Week 4 HW: Protein Design Part 1
Part A: Questions by Shuguang Zhang
How many molecules of amino acids do you take with a piece of 500 grams of meat?
500g divided by 100 Da gives you about 3 × 10²⁴ molecules. So there are roughly 3 trillion trillion amino acids in a single serving of meat.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Digestion breaks everything down to bare amino acids first. The original protein blueprint is completely destroyed. Then our ribosomes rebuild new proteins using our own genetic code, not the cow’s or the fish’s.
Why are there only 20 natural amino acids?
It is probably just a frozen evolutionary accident. Early life found 20 that worked well enough and the genetic code hardwired them in. At that point there is no going back without breaking every living thing on the planet.
Can you make other non-natural amino acids? Design some new amino acids.
You just swap out the side chain for something chemically stable. For example you can put a fluorine where the methyl group is in alanine and get fluoroalanine which is more hydrophobic and harder to degrade. You can add an azide group for click chemistry. You can even shift to beta amino acids by inserting an extra carbon in the backbone which makes them resistant to proteases.
Where did amino acids come from before enzymes that make them, and before life started?
They formed abiotically. The Miller-Urey experiment showed that just mixing early atmospheric gases with lightning produces amino acids spontaneously. They also show up on meteorites, glycine has been found in carbonaceous chondrites. No enzymes needed at all.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
It would be left-handed. Normal L-amino acids form right-handed helices because of their backbone dihedral angle preferences. Mirror the chirality and you mirror the helix.
Can you discover additional helices in proteins?
We already know the 3-10 helix and the pi helix exist beyond the standard alpha helix. With cryo-EM resolution improving and AlphaFold predictions getting better, there are likely more unusual helical conformations hiding in membrane proteins and intrinsically disordered regions.
Why are most molecular helices right-handed?
Because life uses L-amino acids, and L-amino acids have backbone angles that naturally favor a right-handed turn. It traces all the way back to whichever chirality got selected early in evolution and then just stuck.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation? Edge strands have exposed hydrogen bond donors and acceptors sitting there unsatisfied. They are basically sticky edges looking for a partner. The driving force is intermolecular hydrogen bonding combined with hydrophobic burial, and water gets released in the process which makes it entropically favorable too.
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials? When proteins misfold under stress they expose hydrophobic patches that seed beta sheet stacking. Once that nucleus forms it is thermodynamically very stable so more protein keeps piling on. As for materials, amyloid fibers are actually incredibly strong, comparable to silk, and they are self-assembling and tunable. People are already engineering them into scaffolds, nanowires, and hydrogels.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
In the plasmodium of Physarum polycephalum, the F-actin capping activity of the actin-fragmin complex is regulated by phosphorylation of actin, mediated by a novel type of protein kinase with no sequence homology to eukaryotic-type protein kinases. This protein sits at the heart of what makes Physarum behavior fascinating. The oscillatory protoplasmic streaming that drives Physarum’s decision-making and network formation depends on rapid, rhythmic reorganization of the actin cytoskeleton. AFK is the molecular switch that controls it by phosphorylating actin, it determines whether actin filaments are being capped and severed (disrupting the cytoskeleton) or allowed to grow (driving streaming). Studying this kinase is therefore studying the molecular basis of Physarum’s behavioral intelligence. The signalling pathway results in phosphorylation of actin, and stage-dependent phosphorylation of actin is associated with morphological alterations and reorganization of the actin cytoskeleton.
2. Identify the amino acid sequence of your protein.

The protein sequence has a total length of 737 amino acids. The most frequent amino acid is Serine (S) with 84 occurrences (11.40%), followed by Leucine (L) with 58 occurrences (7.87%), and Glycine (G) with 56 occurrences (7.60%). The least frequent is Cysteine (C) with 10 occurrences (1.36%).

Protein family AFK belongs to the eukaryotic protein kinase (ePK) superfamily structurally, but functionally it is classified as the founding member of a unique actin kinase family. It is structurally related to the phosphoinositide kinase superfamily rather than classical Ser/Thr kinases, placing it in an unusual evolutionary position.
3. Identify structure page of your protein
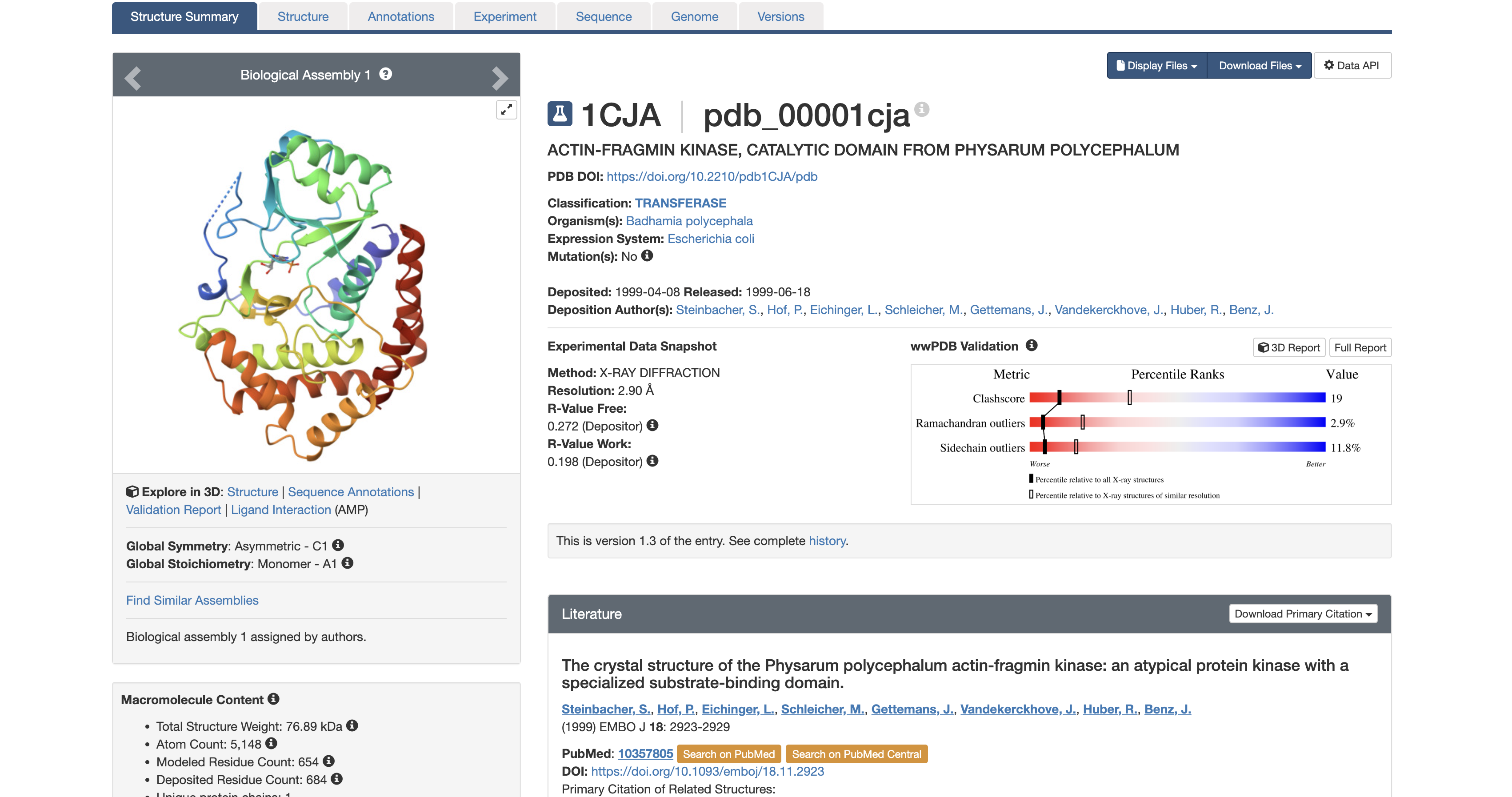
It was solved in 1999. At 2.9 Å, you can reliably identify the backbone fold, secondary structure elements, and the position of the AMP ligand, but side-chain details are slightly less precise than higher-resolution structures.
The structure contains the protein (actin-fragmin kinase) and adenosine monophosphate (AMP). AMP is not a random co-crystal contaminant. AMP occupies the ATP binding pocket of the kinase. This tells you precisely where the nucleotide binding site is and how the kinase is oriented to receive ATP before phosphorylating actin. In the context of Physarum behavior, this pocket is a potential target for disrupting the actin phosphorylation cycle to study what happens to streaming oscillations when AFK is inhibited.
4. Open the structure of your protein in any 3D molecule visualization software
- P.S. There are double protein structures in the screenshots accidentally.
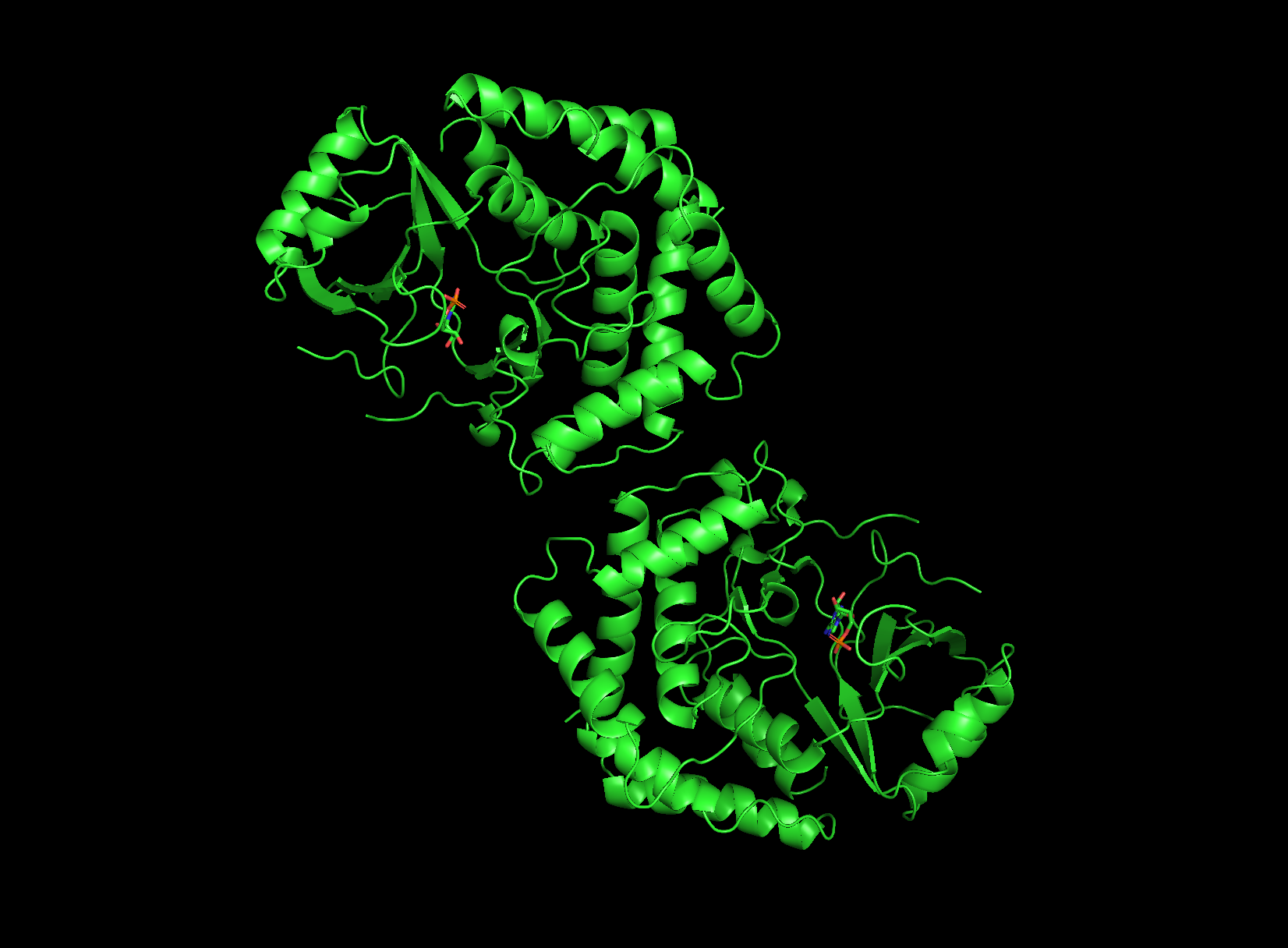
visualizing as ‘cartoon’, ‘ribbon’ and ‘ball and stick’
 cartoon view
cartoon view
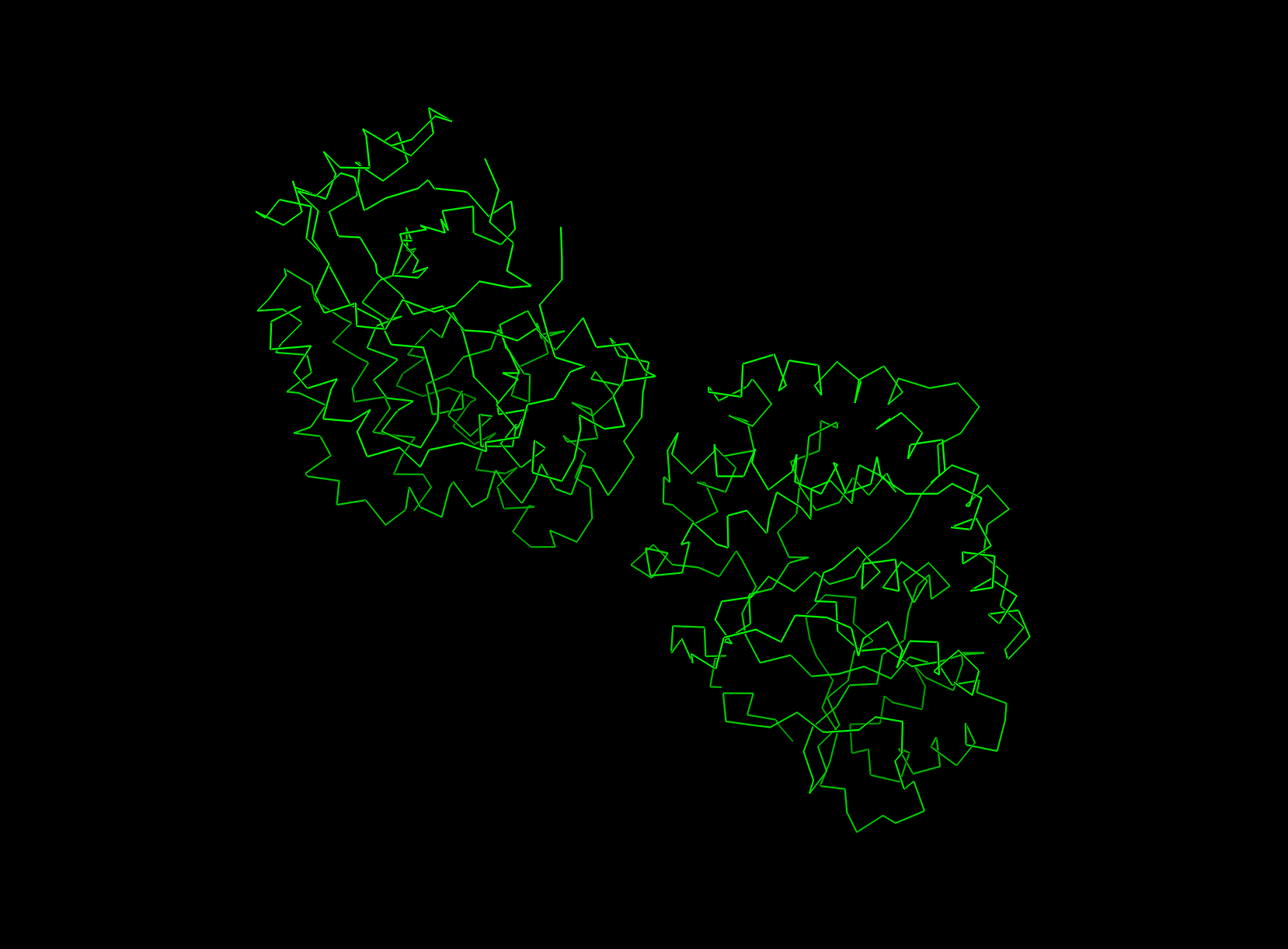 ribbon view
ribbon view
 ball and stick
ball and stick
Looking at the structure image, the catalytic module spans about 160 residues, with the nucleotide binding site and catalytic machinery tucked into the cleft between the two lobes. According to PubMed, there is a pretty balanced mix of alpha helices and beta sheets, which is exactly what you expect from this bilobal kinase fold.
The protein surface is a sea of blue hydrophilic residues, which is what allows it to stay dissolved in the crowded cytoplasm of the cell. In contrast, the protein core is packed with orange hydrophobic residues. These are tucked away from water, creating the internal glue that keeps the entire structure stable and folded correctly. In the AMP binding pocket the hydrophobic patches grip the adenine ring of the nucleotide, while polar residues reach out to coordinate the phosphate groups. This mapping is really the key to Physarum biology. Since the kinase has to dock onto actin filaments, that unique flat substrate recognition domain is covered in hydrophilic patches specifically designed to recognize and stick to actin’s surface chemistry.
First, there’s the ATP/AMP binding pocket, a deep cleft that’s carved right between the N-terminal and C-terminal lobes. Since you can clearly see the yellow AMP ligand tucked inside, it’s obviously the biggest “hole” on the surface and the best place for drug targeting. Second, check out the flat substrate recognition domain. Unlike most kinases that have a narrow groove, AFK uses a remarkably flat, broad surface to dock with the large actin substrate. This unique structural flatness is a huge defining trait for this enzyme.
C1. Protein Language Modelling
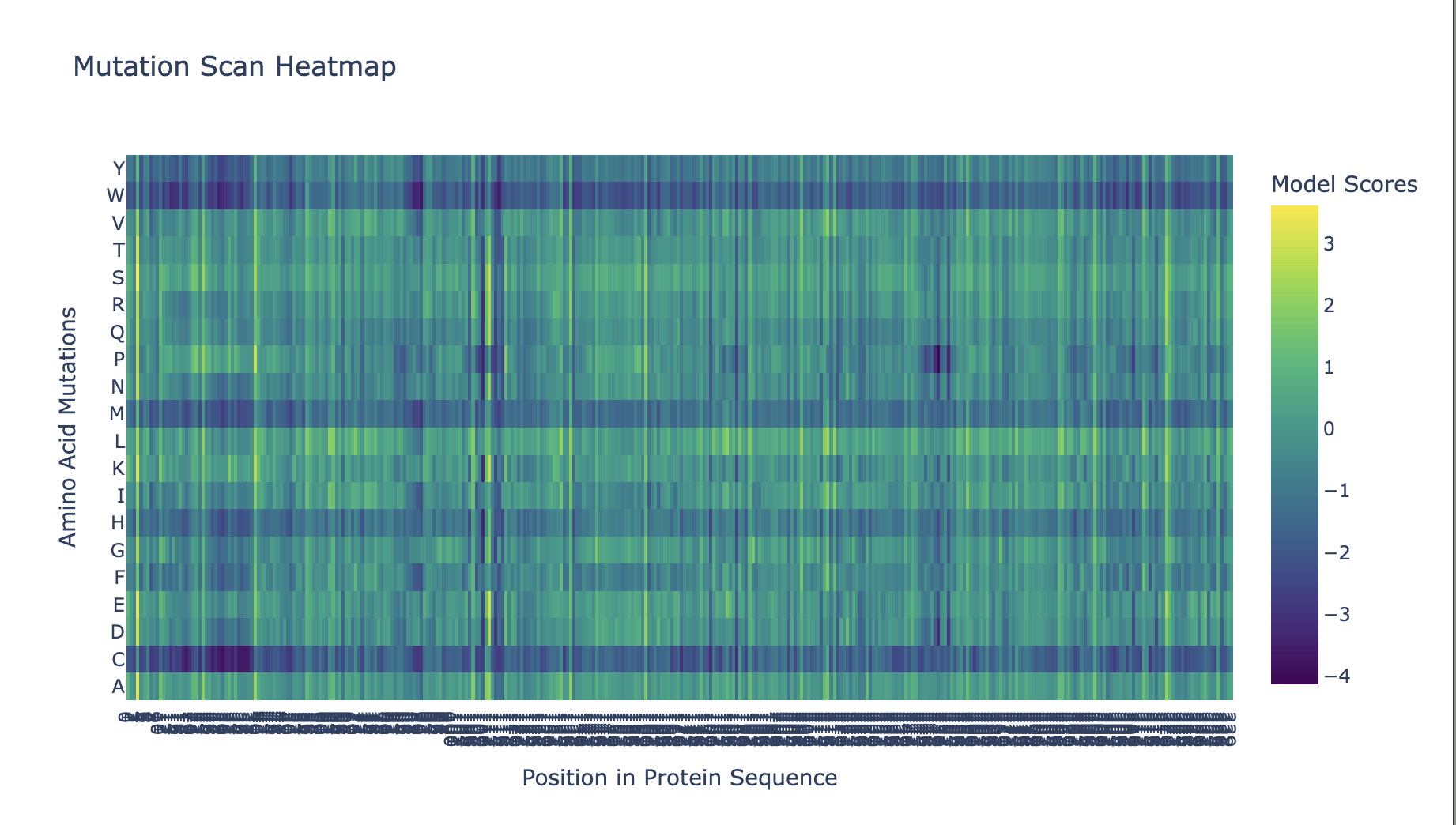
Position 109 (Asp/D) shows the strongest conservation signal in the mutational scan — nearly all substitutions receive strongly negative log-likelihood scores. This is consistent with this residue being the catalytic base in the kinase active site, directly involved in phosphotransfer to actin’s Thr202. Even conservative mutations (D→E) are penalized, suggesting the precise geometry of this aspartate is essential.
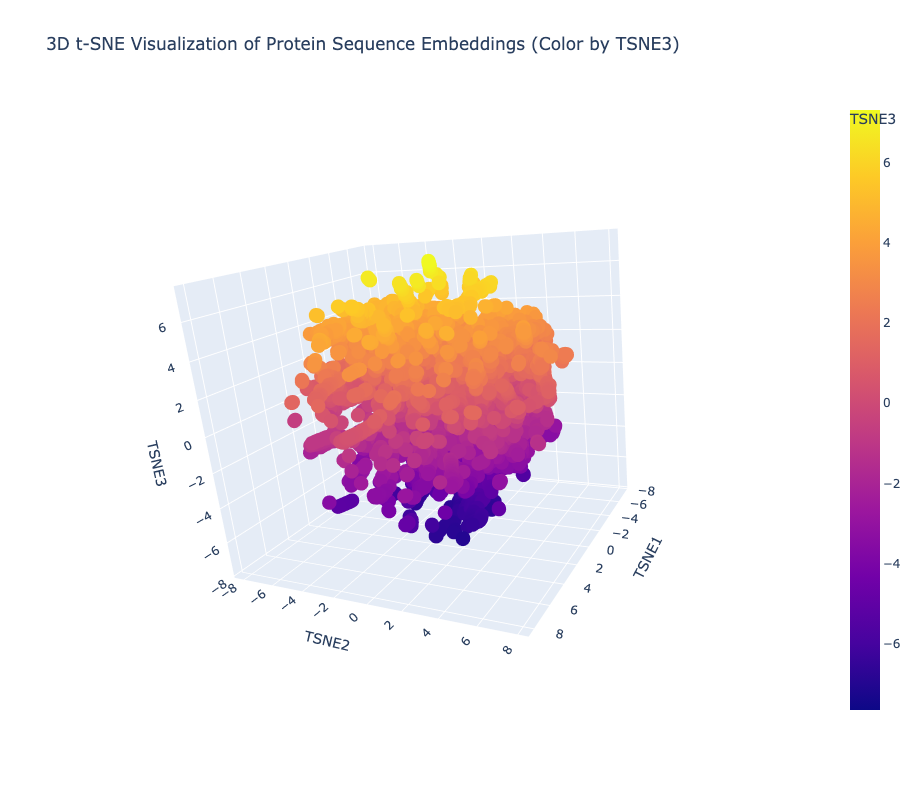
Yes, the t-SNE map forms meaningful neighborhoods where evolutionarily related proteins cluster tightly together, confirming that ESM2 has successfully learned to group biologically similar sequences into shared regions of the latent space.
I ran a request in Gemini to create another 3d t-SNE with the AFK highlighted and this is how it looked
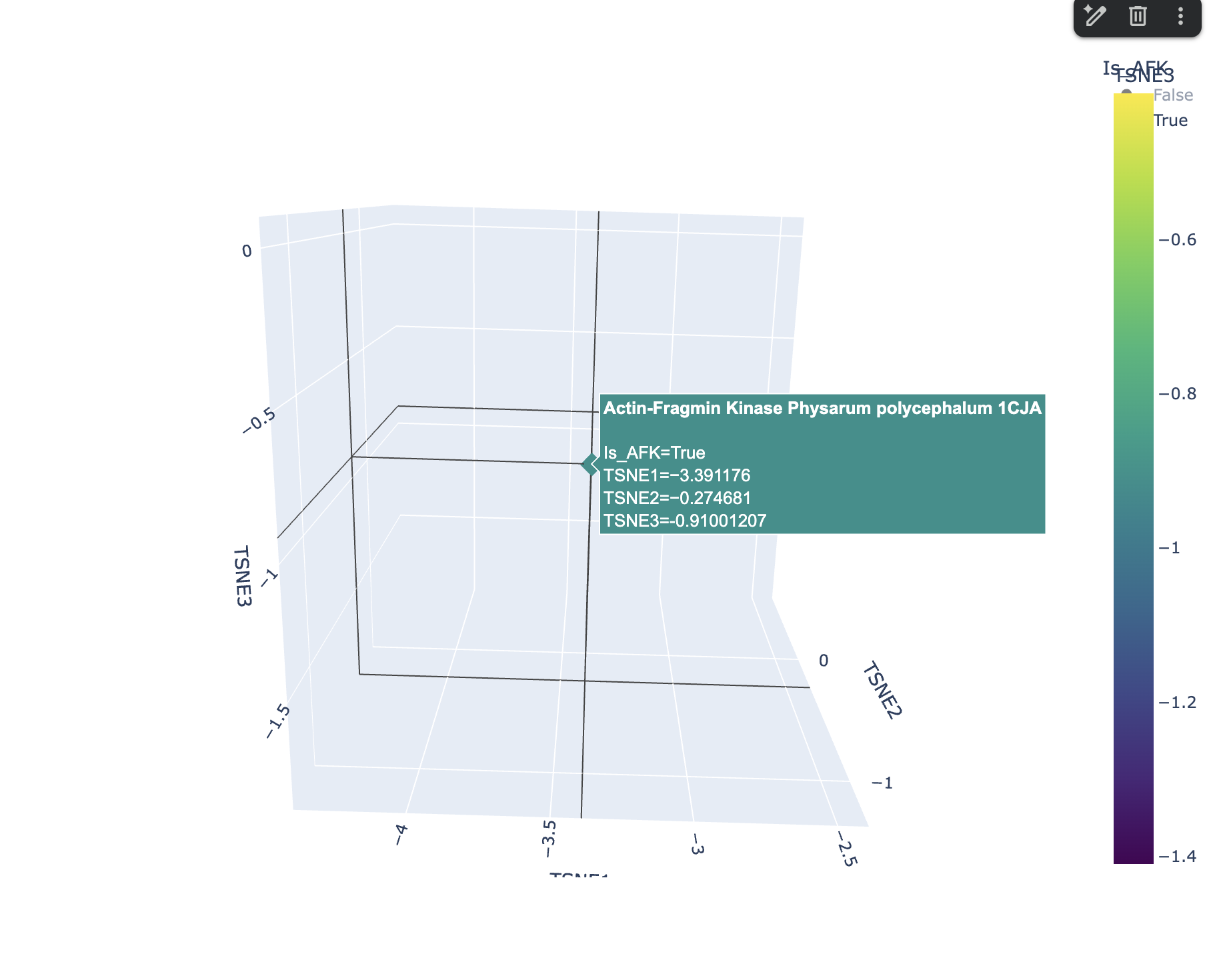
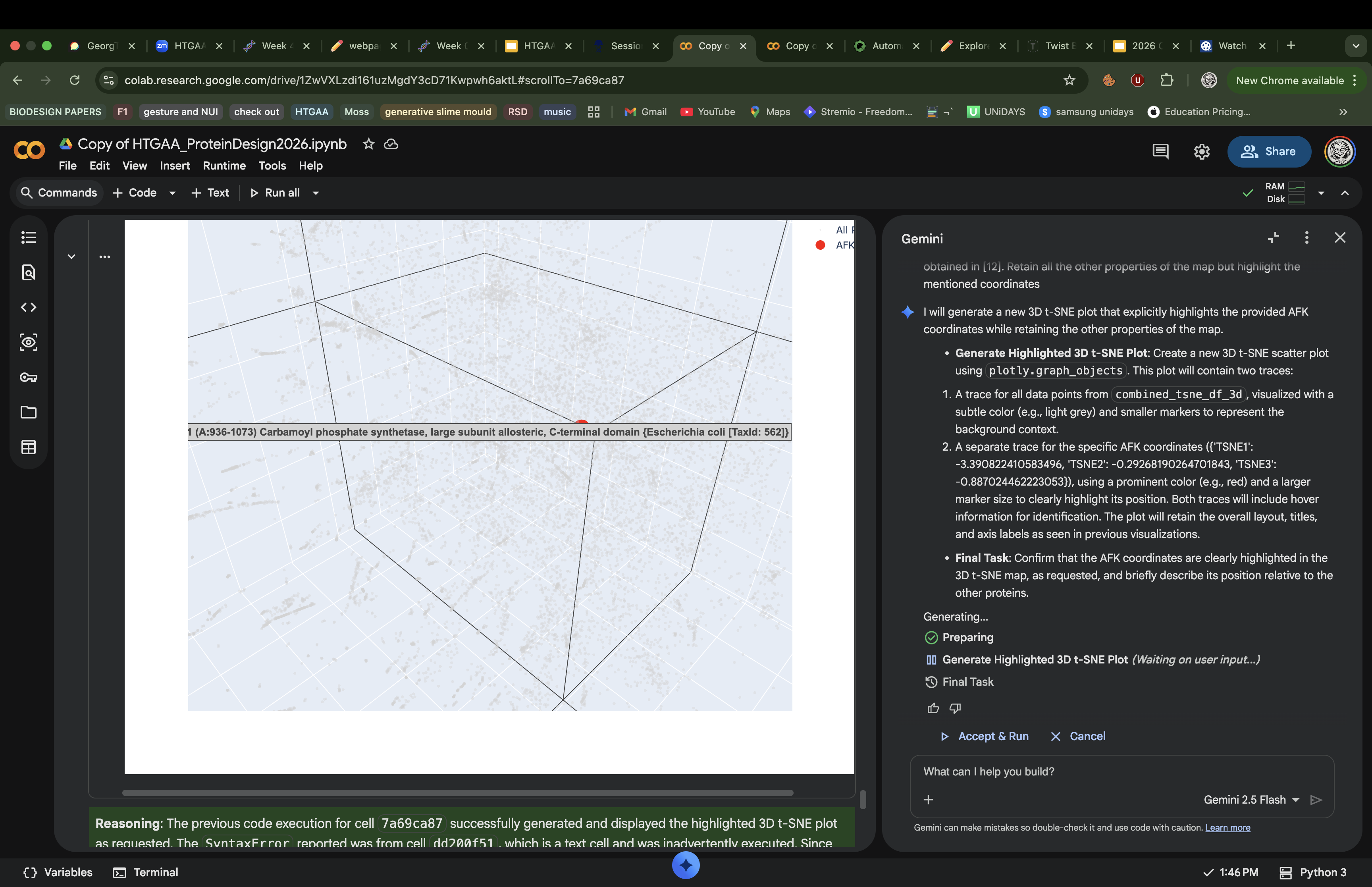
AFK from Physarum polycephalum lands at coordinates (−3.39, −0.29, −0.89) in a sparse, isolated region of the map with no tight cluster, reflecting its status as an evolutionarily unique kinase with no sequence homology to classical eukaryotic protein kinases. Its nearest neighbors are similarly atypical, low-homology proteins rather than mainstream kinases or cytoskeletal proteins like actin
Protein Folding
predicted structure after running it through colab

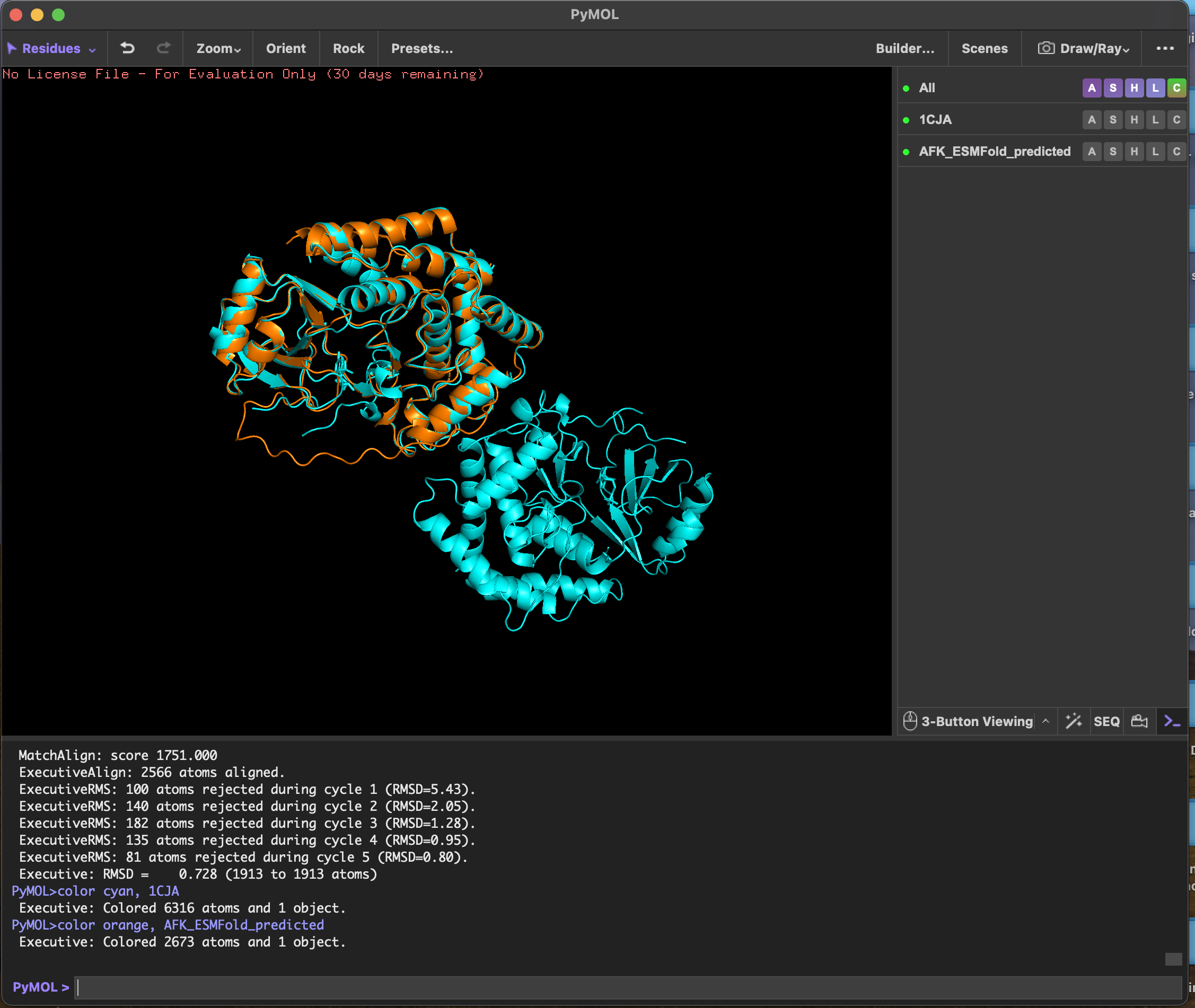 RMSD score Executive: RMSD = 0.728 (1913 to 1913 atoms)
ESMFold predicted the 3D structure of Actin-Fragmin Kinase from Physarum polycephalum using sequence alone, achieving an RMSD of 0.728 Å against the experimentally determined crystal structure 1CJA
RMSD score Executive: RMSD = 0.728 (1913 to 1913 atoms)
ESMFold predicted the 3D structure of Actin-Fragmin Kinase from Physarum polycephalum using sequence alone, achieving an RMSD of 0.728 Å against the experimentally determined crystal structure 1CJA
Mutation 1 - position 45, changed S (Serine) to A (Alanine)
Executive: RMSD = 0.744 (1918 to 1918 atoms) only
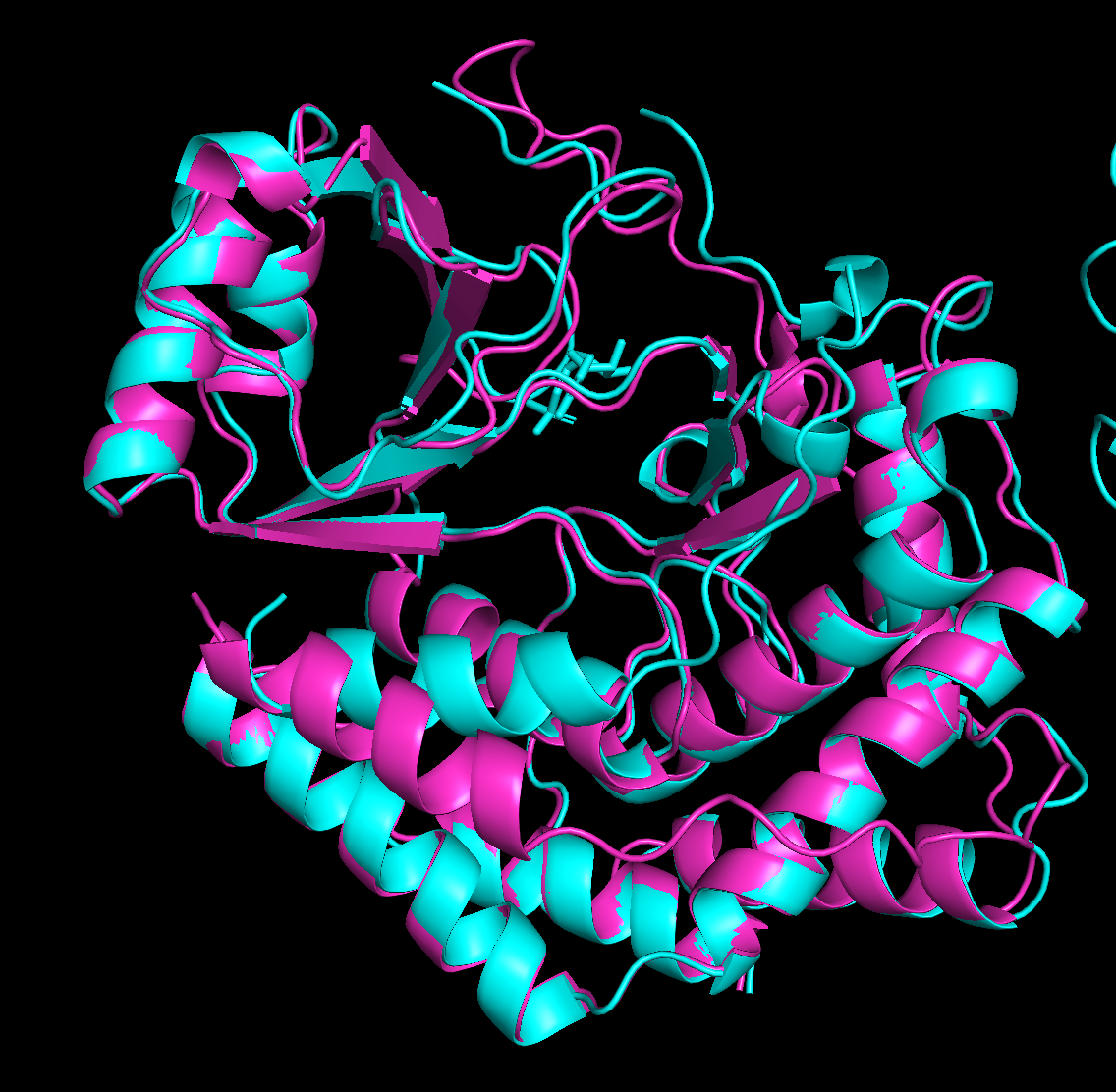
Mutation 2 - changed position 155, which is in the catalytic core. L (Leucine) to P (Proline)
Executive: RMSD = 0.844 (1933 to 1933 atoms)
Inverse Folding
There was an issue with the GPU in my laptop hence I directly did the inverse folding on https://huggingface.co/spaces/simonduerr/ProteinMPNN where for 1CJA i got
cleaned, score=1.6233, fixed_chains=[], designed_chains=[‘A’], model_name=vanilla—v_48_020 AGALWEIEKELFTKLPAPSSAINSHLQPAKPFKVDLSTAVSYNDIGDINWKNLQQFKGIERSEKGTEGLFFVETESGVFIVKRSTNIESETFCSLLCMRLGLHAPKVRVVSSNSEEGTNMLECLAAIDKSFRVITTLANQANILLMELVRGITLNKLTTTSAPEVLTKSTMQQLGSLMALDVIVNNSDRLPIAWTNEGNLDNIMLSERGATVVPIDSKIIPLDASHPHGERVRELLRTLIAHPGHESSQFHSIRDIITLYTGYDVGTEGSISMQEGFLATVRECASFDLDAFERELLSWQESLQKCHNLSISPQAIPFILRMLRIFH
T=0.1, sample=0, score=0.8496, seq_recovery=0.4373 MGRLAALRRELRAKLKPPSDVILPELRPPSPFSVDLSTATPYPDIDRIDWDDLSRFLGIERDPTGHGGDFLVKTKDGVFEVKVEPNPASYVFSTLLALHFGLHAPDVRLVRRDSPEGRALLAALAAIDTSGEFIPTAAPQPVLVLKELVLGIRLDEITAEKAPAILTPETLKQIGKLVAFCDIINDTSRLPLFSDSKGNLGNILLSVRGATVVPTDLDIHPLVGDTPIFEKIKNFLEKLRKDPSKCTPEFQKLGKLIAEATGYDFGEEGCLAIQEGYLELVDKVSKLDLEEFEKFLQEVVDALLRDAGLAIDPDTIPFILKMIKIFK
The sequence recovery of 43.7% means ProteinMPNN retained fewer than half the original residues while designing a backbone-compatible alternative sequence. This is expected for AFK given its unusual fold — the model is not constrained to reproduce the evolutionary sequence, only to find any sequence that satisfies the backbone geometry. Positions it did preserve are likely the most structurally or functionally constrained, such as the catalytic Asp109 and residues lining the nucleotide-binding cleft.
One notable difference: the inverse-folded sequence replaces many of AFK’s surface-exposed serine-rich regions (the native protein is 11.4% serine, unusually high) with a more typical distribution of charged and polar residues. This suggests the high serine content in the native protein is an evolutionary feature — possibly related to phosphorylation regulation of AFK itself — rather than a strict structural requirement for maintaining the fold.