagaagtcgtatgagctgccggacggccaggtgatcaccatcggcaacgaacgcttccgttgccctgccgctgttccagccctcgttcggcatggagagcgccggcatccatgagaccacctacaacagcatcatgaagacctgcgatgtggacatccgcaaggacctgtatggcaacgtggtgctcggcggcaccaccatgttccccggcatcgccgacaggatgcaaaaggagctgaccgccgcgctgccgcccagcaccatgaagatcaagatcatcgcgccgccggagcgtaagtcgtgggtgatcggcggctcgctggcgagcctgagcacgttccagcagatgtggatcagcaaggaggaatacgacgagtcgggcccgagcatcgtgcaccgcaagtgcttcggcaagcgcaagatgaa
3.4 Production Strategy
I would recommend a plasmid-based cloning approach for initial expression work. The optimized DNA can be inserted into a standard expression plasmid and introduced into E. coli via transformation. Once inside the bacterial cells, the plasmid replicates autonomously, allowing the host machinery to transcribe my DNA into mRNA and subsequently translate it into actin protein. However, since actin is a eukaryotic cytoskeletal protein and my sequence lacks the signal peptides and targeting sequences necessary for membrane localization or secretion, the expressed protein will likely accumulate intracellularly. This necessitates cell lysis and downstream purification via affinity chromatography or other protein separation techniques to isolate and characterize my recombinant actin.
Alternatively, the PURE system (Protein synthesis Using Recombinant Elements) presents a compelling option due to its turnaround time. In this cell-free approach, my DNA template is incubated with a defined set of recombinant enzymes and cellular extracts that provide the necessary transcriptional and translational machinery. This in vitro reaction proceeds rapidly without the overhead of maintaining living cells, generating my actin protein directly in the reaction mixture. The resulting product must subsequently be purified via affinity chromatography to obtain homogeneous, functional protein suitable for downstream biochemical investigations.
Part 4: Twist DNA Synthesis Order
I used GFP (from Aequorea victoria, UniProt P42212) as my synthesis target, as it is a useful fluorescent reporter for tracking actin dynamics in Physarum polycephalum tubes. Physarum polycephalum has actin and myosin predominantly, and to understand the movements within the Physarum tubes, fluorescence can help visualize protein dynamics.
Protein sequence (UniProt):
sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria OX=6100 GN=GFP PE=1 SV=1 MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL VTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLV NRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLAD HYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
Reverse translated DNA sequence:
reverse translation of sp|P42212|GFP_AEQVI to a 714 base sequence using https://www.bioinformatics.org/sms2/rev_trans.html atgagcaaaggcgaagaactgtttaccggcgtggtgccgattctggtggaactggatggc gatgtgaacggccataaatttagcgtgagcggcgaaggcgaaggcgatgcgacctatggc aaactgaccctgaaatttatttgcaccaccggcaaactgccggtgccgtggccgaccctg gtgaccacctttagctatggcgtgcagtgctttagccgctatccggatcatatgaaacag catgatttttttaaaagcgcgatgccggaaggctatgtgcaggaacgcaccatttttttt aaagatgatggcaactataaaacccgcgcggaagtgaaatttgaaggcgataccctggtg aaccgcattgaactgaaaggcattgattttaaagaagatggcaacattctgggccataaa ctggaatataactataacagccataacgtgtatattatggcggataaacagaaaaacggc attaaagtgaactttaaaattcgccataacattgaagatggcagcgtgcagctggcggat cattatcagcagaacaccccgattggcgatggcccggtgctgctgccggataaccattat ctgagcacccagagcgcgctgagcaaagatccgaacgaaaaacgcgatcatatggtgctg ctggaatttgtgaccgcggcgggcattacccatggcatggatgaactgtataaa
4.1 Accounts
Created accounts on Twist Bioscience and Benchling.
4.2 Expression Cassette (built in Benchling)
I assembled the following parts in Benchling as a linear DNA sequence, annotating each region:
- Promoter (BBa_J23106):
TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC - RBS (BBa_B0034):
CATTAAAGAGGAGAAAGGTACC - Start Codon:
ATG - Coding Sequence: codon-optimized GFP sequence (see above)
- 7x His Tag:
CATCACCATCACCATCATCAC - Stop Codon:
TAA - Terminator (BBa_B0015):
CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
[Screenshot of annotated Benchling linear map]
4.3–4.6 Twist Order
On Twist, I selected Genes → Clonal Genes and uploaded the FASTA file of my expression cassette. I selected pTwist Amp High Copy as the cloning vector (Ampicillin resistance, high copy origin). I downloaded the full construct as a GenBank file and re-imported it into Benchling to view the complete plasmid map.
[Screenshot of Twist order screen and Benchling plasmid map]
Part 5: DNA Read / Write / Edit
5.1 DNA Read
(i) What DNA would I want to sequence and why?
I would want to sequence the genome of wild-type Physarum polycephalum and compare it against my engineered strain expressing tyrosinase/laccase-type oxidase. Sequencing would allow me to confirm the insertion site, verify no off-target integrations have occurred, and monitor for any spontaneous mutations that emerge over passages. This is directly relevant to the biosafety goals outlined in Week 1 — tracking genomic stability of the GMO is essential to ensuring the narrow functional envelope I described there.
(ii) Sequencing technology: Nanopore long-read sequencing (Oxford Nanopore Technologies)
- Generation: Third-generation sequencing. Nanopore reads single DNA molecules directly without PCR amplification, enabling very long reads (tens of kilobases), which is well suited to assembling the large, complex Physarum genome.
- Input & preparation: Genomic DNA extracted from the plasmodium, end-repaired and adapter-ligated using a ligation sequencing kit. No fragmentation is needed for long-read mode.
- Essential steps & base calling: DNA strands are driven through protein nanopores by an applied voltage. As each base translocates, it causes a characteristic disruption in ionic current. A neural-network basecaller (e.g., Dorado) converts the current signal into a nucleotide sequence.
- Output: Long-read FASTQ files, assembled into a draft genome or aligned to a reference for variant and insertion-site calling.
5.2 DNA Write
(i) What DNA would I want to synthesize and why?
I would synthesize the expression cassette for tyrosinase fused to a secretion signal peptide, optimized for expression in Physarum polycephalum. This is the core genetic payload for my dyeing application — the enzyme needs to be secreted into the extracellular slime trail to catalyze pigment formation on the substrate fabric. The GFP sequence prepared above would also be synthesized as a reporter to co-localize with the enzyme and confirm secretion into the trail.
(ii) Synthesis technology: Phosphoramidite-based gene synthesis (via Twist Bioscience)
- Essential steps: Solid-phase synthesis of oligonucleotides (~200 nt max per oligo), followed by error-correction, oligo assembly via overlap extension or Gibson Assembly into the full gene, and sequence verification by Sanger sequencing before delivery.
- Limitations: Maximum direct synthesis length is ~200 nt per oligo, so longer genes require assembly of multiple fragments introducing additional error-checking steps. Error rate of approximately 1 in 200 bases per oligo necessitates error-correction. Very GC-rich or repetitive sequences (common in some signal peptide sequences) require special synthesis handling.
5.3 DNA Edit
(i) What DNA would I want to edit and why?
I would edit the Physarum polycephalum genome to knock in the tyrosinase expression cassette at a defined safe-harbor locus — a genomic location where insertion causes no disruption to essential genes. Targeted integration is preferable to plasmid-based expression because it provides stable, heritable expression without plasmid loss over successive growth cycles, which is important for a consumer product where batch-to-batch consistency matters.
(ii) Editing technology: CRISPR-Cas9
- How it works: A guide RNA (gRNA) complementary to the target locus directs the Cas9 nuclease to create a double-strand break at a specific site adjacent to a PAM sequence (5’-NGG-3’). The break is repaired via homology-directed repair (HDR) using a donor template containing the tyrosinase expression cassette flanked by homology arms matching the safe-harbor locus.
- Preparation & inputs: Design gRNA targeting the safe-harbor locus using Benchling’s CRISPR design tool, synthesize the gRNA, prepare Cas9 protein or plasmid, and construct an HDR donor template with ~500 bp homology arms flanking the insertion site.
- Limitations: HDR efficiency in Physarum is not well characterized and may require significant optimization of delivery method (electroporation or lipofection). Off-target cuts remain a concern and require whole-genome sequencing to verify clean edits. PAM site availability constrains the exact targetable positions within the locus.