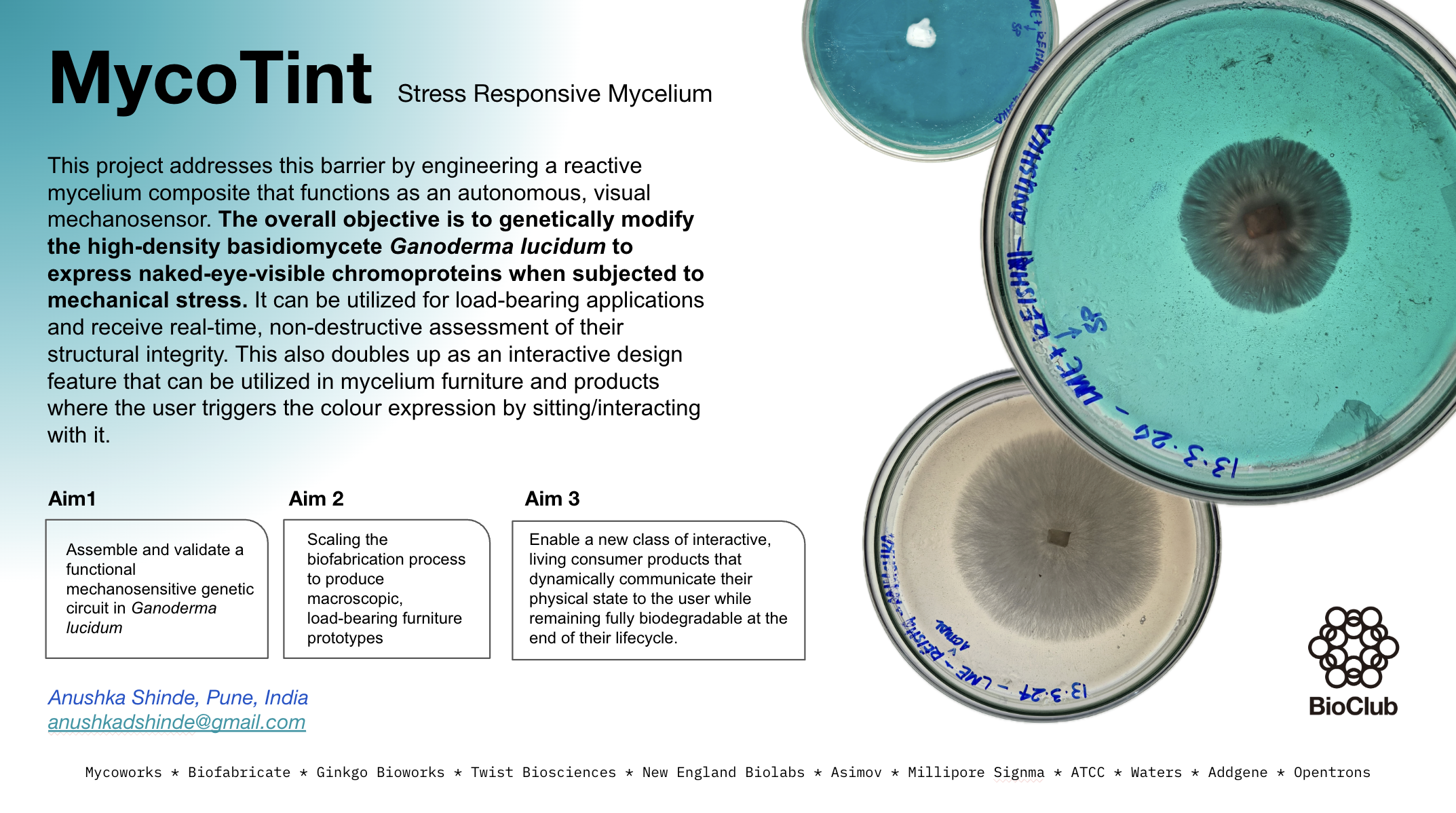
Stress-Chromatic Mycelium: Engineering Ganoderma lucidum to Visually Report Mechanical Stress via Indigoidine Biosynthesis I am carrying over some previous experience working with mycelium over here
Abstract Mycelium-based composites are emerging as sustainable alternatives to conventional materials in furniture and architecture, yet no current bio-based structural material can non-destructively report its internal stress history after fabrication. This project proposes engineering Ganoderma lucidum to function as a living mechanical stress reporter by coupling the cell wall integrity (CWI) signaling pathway to heterologous indigoidine biosynthesis. The central hypothesis is that placing the bpsA gene from Streptomyces lavendulae — encoding the non-ribosomal peptide synthetase (~141 kDa) responsible for indigoidine production — under the control of the experimentally validated GlSwi6B-responsive GL18134 promoter element will drive spatially localized blue pigmentation in hyphal zones experiencing elevated cell wall stress, including mechanical compression. Critically, the CWI pathway responds broadly to osmotic, heat, oxidative, and mechanical stress; demonstrating mechanical specificity over these other stressors is a central experimental objective, not an assumed outcome. Aim 1 establishes proof-of-concept by designing and ordering a genomic-integration construct from Twist Bioscience (pGl_GL18134_bpsA_integration, 8,375 bp), transforming G. lucidum, and measuring indigoidine production under compression versus osmotic and heat stress controls using the Spark Plate Reader. Aim 2 optimizes spatial resolution, stress specificity, and pigment retention through post-processing. Aim 3 envisions deployment of stress-chromatic mycelium panels as a scalable, non-destructive diagnostic platform for bio-based architecture. This work sits at the intersection of synthetic biology, materials science, and sustainable design, with direct relevance to partners including MycoWorks, BioFabricate, Ginkgo Bioworks, and Twist Bioscience.
from Part D — Group Brainstorm on Bacteriophage Engineering (Individual submission — solo student)
Project Goal The primary goal of this project is to increase the structural stability of the MS2 bacteriophage lysis protein (L-protein), with a secondary goal of reducing its dependency on the host chaperone DnaJ, while preserving its capacity to lyse bacterial cells through membrane pore formation.
Subsections of Projects
Individual Final Project
Stress-Chromatic Mycelium: Engineering Ganoderma lucidum to Visually Report Mechanical Stress via Indigoidine Biosynthesis
I am carrying over some previous experience working with mycelium over here
Abstract
Mycelium-based composites are emerging as sustainable alternatives to conventional materials in furniture and architecture, yet no current bio-based structural material can non-destructively report its internal stress history after fabrication. This project proposes engineering Ganoderma lucidum to function as a living mechanical stress reporter by coupling the cell wall integrity (CWI) signaling pathway to heterologous indigoidine biosynthesis. The central hypothesis is that placing the bpsA gene from Streptomyces lavendulae — encoding the non-ribosomal peptide synthetase (~141 kDa) responsible for indigoidine production — under the control of the experimentally validated GlSwi6B-responsive GL18134 promoter element will drive spatially localized blue pigmentation in hyphal zones experiencing elevated cell wall stress, including mechanical compression. Critically, the CWI pathway responds broadly to osmotic, heat, oxidative, and mechanical stress; demonstrating mechanical specificity over these other stressors is a central experimental objective, not an assumed outcome. Aim 1 establishes proof-of-concept by designing and ordering a genomic-integration construct from Twist Bioscience (pGl_GL18134_bpsA_integration, 8,375 bp), transforming G. lucidum, and measuring indigoidine production under compression versus osmotic and heat stress controls using the Spark Plate Reader. Aim 2 optimizes spatial resolution, stress specificity, and pigment retention through post-processing. Aim 3 envisions deployment of stress-chromatic mycelium panels as a scalable, non-destructive diagnostic platform for bio-based architecture. This work sits at the intersection of synthetic biology, materials science, and sustainable design, with direct relevance to partners including MycoWorks, BioFabricate, Ginkgo Bioworks, and Twist Bioscience.
Project Aims
Aim 1 — Experimental: Construct Design, Transformation, and Stress-Induced Indigoidine Detection in G. lucidum
Design and order a genomic-integration construct (pGl_GL18134_bpsA_integration, 8,375 bp) encoding bpsA under the experimentally validated GlSwi6B-responsive GL18134 chitin synthase promoter, flanked by homology arms targeting the G. lucidum leu2 locus. Order from Twist Bioscience. Transform into G. lucidum protoplasts via PEG-mediated transformation. Apply defined mechanical compression, osmotic stress (1 M sorbitol), and heat stress (37°C) as parallel conditions during growth, then measure indigoidine production by visual inspection and absorbance at 590 nm on the Spark Plate Reader. Determine whether mechanical compression produces a statistically distinct indigoidine signal relative to other stress types. This comparison is essential because the CWI pathway — and by extension the GL18134 promoter — responds broadly to any cell wall perturbation, not exclusively to mechanical load; mechanical specificity must be demonstrated empirically, not assumed.
Aim 2 — Medium-Term: Optimization of Stress Specificity, Spatial Resolution, and Post-Processing Retention
Systematically vary compression magnitude, duration, and growth stage to map indigoidine distribution relative to load-bearing zones. Perform ChIP-seq on G. lucidum to validate GlSwi6B binding site occupancy at the engineered promoter under mechanical stress, confirming that the CGCGAAA (SCB) motif is occupied by GlSwi6B in vivo under mechanical conditions and not equivalently under osmotic or heat stress. Validate that blue pigmentation is retained after heat-killing at 80°C and standard composite surface finishing. If mechanical specificity is insufficient, explore synthetic promoter engineering combining multiple SCB motifs with repressor elements active under non-mechanical stress conditions.
Aim 3 — Visionary: Stress-Chromatic Bio-Panels as a Permanent Structural Diagnostic Platform
Deploy engineered stress-chromatic G. lucidum composites as architectural and furniture panels that permanently encode their mechanical loading history in visible color gradients — enabling non-destructive inspection of internal stress distribution in bio-based building materials without instrumentation, at the point of use, anywhere in the world.
Background
Literature Context
Jones et al. (2020) conducted a critical review synthesizing published mechanical property data for mycelium composites derived from Ganoderma and related species, establishing that these materials achieve compressive strengths comparable to expanded polystyrene when grown on lignocellulosic substrates. Importantly, their review identifies the lack of integrated structural health monitoring as an unaddressed challenge in the field — no current mycelium composite product offers any method for post-fabrication stress mapping, representing a clear gap for safety-critical applications. Separately, Wehrse et al. (2018) demonstrated that the bpsA NRPS gene from Streptomyces lavendulae can be functionally expressed in the heterologous host Saccharomyces cerevisiae to produce indigoidine, confirming cross-kingdom portability of the pathway from bacteria to a eukaryotic fungal host. However, S. cerevisiae is an ascomycete yeast; expression in a basidiomycete such as G. lucidum has not been demonstrated, and this represents a key experimental risk that Aim 1 directly addresses.
Innovation
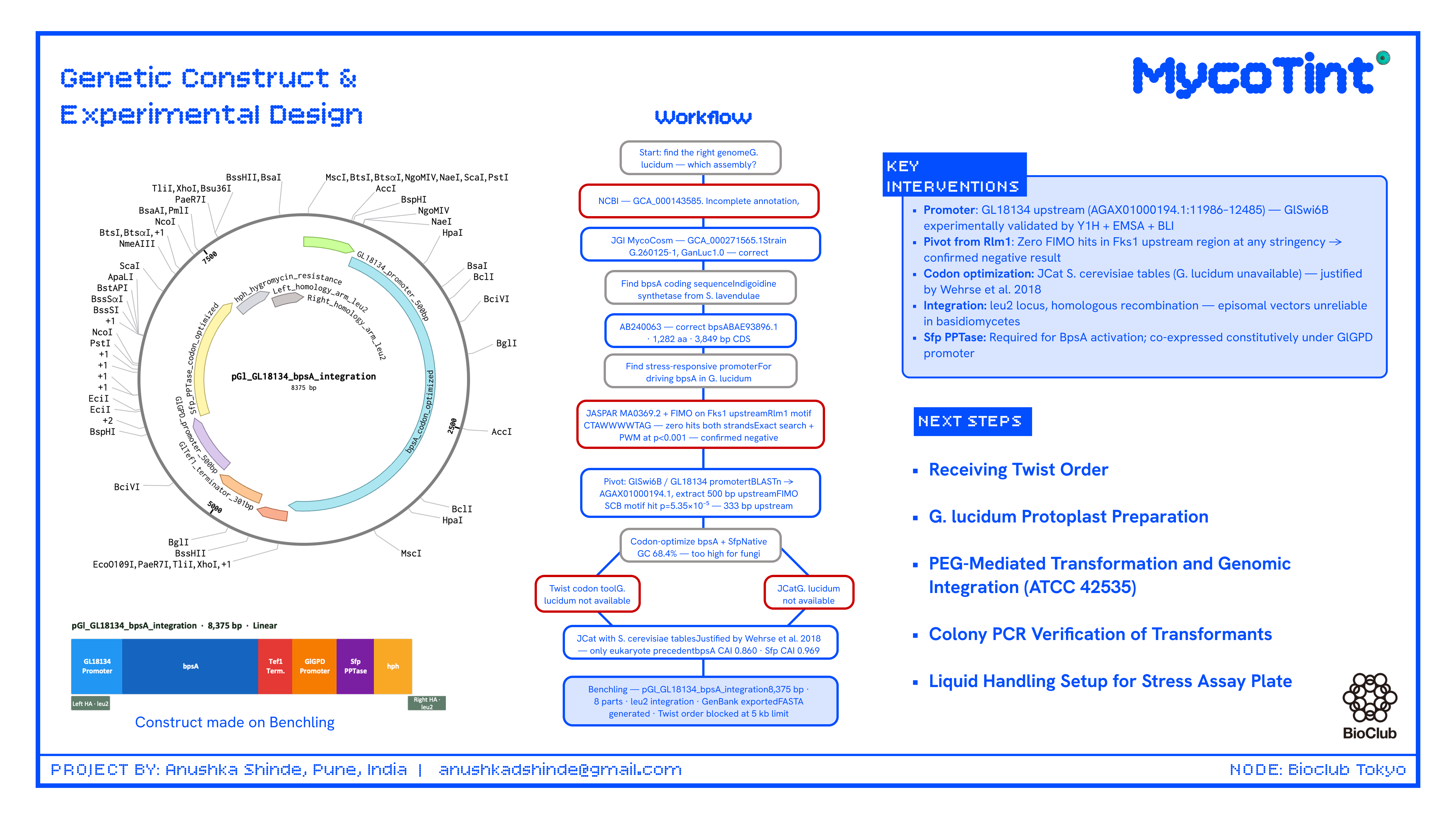
This project introduces the first genetically encoded mechanical stress reporter proposed for a mycelium composite organism, grounded in a promoter element with direct experimental validation in G. lucidum (GlSwi6B/GL18134 by Y1H, EMSA, and BLI assays) rather than cross-species inference. Unlike fluorescent reporters, indigoidine is a stable, non-fluorescent pigment visible to the naked eye in finished materials without UV illumination or instrumentation. The pivot from the originally proposed Rlm1/Fks1 promoter strategy — abandoned after empirical in silico confirmation of zero Rlm1 binding sites in the G. lucidum Fks1 upstream region — to the GlSwi6B/GL18134 system represents a meaningful improvement in biological grounding and constitutes itself a finding documented in this proposal.
Significance
Bio-based structural materials are projected to capture a significant share of the sustainable construction market within the next decade, yet no current product offers integrated structural health monitoring. A mycelium panel that visually records its own stress history could transform quality control in bio-fabrication, reducing the need for destructive testing. This technology is directly relevant to companies like MycoWorks and BioFabricate, who are scaling mycelium leather and composite products. Beyond furniture, stress-chromatic panels could find application in aerospace, packaging, and medical device contexts where load history matters. Finally, this project advances the broader synthetic biology principle that living materials can be programmed to sense and report their physical environment, opening a new design space at the intersection of biology and engineering.
Bioethical Considerations
G. lucidum is a non-pathogenic saprotrophic fungus with a long history of safe human use. Introducing a heterologous pigment biosynthesis gene does not confer pathogenicity or antibiotic resistance to the environment. However, the release of genetically modified fungal spores into open environments during manufacturing must be carefully controlled. Transparency with consumers about the use of engineered organisms in commercial products is an ethical obligation, and labeling standards should be developed in consultation with regulatory bodies before commercialization.
All experimental work will be conducted under BSL-1 containment with standard fungal biosafety protocols. The engineered strain will carry a genetic kill switch — a conditional auxotrophy — to prevent survival outside controlled growth conditions. Finished heat-killed composite panels contain no viable organisms and pose no biological risk to end users. Engagement with SecureDNA’s screening framework is recommended before any scale-up to verify that the bpsA sequence and construct design do not inadvertently encode sequences of concern. The sequence was screened via SecureDNA before Twist submission.
Experimental Design
Step 1 — Bioinformatic Identification of the G. lucidum Stress-Responsive Promoter Element
Purpose: Identify a validated stress-responsive cis-regulatory element to drive bpsA expression in G. lucidum.
What was attempted, what failed, and what worked:
Attempt 1 — Rlm1/Fks1 strategy (AI-suggested initial approach, FAILED):
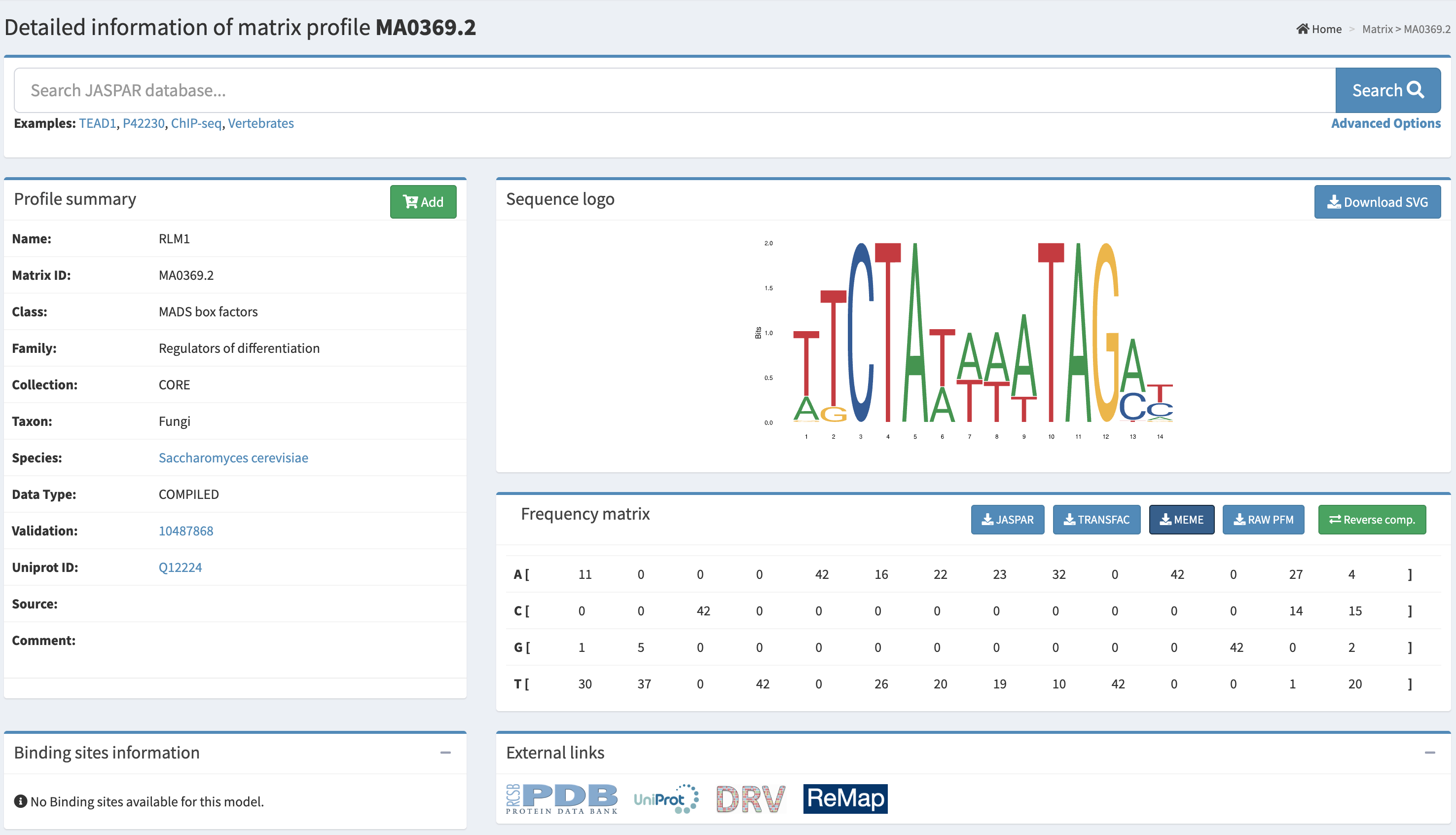
The original proposal, designed with AI assistance, suggested mining the G. lucidum genome for sequences matching the S. cerevisiae Rlm1 consensus motif (CTAWWWWTAG) upstream of the Fks1 glucan synthase homolog. The AI correctly identified this as a hypothesis-generating step with unvalidated cross-species conservation. The student executed this search against the correct genome assembly (GCA_000271565.1, strain G.260125-1, GanLuc1.0). The Fks1 homolog (XLV31108.3, 1,777 aa) was localized to scaffold AGAX01000145.1 at coordinates 154,315–157,050 (+ strand, 78.1% identity, 50.1% query coverage) by tBLASTn. A 1,500 bp upstream region (positions 152,815–154,315) was extracted and subjected to both exact string search for CTAWWWWTAG (including relaxed 3–5 W variants) and PWM-based FIMO scanning (JASPAR MA0369.2, Rlm1, p < 0.001 threshold). Both searches returned zero hits on both strands. This is an empirically confirmed negative result.
Attempt 2 — GlSwi6B/GL18134 strategy ( SUCCEEDED):
The GlSwi6B transcription factor — an APSES-family regulator in the G. lucidum CWI pathway, experimentally shown by Y1H, EMSA, and BLI assays to directly bind the GL18134 chitin synthase promoter — was selected as the new target. The GL18134 chitin synthase protein (QDK64614.1) was localized to scaffold AGAX01000194.1 at coordinates 12,486–13,325 (+ strand) by tBLASTn (99% query coverage, 86.43% identity, E = 0.0). A 500 bp upstream region (positions 11,986–12,485) was extracted and scanned by FIMO using the SCB motif (CGCGAAA). FIMO returned one significant hit at scaffold position 12,147–12,153 (minus strand, score = 13.46, p = 5.35×10⁻⁵), located approximately 333 bp upstream of the predicted gene start — a textbook promoter-proximal position. This result is consistent with published experimental validation of GlSwi6B binding at this locus.
Final promoter selected: GL18134 upstream region, AGAX01000194.1:11986–12485 (500 bp), containing one confirmed CGCGAAA SCB motif (p = 5.35×10⁻⁵).
Automation: NCBI tBLASTn, MEME Suite FIMO (v5.5.9), JASPAR MA0369.2. Timeline: Days 1–3.
Step 2 — Codon Optimization of bpsA and Sfp PPTase for G. lucidum Expression
Purpose: Maximize translation efficiency of the heterologous NRPS gene and its required activating PPTase.
What was attempted, what failed, and what worked:
Twist Bioscience codon optimization tool (FAILED):
The AI initially suggested using the Twist Bioscience integrated codon optimization tool with G. lucidum codon usage tables. The student attempted this on the Twist portal. G. lucidum was not available as a host organism in Twist’s tool. Basidiomycete codon tables are absent from the Twist optimization interface.
JCat with G. lucidum tables (FAILED):
JCat (https://www.jcat.de) was also checked for G. lucidum tables. G. lucidum is not available as a codon optimization host in JCat.
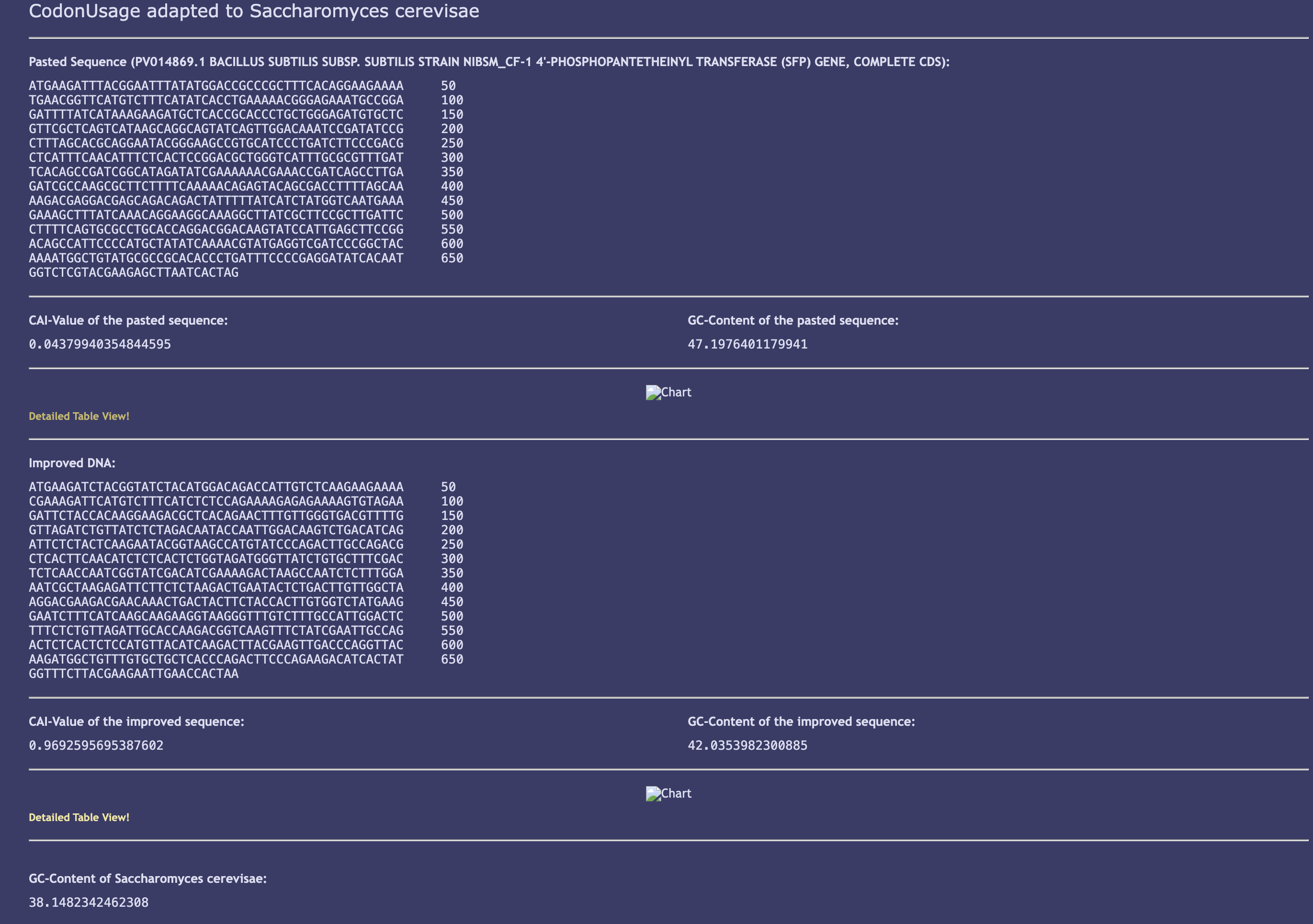
Executed approach — JCat with S. cerevisiae tables (SUCCEEDED, with documented limitation):
bpsA (GenBank AB240063, 3,849 bp CDS, protein BAE93896.1) and Sfp PPTase (GenBank PV014869.1, 678 bp) were both codon-optimized using JCat with S. cerevisiae codon usage tables. This substitution is scientifically justified: Wehrse et al. (2018) demonstrated functional BpsA expression in S. cerevisiae, making it the only published eukaryotic bpsA expression system and a validated reference point. The limitation (divergence from G. lucidum-optimal codons) is documented and flagged for iterative optimization in Aim 2.
bpsA optimization results:
Parameter
Native bpsA
Optimized (JCat, S. cerevisiae)
Status
Length
3,849 bp
3,849 bp
✅ Match
GC content
68.4%
44.2%
✅ Reduced toward fungal range
CAI value
~0.3 (estimated)
0.860
✅ Excellent
Start codon
ATG
ATG
✅
Stop codon
TGA
TAA
✅ (both valid)
Sfp PPTase optimization results:
Parameter
Native Sfp
Optimized (JCat, S. cerevisiae)
Status
Length
678 bp
678 bp
✅ Match
GC content
~52% (B. subtilis)
42.0%
✅ Shifted toward fungal range
CAI value
~0.6 (estimated)
0.969
✅ Exceptional
Start codon
ATG
ATG
✅
Stop codon
TAA
TAA
✅
Protein length
224 aa
224 aa
✅
Timeline: Days 3–4.
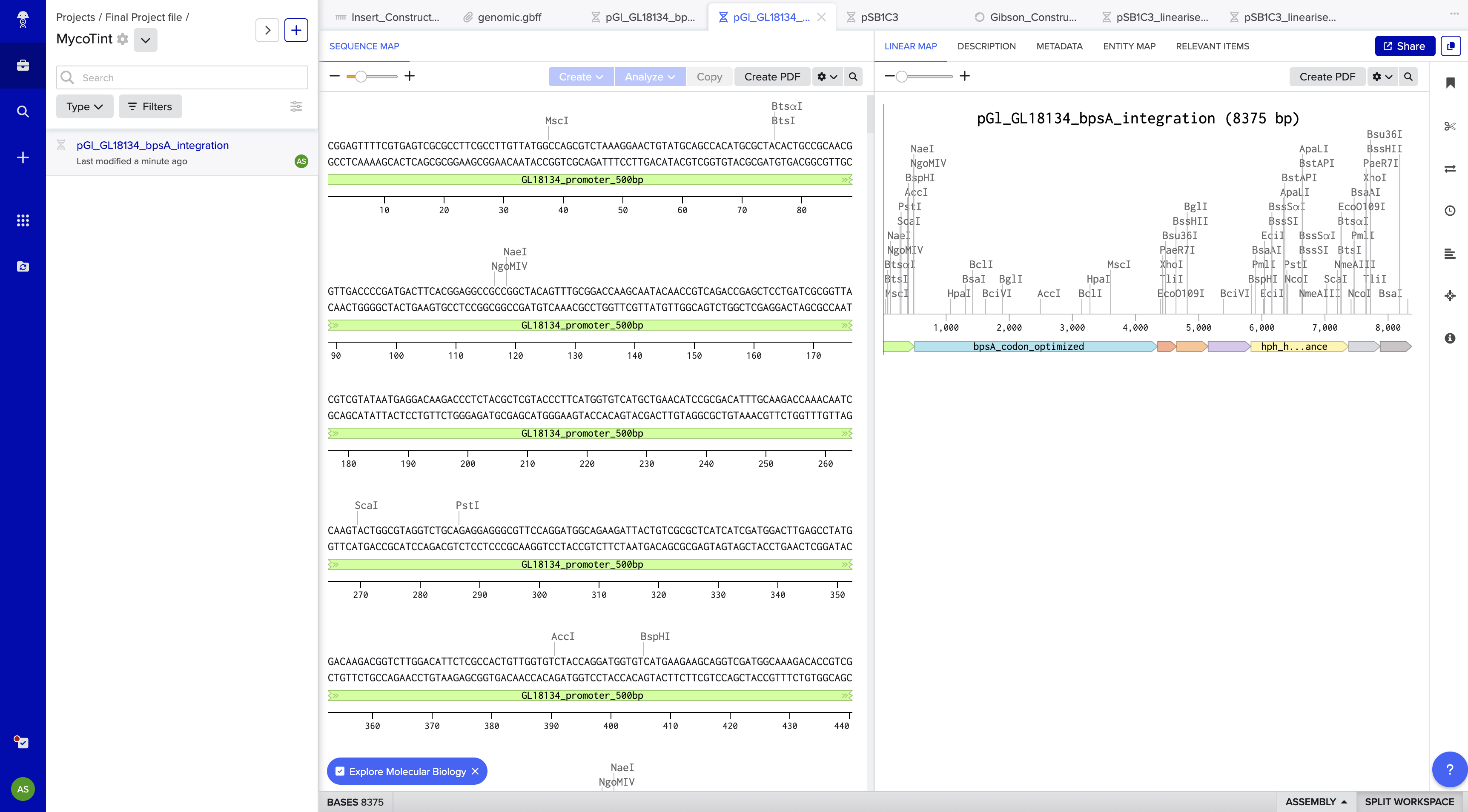
Step 3 — Construct Design and GenBank File Preparation in Benchling
Purpose: Assemble the complete integration construct map in Benchling for Twist ordering.
Method: The construct was designed in Benchling with the following finalized architecture, targeting the G. lucidum leu2 locus by homologous recombination. ARS/CEN episomal origins were explicitly excluded — episomal vectors are unreliable in basidiomycetes and integration is the established approach for G. lucidum transformation.
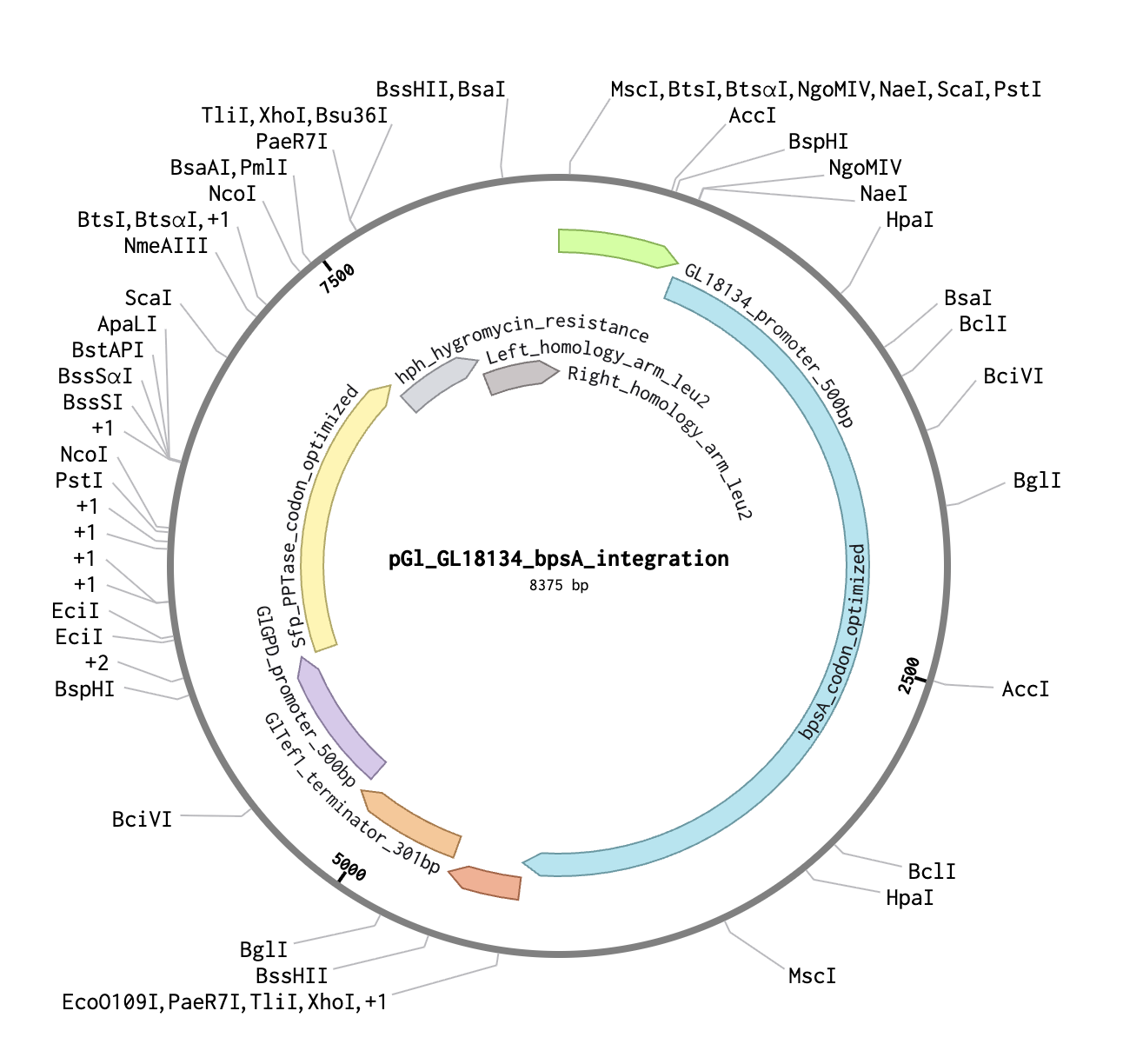
Construct name: pGl_GL18134_bpsA_integration Topology: Linear (integration construct) Total length: 8,375 bp File exported: MycoTint_-_all_DNA_RNA.gb (Benchling GenBank export)
Part map:
Part
Positions
Length
Genomic Source
GL18134 promoter
1–500
500 bp
AGAX01000194.1:11986–12485
bpsA codon-optimized
501–4,349
3,849 bp
AB240063, JCat optimized
GlTef1 terminator
4,350–4,650
301 bp
AGAX01000163.1:c770696–770396
GlGPD promoter
4,651–5,150
500 bp
AGAX01000011.1:c116036–115537
Sfp PPTase codon-optimized
5,151–5,828
678 bp
PV014869.1, JCat optimized
hph hygromycin resistance
5,829–7,375
1,547 bp
V01499.1 (aph(4))
Left homology arm (leu2)
7,376–7,875
500 bp
AGAX01000176.1:c717491–716992
Right homology arm (leu2)
7,876–8,375
500 bp
AGAX01000176.1:c715775–715276
Signal transduction logic (AI-clarified): Mechanical stress → cell wall deformation → CWI/MAPK cascade (GlSlt2) → GlSwi6B phosphorylation → GlSwi6B binds CGCGAAA motif in GL18134 promoter → bpsA transcription → BpsA + Sfp → indigoidine (blue pigment). Sfp PPTase is constitutively expressed under GlGPD to ensure BpsA is always post-translationally activated and competent.
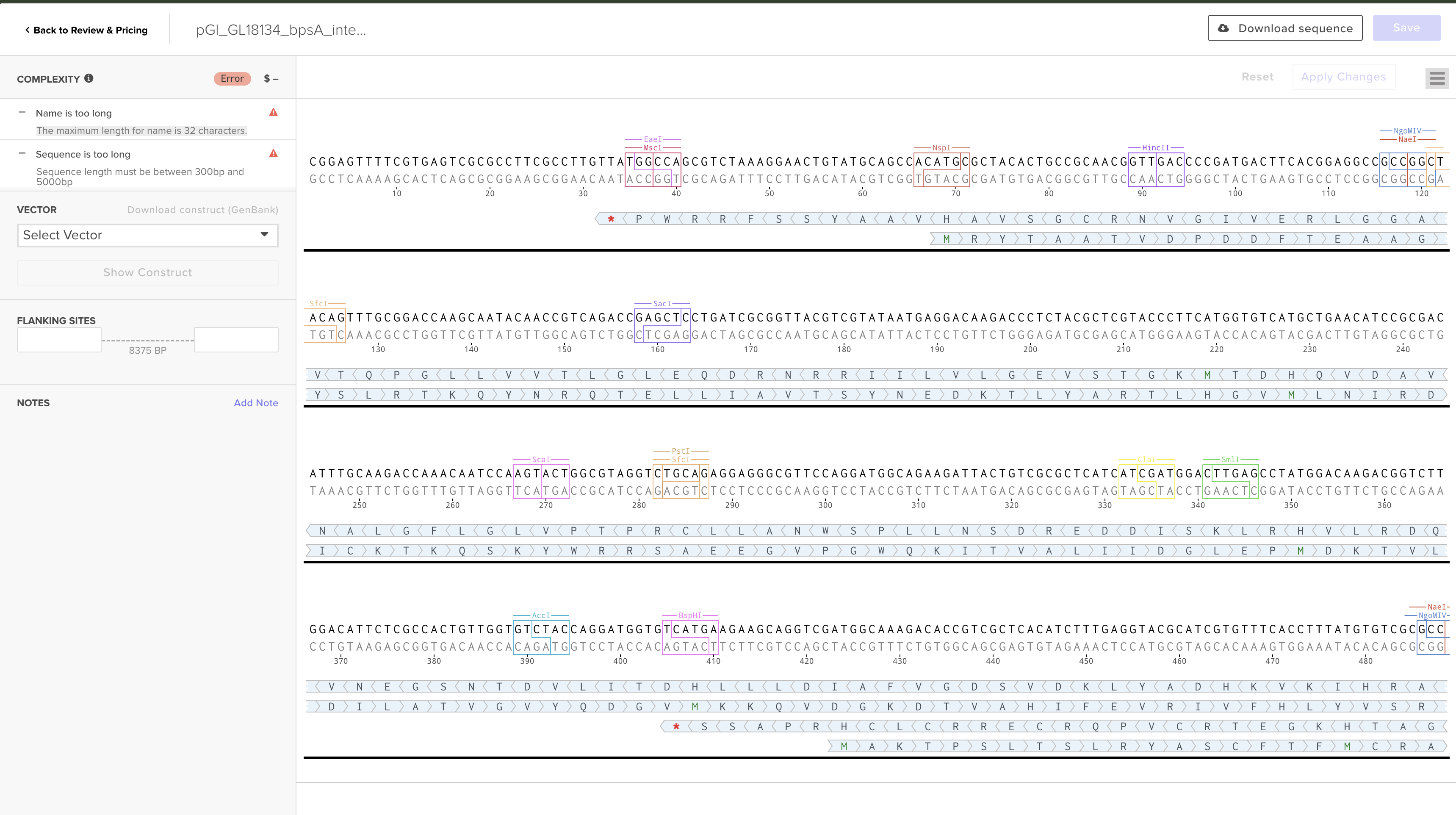
GenBank snippet (exported from Benchling):
LOCUS pGl_GL18134_bpsA_integr 8375 bp ds-DNA linear 10-MAY-2026
DEFINITION Stress-inducible indigoidine genomic integration construct for G. lucidum.
FEATURES Location/Qualifiers
Promoter 1..500
/label="GL18134_promoter_500bp"
/note="GlSwi6B-responsive; CGCGAAA SCB motif at pos 12147-12153
(p=5.35e-05); AGAX01000194.1:11986-12485"
CDS 501..4349
/label="bpsA_codon_optimized"
/note="AB240063, 3849 bp, JCat S. cerevisiae CAI=0.860, GC=44.2%"
Terminator 4350..4650
/label="GlTef1_terminator_301bp"
Promoter 4651..5150
/label="GlGPD_promoter_500bp"
/note="Constitutive; drives Sfp PPTase"
CDS 5151..5828
/label="Sfp_PPTase_codon_optimized"
/note="PV014869.1, 678 bp, JCat S. cerevisiae CAI=0.969, GC=42.0%"
CDS 5829..7375
/label="hph_hygromycin_resistance"
/note="V01499.1, aph(4), 50 µg/mL hygromycin B selection"
Misc feature 7376..7875
/label="Left_homology_arm_leu2"
/note="AGAX01000176.1:c717491-716992"
Misc feature 7876..8375
/label="Right_homology_arm_leu2"
/note="AGAX01000176.1:c715275-715276"
Timeline: Days 4–5.
Step 4 — Twist Bioscience Order Submission
Purpose: Obtain sequence-verified integration construct for transformation.
What was attempted, what failed, and current status:
Attempted — Clonal Gene product :
Uploaded the construct as a Clonal Gene order. Two errors were encountered:
Name too long: Twist maximum is 32 characters. The original name pGl_GL18134_bpsA_integration_8375bp exceeded this. Fixed: renamed to pGl_bpsA_leu2_integration (25 characters).
Sequence too long: Twist Clonal Gene (and Gene Fragment) products are limited to 0.3–5 kb. The construct is 8,375 bp. This error cannot be resolved within the current product tier.
Vector selected: pTwist Amp High Copy (preferred over pTwist Chlor High Copy — ampicillin is the standard for cloning workflows; construct already carries hygromycin resistance for fungal selection, so avoiding a third antibiotic keeps the system cleaner).
Note: The pTwist backbone is for E. coli propagation only. Before G. lucidum transformation, the plasmid is linearized by restriction digest within one homology arm; the backbone does not enter the fungus.
Current status: Order saved as draft on Twist portal. Blocked pending resolution of size limit.
Option B: Split into 3 overlapping ~2.8 kb Gibson assembly fragments with 40 bp overlaps; order as Gene Fragments; assemble via NEB HiFi Assembly
HTGAA Ordering Sheet fields prepared:
Field
Value
Order Type
Clonal Gene
Vector
pTwist Amp High Copy
Insertion Point
Default
Length
8,375 bp
Cost (estimate)
~$750
Order Date
May 11, 2026
Notes
Size exceeds 5 kb limit — contact Twist support or split into Gibson fragments
Expected Result: Sequence-verified construct delivered in 10–14 business days. Timeline: Days 5–6 (submission); Days 15–20 (receipt).
Step 5 — G. lucidum Protoplast Preparation
Purpose: Generate transformation-competent cells. Method: Grow G. lucidum (ATCC 42535) in PDB liquid culture for 5 days. Digest hyphal cell walls with Lysing Enzymes (Sigma L1412) in osmotic stabilizer (1.2 M sorbitol). Pellet protoplasts by centrifugation on HiG Centrifuge. Microplate: 96-v-eppendorf-951033502-deep (centrifugation steps). Timeline: Days 20–22.
Step 6 — PEG-Mediated Transformation and Genomic Integration
Purpose: Introduce the linearized Twist-synthesized construct into G. lucidum for integration at the leu2 locus. Method: Linearize the construct by restriction digest within one homology arm. Mix protoplasts with linearized DNA and PEG4000/CaCl₂ solution. Plate on regeneration medium with hygromycin B selection (50 µg/mL). Timeline: Days 22–23; colonies visible Days 30–35.
Step 7 — Colony PCR Verification of Transformants
Purpose: Confirm genomic integration at the leu2 locus. Method: Pick 24 hygromycin-resistant colonies. Extract genomic DNA. Run PCR with one primer outside the homology arm (genomic) and one primer inside bpsA — a band forms only if integration occurred at the correct locus. Automation: ATC Thermal Cycler (PCR); CFX Opus (qPCR, single-copy integration confirmation). Microplate: 96-Armadillo-PCR-AB2396X. Expected Result: ~800 bp junction band in correctly integrated transformants. Timeline: Days 35–37.
Step 8 — Liquid Handling Setup for Stress Assay Plate
Purpose: Prepare replicate cultures for multi-condition stress assay in a standardized format. Method: Use Tempest liquid handler to dispense 50 µL PDB medium per well. Inoculate with verified transformant spore suspension via Echo525. Automation: Echo525, Tempest. Microplate: 384 Greiner black-well clear-bottom. Timeline: Day 38.
Step 9 — Multi-Condition Stress Application
Purpose: Compare indigoidine induction across mechanical, osmotic, and heat stress to assess CWI pathway specificity. This is the critical experiment — mechanical specificity is not assumed, it must be empirically demonstrated. Method: Divide the 384-well plate into four quadrants: (A) mechanical compression (10 kPa via custom jig), (B) osmotic stress (1 M sorbitol), (C) heat stress (37°C), (D) no-stress negative control. Wild-type G. lucidum (no bpsA) is included in each quadrant as an additional negative control. Automation: Cytomat (28°C standard incubation); Inheco Plate Incubator (37°C heat stress wells). Timeline: Days 38–40.
Step 10 — Plate Sealing and Incubation
Purpose: Prevent evaporation and contamination during stress incubation. Method: Seal plates with Plateloc thermal sealer. Remove seal with XPeel before reading. Automation: Plateloc, XPeel. Timeline: Day 38 (seal), Day 40 (unseal).
Step 11 — Visual Inspection of Pigmentation
Purpose: Qualitative confirmation of stress-induced blue coloration across conditions. Method: Photograph plates under white light. Score wells for blue pigmentation (0 = none, 1 = faint, 2 = strong) across all stress conditions and wild-type controls. Expected Result: Blue coloration in stressed wells; no color in wild-type or no-stress controls. Timeline: Day 40.
Step 12 — Quantitative Absorbance Measurement at 590 nm
Purpose: Quantify indigoidine production per well across all stress conditions. Method: Read plates on Spark Plate Reader at 590 nm. Include standard curve of purified indigoidine (0–100 µM) in columns 1–2. Automation: Spark Plate Reader. Microplate: 384 Greiner black-well clear-bottom. Expected Result: Elevated A590 in stressed conditions vs. no-stress control; statistical comparison to assess whether mechanical compression is distinguishable from osmotic/heat stress.
Example Assay Plate Layout (384-well):
Columns 1–2: Indigoidine standard curve (0, 1, 5, 10, 25, 50, 100 µM)
Columns 3–8: Mechanical compression — engineered transformant (n=16/row)
Columns 9–14: Osmotic stress (1 M sorbitol) — engineered transformant
Columns 15–20: Heat stress (37°C) — engineered transformant
Columns 21–22: No-stress negative control — engineered transformant
Columns 23–24: Wild-type G. lucidum (no bpsA) — all stress conditions
Rows A–P: Biological replicates (16 replicates per condition)
Timeline: Day 40.
Step 13 — Data Analysis and Statistical Comparison
Purpose: Determine whether stress induction is statistically significant and whether mechanical stress is distinguishable from other CWI stressors. Method: Export Spark data to CSV. Calculate mean A590 ± SD per condition. Run one-way ANOVA with Tukey’s post-hoc test across all four stress conditions. Generate bar graphs with error bars. Success criterion (AI-suggested explicit threshold): Mechanical compression A590 must be ≥1.5-fold higher than both osmotic and heat stress conditions (p < 0.05 by Tukey’s) to claim mechanical enrichment, not merely pathway activation. Timeline: Day 41.
Step 14 — Heat-Killing and Pigment Retention Test
Purpose: Confirm indigoidine survives the composite finishing process. Method: Transfer mycelium from positive wells to agar slabs. Heat-kill at 80°C for 2 hours in Inheco Plate Incubator. Re-photograph and re-read A590 on Spark. Expected Result: >80% A590 signal retained post-heat-killing. Timeline: Days 42–43.
Step 15 — Macroscale Compression Jig Validation
Purpose: Confirm spatial stress mapping in a furniture-scale prototype. Method: Grow engineered G. lucidum on a 10×10 cm lignocellulosic substrate block. Apply point loads at defined positions using a mechanical press. Heat-kill, section, and photograph cross-sections to visualize blue pigmentation distribution relative to load points. Expected Result: Blue zones co-localize with applied load positions; spatial resolution assessed by image analysis. Timeline: Days 44–55.
Techniques, Tools, and Technology
Course Technique Checklist
DNA design and synthesis (Benchling + Twist Bioscience Clonal Gene)
Non-ribosomal peptide synthetases are large, modular enzyme complexes that synthesize bioactive small molecules independently of the ribosome. The bpsA gene (AB240063) encodes a single-module NRPS that condenses two glutamine molecules into indigoidine, a vivid blue pigment with peak absorbance at 590 nm; the protein is 1,282 amino acids (~141 kDa) and requires the 4’-phosphopantetheine cofactor for activity, added post-translationally by a dedicated phosphopantetheinyl transferase (PPTase). Expressing a multi-domain NRPS of this size in a heterologous basidiomycete host is non-trivial — codon optimization, correct folding, and Sfp PPTase co-expression are all critical prerequisites that must be addressed simultaneously. In this project, bpsA is co-expressed with codon-optimized Sfp PPTase (PV014869.1) under the constitutive GlGPD promoter, ensuring BpsA is post-translationally activated and competent for indigoidine synthesis; the closest published precedent for this cross-kingdom expression strategy is Wehrse et al. (2018), who demonstrated functional BpsA expression in S. cerevisiae, though basidiomycete expression remains to be established and constitutes a primary experimental risk of Aim 1.
Expanded Technique 2 — Stress-Responsive Promoter Engineering in Fungi
The cell wall integrity (CWI) pathway in fungi is a conserved MAPK signaling cascade that responds to a broad range of stresses — mechanical perturbation, osmotic shock, heat, oxidative stress, and cell-wall-damaging agents — and is not a dedicated mechanosensory system; this breadth of activation is the central design constraint of this project. The original design (Rlm1/Fks1 strategy) was abandoned after empirical in silico confirmation that the G. lucidum Fks1 upstream region contains zero Rlm1 binding sites detectable by exact motif search or PWM-based FIMO scanning at any stringency, representing an important negative result. The pivot to GlSwi6B/GL18134 is grounded in published experimental validation (Y1H, EMSA, BLI) of direct GlSwi6B binding at this locus, with a FIMO-confirmed CGCGAAA SCB motif (p = 5.35×10⁻⁵) in the 500 bp upstream region. Confirming that GlSwi6B occupies the engineered GL18134 promoter preferentially under mechanical stress — and not equally under all CWI-activating conditions — is the central experimental question of Aim 2, to be addressed by ChIP-seq and multi-condition reporter assays.
Project Validation
10a — Validation Choice
The chosen validation is DNA construct design and cell-free expression testing: the GL18134-bpsA integration plasmid is designed in Benchling, ordered from Twist Bioscience, and a parallel T7-driven TXTL validation construct is tested for functional bpsA expression using a cell-free transcription/translation (TXTL) system dispensed by an Opentrons OT-2 robot. This directly validates the most critical molecular assumption of the project — that the designed bpsA coding sequence and Sfp PPTase co-expression logic can drive indigoidine biosynthesis — before committing to the full fungal transformation workflow, which has a longer timeline and more experimental risk.
10b — Step-by-Step Validation Protocol
Open Benchling and create a new plasmid file named pGl_T7_bpsA_TXTL. Replace the GL18134 fungal promoter with a T7 promoter (the fungal promoter is not recognized by prokaryotic TXTL kits). This is a parallel validation construct; the GL18134-driven version remains the primary experimental construct for Aim 1.
Arrange construct elements: T7 promoter → codon-optimized bpsA → T7 terminator → codon-optimized Sfp PPTase under a second T7 promoter → T7 terminator. Annotate all features.
Export as GenBank (.gb) from Benchling.
Submit to Twist Bioscience as a Clonal Gene order alongside the primary integration construct.
Upon receipt, resuspend in nuclease-free water to 50 ng/µL.
Prepare TXTL master mix using NEB PURExpress or myTXTL kit.
Program Opentrons OT-2 to dispense 7 µL TXTL master mix + 1 µL plasmid (50 ng/µL) + 2 µL nuclease-free water per well into a 96-well plate. Include three conditions in triplicate: (a) T7-bpsA-Sfp plasmid, (b) GFP positive control (kit-supplied), (c) no-DNA negative control.
Seal plate and incubate at 29°C for 4–6 hours.
Visual inspection: photograph under white light — blue = active bpsA, green = GFP control, colorless = negative.
Read plate on Spark Plate Reader at 590 nm (indigoidine) and 488/510 nm (GFP).
Export CSV, calculate mean A590 ± SD per condition, run one-way ANOVA with Tukey’s post-hoc test.
Success criterion: ≥2-fold increase in A590 in bpsA wells vs. no-DNA negative control (p < 0.05).
10c — Techniques Used
This validation integrates four course techniques. DNA construct design is performed in Benchling using its sequence editor and annotation tools to produce an annotated plasmid map ready for synthesis. DNA synthesis via Twist Bioscience Clonal Gene ordering ensures the construct is sequence-verified before any expression testing, eliminating assembly errors as a confounding variable. Cell-free expression (TXTL) tests whether the bpsA coding sequence and Sfp PPTase co-expression logic drive indigoidine synthesis in a cell-free environment without requiring fungal transformation; the T7 promoter substitution is an explicit design choice to ensure compatibility with prokaryotic TXTL kits, and the result validates the coding sequence logic independently of the fungal promoter. Automated liquid handling via the Opentrons OT-2 ensures precise, reproducible dispensing across all wells, minimizing pipetting variability and enabling triplicate measurements for statistical confidence.
10d — Hypothetical Data
Simulated TXTL validation data:
Condition
Mean A590
SD
T7-bpsA-Sfp (indigoidine expression)
0.48
±0.04
GFP positive control
0.09
±0.02
No-DNA negative control
0.07
±0.01
The bpsA condition shows a ~6.9-fold increase in A590 over the no-DNA negative control. One-way ANOVA with Tukey’s post-hoc confirms the bpsA condition is significantly different from both controls (p < 0.001).
The large size of BpsA (~141 kDa) may strain the finite ATP and amino acid pools of a cell-free TXTL system, resulting in incomplete translation and low indigoidine yield; this can be mitigated by extending incubation to 6–8 hours and supplementing with an energy regeneration buffer. If PPTase is expressed at lower levels than BpsA, the proportion of active enzyme will be insufficient for detectable pigment; the fix is to increase the Sfp:bpsA plasmid molar ratio to 3:1 in the TXTL reaction. A critical limitation is that this TXTL validation tests only the bpsA coding sequence and PPTase co-expression logic — it does not validate the GL18134 promoter, which remains the key unvalidated assumption addressed only by the full Aim 1 fungal transformation experiment. The Twist order for the integration construct is currently blocked at 8,375 bp due to the 5 kb size limit of Clonal Gene and Gene Fragment products; resolution requires either contacting Twist support for a custom large construct order or splitting into three overlapping Gibson assembly fragments — this is a real timeline risk that must be resolved before Days 5–6.
HTGAA Slides for final presentation
Additional Information
Industry Partner Connections
Twist Bioscience — Clonal Gene synthesis of pGl_GL18134_bpsA_integration and pT7_bpsA_Sfp TXTL validation construct
MycoWorks / BioFabricate — direct application partners for stress-chromatic mycelium composites in leather and furniture
Ginkgo Bioworks — automation platform (Echo525, Tempest, Spark, Cytomat) for scaled transformation and screening workflows
SecureDNA — sequence screening of bpsA construct before synthesis submission
Jones, M., Mautner, A., Luenco, S., Bismarck, A., & John, S. (2020). Engineered mycelium composite structures from fungal biorefineries: A critical review. Materials & Design, 187, 108397. https://doi.org/10.1016/j.matdes.2019.108397
Wehrse, E., et al. (2018). Heterologous production of indigoidine in Saccharomyces cerevisiae by expression of the non-ribosomal peptide synthetase BpsA from Streptomyces lavendulae. Microbial Cell Factories, 17(1), 200. https://doi.org/10.1186/s12934-018-1048-1
Levin, D. E. (2011). Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics, 189(4), 1145–1175. https://doi.org/10.1534/genetics.111.128264
Xu, F., Gage, D., & Zhan, J. (2015). Efficient production of indigoidine in Escherichia coli. Journal of Industrial Microbiology & Biotechnology, 42(7), 1083–1090. https://doi.org/10.1007/s10295-015-1618-5
Chen, S., et al. (2012). Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nature Communications, 3, 913. https://doi.org/10.1038/ncomms1923
Zhang, Y., et al. (2017). The MAPK kinase GlSlt2 governs cell wall integrity in Ganoderma lucidum. Fungal Genetics and Biology, PMID 28435030.
Stamets, P., & Zwickey, H. (2014). Medicinal mushrooms: Ancient remedies meet modern science. Integrative Medicine, 13(1), 46–47.
Group Final Project
from Part D — Group Brainstorm on Bacteriophage Engineering
(Individual submission — solo student)
1. Project Goal
The primary goal of this project is to increase the structural stability of the MS2 bacteriophage lysis protein (L-protein), with a secondary goal of reducing its dependency on the host chaperone DnaJ, while preserving its capacity to lyse bacterial cells through membrane pore formation.
The MS2 L-protein is a 75-residue single-gene lysis toxin. Its architecture divides cleanly into two functional regions:
Soluble N-terminal domain (residues 1–40): intrinsically disordered, interacts with DnaJ, and is responsible for chaperone-dependent folding and activation
Transmembrane C-terminal domain (residues 41–75): forms a hydrophobic helix that inserts into the inner bacterial membrane, drives oligomerization into pore complexes, and executes lysis
A key E. coli resistance mechanism is a single point mutation in DnaJ (P330Q) that prevents it from interacting with the L-protein, blocking lysis. Engineering the L-protein to fold and function without DnaJ would directly circumvent this resistance route. Since the lytic activity resides in the transmembrane domain not the soluble domain that DnaJ binds. There is a credible path to separating folding assistance from lytic function through targeted mutagenesis of the N-terminal region.
The engineering strategy therefore focuses on three things simultaneously:
Stabilizing the soluble domain so it folds autonomously without DnaJ
Maintaining the transmembrane helix integrity for membrane insertion and pore formation
Preserving the conserved L48–S49 dipeptide motif and neighboring residues that are essential for function
2. Computational Tools and Approaches
A multi-step computational pipeline combining sequence analysis, protein language model mutagenesis, and structural prediction will be used.
2.1 BLAST — Homolog Discovery
BLAST is used first to find homologous lysis proteins from related bacteriophages across sequence databases.
Purpose:
Identify which positions across the protein are evolutionarily conserved vs. variable
Collect natural sequence diversity for multiple sequence alignment
Understand which parts of the L-protein have tolerated substitutions in nature, giving prior evidence that those positions can be mutated without destroying function
The BLAST results feed directly into the next step.
Homologous sequences retrieved from BLAST are aligned using Clustal Omega.
Purpose:
Map fully conserved positions (* in the alignment) — these must not be mutated
Identify partially conserved positions (:) where only similar-chemistry substitutions are tolerated
Confirm that the L48–S49 motif and surrounding residues are conserved, protecting them from mutagenesis
A key finding from the MSA of MS2 L-protein homologs is that all conserved positions cluster in the soluble domain (residues 1–40), specifically at positions 21, 25, 28–29, 33, 35–37, and 40. This is biologically meaningful these positions likely form the DnaJ-binding epitope and the structural core of the soluble domain. The transmembrane region (41–75) is less conserved, making it more accessible for hydrophobicity-enhancing substitutions.
2.3 ESM Protein Language Models — In Silico Deep Mutational Scan
The ESM2 protein language model is used to generate a log-likelihood ratio (LLR) score for every possible single-point substitution at every position in the L-protein.
Purpose:
Produce a mutation heatmap across the full 75-residue sequence
Identify substitutions the model predicts as tolerated or stabilizing (positive LLR) vs. harmful (negative LLR)
Guide rational mutation selection rather than random or intuition-based choices
Importantly, LLR scores reflect evolutionary plausibility and structural stability — they do not directly predict lytic function. Cross-referencing against the experimental lysis dataset (Chamakura et al., 2017) is therefore essential to exclude mutations that score well computationally but have been shown to abolish lysis in the wet lab.
2.4 ESMFold — Structure Prediction for Candidate Mutants
Promising mutations identified from the ESM scan are input into ESMFold to predict the 3D structure of the mutant L-protein monomer.
Purpose:
Assess predicted confidence (pLDDT) of the mutant structure vs. wild-type
Confirm the transmembrane helix remains intact in the TM-domain mutants
Identify mutations that significantly distort the backbone and discard them
A known limitation here is that ESMFold, like most structure predictors, performs less well on small intrinsically disordered proteins like the L-protein soluble domain. Low pLDDT scores in the N-terminal region may reflect genuine disorder rather than bad mutations — this ambiguity is a recognized pitfall of the approach.
2.5 AlphaFold Multimer — Oligomerization and DnaJ Interaction
AlphaFold Multimer is used for two separate runs per mutant:
Run A — 8-mer pore assembly: Eight copies of the mutant L-protein are submitted as separate chains to test whether the protein retains the capacity to oligomerize into the cylinder-like transmembrane pore that drives lysis.
Run B — DnaJ co-fold: The mutant L-protein is submitted alongside the DnaJ sequence to assess whether soluble-domain mutations reduce the predicted interaction interface between the two proteins.
A key insight from the reference implementation is that all five designed mutants, as well as a known experimentally validated lytic mutant (R30Q), returned very low pLDDT scores (<50) and low-confidence PAE plots for inter-chain contacts. This confirms a systematic limitation of AlphaFold for this class of small membrane-disrupting proteins — low confidence does not rule out functional lysis activity. All five mutants remain viable candidates for wet lab validation.
Based on the pipeline above, the following five mutations were selected:
#
Position
Wild-type AA
Mutant AA
Domain
LLR Score
Rationale
1
39
Y
L
Soluble
2.24
Highest LLR in soluble domain; non-conserved
2
9
S
Q
Soluble
2.01
High LLR; tests N-terminal stability
3
50
K
L
TM
2.56
Removes charged residue from TM helix; improves membrane insertion
4
53
N
L
TM
1.86
Removes polar residue from TM core
5
52
T
L
TM
1.81
High LLR; non-overlapping with coat/replicase genes
All five avoid the fully conserved positions (21, 25, 28–29, 33, 35–37, 40) and the three mutations that appeared on both the ESM heatmap and the experimental sheet with a lysis score of zero.
5. Expected Outcomes
The engineered variants are expected to produce:
Increased intrinsic structural stability in the soluble domain, particularly for Y39L and S9Q, reducing dependence on DnaJ for folding
Improved membrane insertion kinetics for K50L, N53L, and T52L, by replacing polar/charged residues with leucine in the hydrophobic TM helix, potentially producing faster or more efficient lysis
Retention of the pore-forming oligomeric assembly, since the transmembrane domain is not disrupted at the conserved functional core
A DnaJ-independent folding pathway in the best-case scenario for the soluble-domain mutants, enabling the phage to overcome the P330Q DnaJ resistance mutation in E. coli
6. Potential Pitfalls
6.1 Limited training data for phage proteins
ESM2 and ESMFold are trained predominantly on globular, well-characterized proteins. Short transmembrane phage toxins like the MS2 L-protein are under-represented in training data. This likely reduces prediction accuracy and may explain why even experimentally validated lytic mutants return low pLDDT and PAE scores from AlphaFold Multimer.
6.2 LLR scores predict stability, not function
The ESM heatmap captures evolutionary plausibility and structural fitness, not lytic activity. Three mutations that had high LLR scores were found to abolish lysis completely in the experimental dataset. This confirms that computational stability predictions must always be cross-referenced against functional data — a lesson that the broader field of computational protein design is still learning to internalize.
6.3 Risk of over-stabilization
Mutations that rigidify the soluble domain too much could prevent the conformational changes needed for membrane insertion or DnaJ dissociation. A protein that is too stable may be non-functional even if it folds correctly.
6.4 Poor annotation of amurin-class proteins
Single-gene lysis proteins (amurins) are a poorly annotated class. Homolog discovery via BLAST retrieves relatively few high-quality sequences, which limits the power of the MSA for identifying truly conserved vs. mutable positions.
6.5 Host protease sensitivity
New surface-exposed residues created by the soluble-domain mutations may accidentally introduce protease cleavage sites, reducing the effective concentration of functional L-protein inside infected bacteria and blunting lytic efficacy.
7. Literature Summaries
MS2 Lysis of E. coli Depends on Host Chaperone DnaJ (Chamakura et al., 2017)
This study demonstrates that the L-protein requires the host chaperone DnaJ for efficient lysis. A single missense mutation (P330Q) in DnaJ’s C-terminal domain blocks L-mediated lysis at 30°C, establishing the mechanistic basis of the resistance strategy this project aims to overcome. Genetic suppressor screening found that truncated L-proteins lacking the basic N-terminal domain can bypass DnaJ entirely, directly motivating the idea of engineering the soluble domain to achieve chaperone independence.
Mutational Analysis of the MS2 Lysis Protein L (Chamakura & Young, 2018)
Comprehensive random mutagenesis of all 75 residues showed that most loss-of-function mutations cluster in the C-terminal half, particularly around the conserved L48–S49 dipeptide. Many inactivating mutations were conservative substitutions that still allowed protein accumulation and membrane association, suggesting that lysis depends on specific protein–protein interactions rather than nonspecific membrane disruption. This explains why ESM structural scores are insufficient predictors of lytic activity — function is more sensitive than stability.
In Vitro Characterization of the Phage Lysis Protein MS2-L (Arulandu et al., 2023)
This study shows that MS2-L assembles into high-order oligomeric complexes (≥10 monomers) after insertion into lipid nanodiscs, driven primarily by the transmembrane domain. DnaJ interacts with the N-terminal domain but is not required for membrane insertion or oligomerization itself, suggesting its role is primarily as a folding or stability partner. This supports the feasibility of engineering DnaJ-independent variants — if the TM domain can self-insert and oligomerize, then eliminating DnaJ dependence through N-terminal modifications should not impair pore formation.
Phage Therapy: From Biological Mechanisms to Future Directions (Gordillo Altamirano & Barr, 2023)
This review surveys therapeutic phage applications and engineering strategies. It highlights that phage resistance — including via host factor mutations — remains a central challenge, and that engineered phages with modified lysis proteins represent a promising avenue for overcoming bacterial adaptation. The L-protein engineering effort directly addresses one of the most common and fastest-arising resistance mechanisms identified in clinical phage therapy.
8. Future Wet Lab Validation Steps
If promising computational mutants are identified, the following experimental steps would be required before drawing biological conclusions:
Chemical synthesis of the mutant L-protein gene via Twist Bioscience
Cloning into an expression plasmid using Gibson Assembly
Expression in wild-type and DnaJ-mutant (P330Q) E. coli strains
Plaque assays to measure lysis activity and compare to wild-type L-protein
Western blot to confirm protein accumulation levels are not affected by the mutations
Thermal shift assays (DSF) to directly measure whether the soluble-domain mutants show higher melting temperatures, confirming computational stability predictions